

Abstract
Ferroplasma acidiphilum strain Y (DSM 12658), a ferrous iron-oxidizing, acidophilic and mesophilic archaeon, was found to produce a membrane-bound α-glucosidase (αGluFa) showing no significant similarity to any of the known glycoside hydrolases classified in different families and having an unusual catalytic site consisting of a threonine and a histidine residue. The highest α-glucosidase activity was found at low pH, 2.4–3.5, and the substrate preference order was: sucrose>maltose>maltotriose ≫maltotetraose≫malto-oligosaccharides from maltopentaose to maltoheptaose⋙soluble starch (kcat/Km was 293.0, 197.0, 18.8, 0.3 and 0.02 s−1·mM−1 respectively). The enzyme was able to transfer glucosyl groups from maltose as donor, to produce exclusively maltotriose (up to 300 g/l). Chemical modification and electrospray ionization MS analysis of 5-fluoro-α-D-glucopyranosyl-enzyme derivatives, coupled with site-directed mutagenesis, strongly suggested that the putative catalytic nucleophile in this enzyme is Thr212. Iron was found to be essential for enzyme activity and integrity, and His390 was shown to be essential for iron binding. These results suggest that the metalloenzyme αGluFa is a new member of the glycosyl hydrolase family that uses a novel mechanism for sugar glycosylation and/or transglycosylation.
Keywords: α-glucosidase, catalytic nucleophile, Ferroplasma acidiphilum, glucosyl transferase, glycosyl hydrolase, transglycosylation
Abbreviations: iPr2P-F, di-isopropyl fluorophosphate (‘DFP’); EDC, 1-ethyl-3-[3-(dimethylamino)propyl]carbodi-imide; ESI-MS, electrospray ionization MS; 5FαGlcF, 5-fluoro-α-D-glucopyranosyl fluoride; GH, glycoside hydrolase; ICP-MS, inductively coupled plasma MS; IPTG, isopropyl β-D-thiogalactoside; LB, Luria–Bertani; PCMPS, p-chloromercuriphenylsulphonic acid; TNM, tetranitromethane; αGluFa, α-glucosidase from Ferroplasma acidiphilum strain YT
INTRODUCTION
Extremophilic microorganisms capable of thriving in harsh environments are widely distributed in Nature and have become the subject of intense investigation in recent years [1–7]. They produce unique stress-tolerant enzymes, extremozymes, involved in metabolic and cellular adaptation to the prevailing extreme environmental conditions. These tolerances and preferences (e.g. thermophily, psychrophily, acidophily, alkaliphily and halophily) confer upon extremozymes particular advantages for many industrial applications [8,9]. We have recently reported the isolation and characterization of Ferroplasma acidiphilum strain Y, an archaeon that grows under extremely acidic conditions (pH range of growth 1.3–2.2), oxidizes ferrous iron as its sole energy source and fixes inorganic carbon as the sole source of carbon [10]. We have also demonstrated that five intracellular and membrane-bound enzymes cloned from F. acidiphilum strain Y had optimum pH values much lower than the mean intracellular pH value of 5.6 (O. V. Golyshina, P. N. Golyshin, K. N. Timmis and M. Ferrer, unpublished work).
The present study focuses on glycosidases, including amylases, α-glucosidases, glucoamylases, pullulanases and cyclodextrin glucosyltransferases, enzymes that catalyse the hydrolysis of glycosidic bonds via a general acid catalysis involving a proton donor and a nucleophile/base [11]. In all cases, the carboxylic side chains of glutamic and aspartic residues are involved in catalysis. α-Glucosidases (EC 3.2.1.20; α-D-glucoside glucohydrolases) catalyse the liberation of glucose from non-reducing ends of short oligosaccharide substrates [12]. Some α-glucosidases preferentially hydrolyse α-linked di-, oligo- and/or polyglucans, while others prefer heterogeneous substrates such as sucrose and aryl glucosides [13]. They also mediate transglycosylation reactions, activities (e.g. those from buckwheat [13], Aspergillus niger [14], Bacillus stearothermophilus or brewer's yeast [15]) that are exploited in biotechnology to produce food oligosaccharides [16,17] or to conjugate sugars with biologically useful materials [18]. In the present study, we describe a membrane-bound α-glucosidase from F. acidiphilum strain Y, which shows no significant similarity to other known glycoside hydrolases classified in different families and that, unusually, has a catalytic centre involving threonine and histidine residues.
MATERIALS AND METHODS
Full details of all experimental methods are given in the Supplementary Materials and methods section at http://www.BiochemJ.org/bj/391/bj3910269add.htm.
Materials and strains of microorganisms
F. acidiphilum strain Y (DSMZ 12658) and Escherichia coli strains (i) XL1-Blue MRF′ (Stratagene, La Jolla, CA, U.S.A.), for library construction and screening, (ii) XLOLR (Stratagene), for expression of the α-glucosidase from phagemids, and (iii) DH5α, for site-directed mutagenesis and expression of mutant enzymes (Invitrogen, Carlsbad, CA, U.S.A.), were maintained and cultivated, if not mentioned otherwise, according to the manufacturer's instructions and the standard methods described previously [10,19]. In some cases, additions of 1 g/l sucrose, maltose or glucose were also made to cultures of F. acidiphilum grown in the medium 9K. FαGlcF (5-fluoro-α-D-glucopyranosyl fluoride) was synthesized as described by McCarter and Withers [20]. DNA restriction and modification enzymes were from New England Biolabs (Beverly, MA, U.S.A.).
Cloning, expression of αgluFa from F. acidiphilum strain YT and purification of the recombinant protein
An expression library of the F. acidiphilum genome was established in the bacteriophage lambda ZAP vector using the ZAP Express kit (Stratagene), and the library was used to infect XL1-Blue MRF′ cells, which were plated in NZY soft agar containing 2% (w/v) sucrose and 10 μM FeCl2 over a bottom layer of NZY agar [19] also containing sucrose and FeCl2. The 22.5 cm×22.5 cm plates containing approx. 10000 phage clones were incubated overnight and then overlaid with 50 ml of iodine solution (Sigma). Positive clones exhibiting a violet halo were picked and purified by serial dilution. The pBKGluFa phagemid was generated from one of the selected phage colonies by the helper phage excision procedure (Stratagene) and transferred to E. coli XLOLR cells. The complete nucleotide sequence of the DNA fragment, coding for the αgluFa enzyme described in the present study has been deposited in DDBJ, EMBL, GenBank® and GSDB Nucleotide Sequence Databases under the accession number AJ717661.
For the expression of αgluFa, E. coli cells containing pBKGluFa were grown at 37 °C in LB (Luria–Bertani) medium containing 50 μg of kanamycin/ml and 10 μM FeCl2. When the absorbance A600 reached 1.0, IPTG (isopropyl β-D-thiogalactoside) was added to a final concentration of 1 mM to induce expression. Cells were harvested 3 h after induction, and resuspended in buffer A (10 mM sodium citrate buffer, pH 3.0) containing one protease inhibitor cocktail tablet (Roche) and 10 μg/ml DNase I grade II (Roche), incubated on ice for 30–45 min and then sonicated for a total of 4 min. The soluble fraction was separated from insoluble debris by centrifugation (10000 g, 30 min and 4 °C), dialysed overnight against buffer A, concentrated to 1 ml by ultrafiltration on a Centricon YM-10 membrane (Amicon, Millipore, Billerica, MA, U.S.A.), and the αGluFa (α-glucosidase from F. acidiphilum strain YT) was purified as follows. The sample was applied to a HiPrep 16/10 SP XL (Amersham Biosciences, Little Chalfont, U.K.) column, which was washed with buffer A and subsequently eluted with a linear gradient of NaCl (total volume, 200 ml; 0–0.2 M). Active fractions were pooled and dialysed against 50 mM sodium citrate (pH 3.0) and 1 M (NH4)2SO4, concentrated to 1 ml on a Centricon YM-10 membrane and filtered using a 0.22 μm filter. The αGluFa-containing fractions were loaded on to a Resource 15PHE hydrophobic chromatography column (PE 4.6/100) previously equilibrated with the same buffer. After washing with the equilibration buffer [50 mM sodium citrate, pH 3.0 and 1 M (NH4)2SO4], αGluFa was eluted with a linear gradient of (NH4)2SO4 (total volume 25 ml; 1.0–0 M). The eluted enzyme was dialysed against buffer A overnight, concentrated to 1 ml by ultrafiltration and applied on to a Superose 12 HR 10/30 gel-filtration column pre-equilibrated with 10 mM sodium citrate (pH 3.0) and 150 mM NaCl. Separation was performed at 4 °C at a flow rate of 0.5 ml/min. The purified recombinant α-glucosidase was dialysed against buffer A overnight and stored at −20 °C at a concentration of 10 mg/ml until use.
Hydrolytic assays
Unless otherwise indicated, the hydrolytic activity towards sucrose, starch, amylose, amylopectin, pullulan and dextrin was determined by measuring the release of reducing sugars from 1% (w/v) substrate solutions using the dinitrosalicylic acid method [21]; hydrolytic activity towards kejibiose, nigeriose, iso-maltose, iso-maltotriose, trehalose and malto-oligosaccharides from maltotetraose to maltoheptaose was measured by HPLC analysis (see below) in reaction mixtures containing the substrate (1%, w/v). Reactions were stopped by heating for 15 min at 80 °C. Activity towards p-nitrophenyl-α/β-D-glucopyranoside (2 mM) was measured spectrophotometrically in a UV/visible spectrophotometer by following the increase in absorbance at 346 nm (ε346 4800 M−1·cm−1). Unless otherwise indicated, hydrolytic activities were routinely measured by incubating the purified enzyme (5 μg/ml) with a substrate at various concentrations in 100 mM sodium citrate buffer (pH 3.0) at 50 °C for an assay time of 30 min. All values were determined in triplicate and corrected for autohydrolysis of the substrate. One enzyme unit was defined as the amount of enzyme liberating 1 μmol of glucose (or equivalent reducing sugar) or p-nitrophenol per min. The standard assay used in the present study was performed at 50 °C in 100 mM sodium citrate buffer (pH 3.0) and 1% sucrose as substrate.
Kinetic parameters (kcat and Km) were determined at 50 °C in 100 mM sodium citrate buffer (pH 3.0). Substrate concentration was varied in the range 0.1–20.0 mM and the activity was measured as described above. Kinetic parameters were calculated by fitting the initial rate values to the Hanes–Woolf transformation of the Michaelis–Menten equation.
Transglycosylation assay and HPLC conditions
The transglycosylation assay was carried out at 50 °C in 0.2 M sodium citrate buffer (pH 3.0) containing 5 μg of purified αGluFa/ml and 600 g/l maltose. Aliquots were taken at intervals for a 180 min period, heated at 80 °C for 15 min, diluted 1:5 (v/v) with water, centrifuged and then filtered using Ultrafree-MC filter (0.45 μm) devices (Millipore). Substrate–product analyses were carried out by HPLC using a 4.6 mm×250 mm Lichrospher-NH2 column (Merck, Darmstadt, Germany) and acetonitrile/water (75:25, v/v) as the mobile phase at 0.7 ml/min and a refractive index detector (Varian, Basel, Switzerland). The column was kept constant at 25 °C. Integration was carried out using the Millennium software (Waters).
Inactivation kinetics
The enzyme (final concentration, 0.1 mg/ml) was incubated with compounds to be tested as inhibitors over the concentration range 0–10.0 mM at 50 °C in 100 mM sodium citrate buffer (pH 3.0). Aliquots (50 μl) were withdrawn at time intervals, chilled on ice and analysed by the standard α-glucosidase assay. Residual activity was expressed as the percentage of the control value obtained without the addition of chemical. Values for the inactivation rate constants (ki) and the dissociation constants for the inactivators (Ki) were determined by fitting to the equation
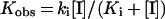 |
(1) |
where Kobs is the measured rate constant and [I] is the concentration of the inhibitor.
Chemical modification of αGluFa
α-Glucosidase was subjected to modification of specific amino acids with PCMPS (p-chloromercuriphenylsulphonic acid), iPr2P-F (di-isopropyl fluorophosphate; ‘DFP’), TNM (tetranitromethane) and EDC {1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide}, using standard methods [22–24]. Unless otherwise indicated, chemical modifications were routinely performed by incubating purified recombinant αGluFa (100 μg) either in 10 mM sodium citrate buffer (pH 3.0) (for PCMPS, iPr2P-F and TNM) or in 10 mM Mes buffer (pH 4.0) containing 10 mM KCl (for EDC), with the corresponding chemical. Activity measurements were done both immediately and after 30 min of incubation. In all cases, residual activity after modification was measured immediately, in three independent assays, and expressed as percentage of the control value obtained without the addition of the inhibitor. Full details are given in the Supplementary Materials and methods section (http://www.BiochemJ.org/bj/391/bj3910269add.htm).
Construction of αGluFa protein variants containing Glu, Asp and His mutations
Point mutations were introduced into the phagemid pBKGluFa using the QuikChange SDM procedure (Stratagene) with the oligonucleotide pairs synthesized at Sigma-Genosys (Pampisford, Cambs., U.K.). Eight Glu residues at positions 54, 258, 311, 426, 450, 452, 516 and 531 were replaced by Gln residues (αGluFa E/Q), 17 Asp residues at positions 40, 125, 172, 179, 185, 202, 250, 276, 334, 346, 391, 397, 428, 476, 502, 504 and 510 were replaced by Gln (αGluFa D/Q), and eight His residues at positions 9, 41, 47, 243, 275, 325, 377 and 390 were replaced by Ala (αGluFa H/A). The oligonucleotides used for mutagenesis are listed in Supplemental Table S1 at http://www.BiochemJ.org/bj/391/bj3910269add.htm. The resulting mutated plasmids were transformed into E. coli DH5α electrocompetent cells (Invitrogen), which were plated on to LB agar supplemented with 50 μg/ml kanamycin. Mutations were confirmed by DNA sequencing using the sequencing primers 5′-AACTCATTATATATATTGAATC-3′ (CH4) and 5′-ATTAGTGTTCCATGACGGTAAA-3′ (CH1158). Mutant proteins were purified using the same method as for the wild-type protein.
Detection of the catalytic nucleophile
To identify the catalytic nucleophile of F. acidiphilum α-glucosidase, the enzyme was labelled with 5FαGlcF and then subjected to proteolysis and ESI-MS (electrospray ionization MS) to identify labelled peptides, as described by Lee et al. [25], although with small modifications. Briefly, a stock solution of the enzyme (50 μl; 10 mg/ml) was incubated with 5FαGlcF (50 μl; 10 mM) at 37 °C for 5 min. The sample was diluted with 200 μl of 50 mM phosphate buffer (pH 2.0) and incubated with 15 μl of pepsin (1 mg/ml) for 15 min at room temperature (25 °C). The sample was then quickly frozen and analysed immediately upon thawing. A control sample not treated with 5FαGlcF was prepared according to the same procedure. Mass spectra were recorded on a VG AutoSpec spectrometer equipped with an ESI ion source. ESI-MS in the negative mode was performed in a QTOF (quadrupole/time-of-flight) mass spectrometer. Neon served as the collision gas for high-energy collision-induced dissociation. Peptides were separated by reversed-phase HPLC using a C18 column (4.6 mm×150 mm; Análisis Vínicos, Tomelloso, Spain) and a refractive index detector (Spectra-Physics, San Jose, CA, U.S.A.) and eluted with a gradient of 0–60% eluting solvent [0.045% trifluoroacetic acid/80% (v/v) acetonitrile in water] for 60 min at a flow rate of 100 μl/min. The temperature of the column was kept constant at 25 °C. Total ion chromatograms of the labelled and unlabelled enzyme digests were compared to find the fraction containing the labelled peptide fragments, which were collected and analysed by MS/MS fragmentation analysis.
CD and ICP-MS (inductively coupled plasma MS)
CD spectra of αGluFa at a concentration of 10 mg/ml and pH in the range 1.0–7.0 were measured with a Jasco J-720 spectropolarimeter equipped with a constant-temperature cell holder (40 °C), and 0.1 cm cell. Spectra were measured in the following buffers (100 mM): sodium citrate (pH 1.0–4.5), sodium acetate (pH 4.5–5.5), Mes (pH 5.5–7.0) and Hepes (pH 7.0). When necessary, the enzyme solution was incubated with 1 mM EDTA before absorption spectra were recorded. The metal ion content of αGluFa variants was determined using a PerkinElmer Life Sciences ICP mass spectrometer (model PE ELAN 6100 DRC). The metal content was determined by dilution of 50 μg of enzyme with 5 ml of 0.5% (v/v) HNO3 to digest the protein and release the metal ions, and this solution was used without any further manipulation.
RESULTS AND DISCUSSION
Cloning of F. acidiphilum αgluFa gene and sequence comparison
An F. acidiphilum genome expression library was generated in E. coli using the phage lambda ZAP vector (see the Materials and methods section). Activity-based screening of the library led to the detection of three different clones with α-glucosidase activity. One, designated αGluFa, was studied further after recloning its gene in the phagemid vector. Although the level of expression of the recombinant protein was approx. 1.4% (w/w) after induction with 1 mM IPTG, α-glucosidase activity was undetectable in freeze-dried cell extracts (<0.01 unit/mg, using sucrose or maltose as substrate). High enzymatic activities were, however, obtained by supplementation of the growth medium with Fe(II) at concentrations exceeding 10 μM and extensive dialysis of cell extracts against 10 mM sodium citrate buffer at pH 3.0. αGluFa was purified to homogeneity, as described in the Materials and methods section, from a cell-free extract of E. coli grown in LB medium supplemented with 100 μM FeCl2 and 50 μg of kanamycin/ml. We obtained 6.2 mg of pure enzyme from 1 litre of culture, which had specific activities of 99, 65, 42 and 0.15 units/mg of protein for the hydrolysis of maltose, sucrose, maltotriose and maltotetraose respectively at 50 °C and pH 3.0 in 10 mM sodium citrate buffer.
The αgluFa α-glucosidase gene derived from the phagemid clone pBKGluFa (GenBank® accession no. AJ717661) encodes a protein of 531 amino acids, having a theoretical molecular mass of 57300 Da and a deduced pI of 6.42. The sequence of the cloned fragment showed no convincing BlastP hits in the SwissProt/TrEMBL and NCBInr databases, and showed no significant similarity to sequences of known glycosyl hydrolases. Moreover, αGluFa does not show any significant homology to the members of the GH (glycoside hydrolase) family GH 13 [the α-amylase family, formed by a large group of homologous (β/α)8-barrel proteins that degrade starch, such as α-amylases, α-glucosidases, pullulanases and isoamylases] and GH 31 (comprising most α-glucosidases) of Coutinho and Henrissat [26] (Carbohydrate-Active Enzymes server: http://afmb.cnrs-mrs.fr/~cazy/CAZY/index.html).
Biochemical characterization
The αgluFa gene was expressed in E. coli XLOLR and the activity of its purified recombinant product with various substrates was determined. The molecular mass of αGluFa was estimated to be 57000 Da on the basis of relative mobility on the SDS and native polyacrylamide gels (results not shown).
Kinetic parameters
The kinetic constants (kcat, Km and kcat/Km) of the purified recombinant F. acidiphilum αGluFa for sugar-based substrates were calculated. As shown in Table 1, the substrate preferences of the αGluFa were: sucrose>maltose≫maltotriose≫p-nitrophenyl-α-D-glucopyranoside≫maltotetraose⋙malto-oligosaccharides from maltopentaose to maltoheptaose (kcat/Km values of 293.0, 197.0, 18.8, 0.3 and 0.02 s−1·mM−1 respectively). Other glycosides, such as soluble starch, pullulan, kejibiose, nigeriose, isomaltose, iso-maltotriose, trehalose and p-nitrophenyl-β-D-glucopyranoside, were not substrates of αGluFa.
Table 1. Kinetic parameters of α-glucosidase from F. acidiphilum for hydrolysis of sugar-based substrates.
Activity was measured at 50 °C in 100 mM sodium citrate buffer (pH 3.0).
| Substrate | kcat (s−1) | Km (mM) | kcat/Km (s−1·mM−1) |
|---|---|---|---|
| Sucrose [α-D-Glc-(1→2)-α-D-Fru] | 94.0±11.0 | 0.32±0.06 | 293.0±44.0 |
| Maltose [α-D-Glc-(1→4)-D-Glc] | 126.0±8.0 | 0.64±0.09 | 197.0±20.0 |
| Maltotriose [α-D-Glc-(1→4)-D-Glc-(1→4)-D-Glc] | 13.0±1.1 | 0.69±0.08 | 18.8±1.8 |
| Maltotetraose [α-D-Glc-(1→4)-D-Glc-(1→4)-D-Glc-(1→4)-D-Glc] | 2.8±0.1 | 9.30±0.40 | 0.3±0.1 |
| p-Nitrophenyl α-D-glucopyranoside | 22.5±3.5 | 5.00±0.10 | 4.5±0.6 |
Cation requirement
αGluFa was shown to contain 1.04±0.08 mol of iron/mol of αGluFa (ICP-MS analysis). Alternative bivalent cations Ni(II), Co(II), Mn(II), Ca(II) and Mg(II) were not detected in αGluFa and did not stimulate α-glucosidase activity. Dialysis of αGluFa against a solution containing 1 mM EDTA or EGTA to remove iron inactivated the enzyme (Figure 1A). The structural integrity of α-glucosidase was examined by CD spectroscopy. As shown in Figure 1(B), αGluFa exhibited minima at approx. 210–220 nm, characteristic of β-sheet structure. However, when αGluFa was depleted of iron through the action of a chelator, the CD spectrum changed to resemble those of denatured proteins (Figure 1B). This suggests that iron plays a significant functional role in the structural stability of the enzyme, its local or general conformation and thereby its activity.
Figure 1. Dependence of αGluFa activity on iron content.

(A) Influence of iron removal on αGluFa activity. Purified enzyme (50 μM in 100 mM citrate buffer, pH 3.0) was incubated with 1 mM EDTA. At different time intervals, two aliquots from the mixture were taken. One of these was used to determine the ability to hydrolyse sucrose using the standard methods, and the second one was used to determine the content of iron by ICP-MS. Hydrolytic activity was measured in 100 mM sodium citrate buffer (pH 3.0) using sucrose as substrate at 50 °C, and is expressed as a percentage of that found in the absence of EDTA. (B) CD of αGluFa. The secondary-structure CD was measured at wavelengths between 200 and 250 nm. The CD spectra were measured for recombinant (------) and EDTA-dialysed αGluFa (50 μM) in 100 mM sodium citrate buffer (pH 3.0) for 6, 11 and 24 h (corresponding to 0.76, 0.48 and 0.08 mol of iron/mol of αGluFa respectively). The metal ion content of αGluFa was determined using a PerkinElmer Life Sciences ICP-MS before CD was recorded.
Effect of pH and temperature
The optimum pH of the purified αGluFa measured with sucrose as substrate was 2.4–3.5 (Figure 2), although it retains >92% activity at pH 2.0 and >74% at pH 1.5. No measurable activity was detected at pH values above 7.0 and below 1.0. At 50 °C, α-glucosidase was highly stable at pH 2.4–3.5 (t1/2=34 min at pH 2.5; t1/2=9.5 min at pH 6.0). As shown in Figure 2, the molar ellipticity at 210 nm, and thus the helical structure content of αGluFa, was maximum at the pH where maximal activity was achieved; however, at pH values <2.0 or >5.0, the enzyme started to unfold. The highest specific activity of the enzyme was observed to be at 55–60 °C for sucrose hydrolysis, and its thermostability was <5 min at 65 °C, 32 min at 60 °C, 57 min at 55 °C, 190 min at 50 °C and >400 min below 50 °C. No measurable activity was detected above 85 °C.
Figure 2. Effect of pH on activity and peptide ellipticity of αGluFa.

The optimal pH for enzyme activity (□; right axis) was measured by the dinitrosalicylic acid method at 50 °C using sucrose as substrate. Secondary structure by far-UV CD was monitored by measuring the molar ellipticity at 210 nm (○; left axis). The following 100 mM buffers were used: citrate (pH 0.8–4.5), acetate (pH 4.5–5.5), Mes (pH 5.5–7.0) and Hepes (pH 7.0).
α-Glucosidase from F. acidiphilum transfers glucosyl groups from maltose to produce maltotriose selectively
The formation of a glycosidic bond (transglycosylation) and its hydrolysis are two variants of the same catalytic process [27]. This duality allows the use of sugar hydrolytic enzymes in synthetic processes [15]. Although previously characterized glycosidases (EC 3.2) are being applied for synthetic purposes, their applications are often limited by low yields and poor regioselectivity [28,29]. Thus most α-glucosidases transfer glucose units to the 6-OH group of the acceptor, yielding products such as isomaltose or panose, although transfer to other hydroxy groups (2-OH, 3-OH and 4-OH) also usually takes place [12–15]. As a result, a mixture of oligosaccharides consisting of α-1,3, α-1,4 and α-1,6 linkages is produced [30]. To assess whether αGluFa could be useful for the synthesis of oligosaccharides, its transglycosylation capability was investigated. We found that αGluFa catalyses the transglycosylation of glucosyl moieties of maltose, selectively producing the trisaccharide maltotriose (Figure 3; also see Supplemental Figure S2 at http://www.BiochemJ.org/bj/391/bj3910269add.htm). Also see Supplemental Table S2, which shows the composition of the reaction mixture over time. In the presence of 5 μg of αGluFa/ml and 600 g/l maltose, the transferase reaction yielded more than 300 g/l maltotriose after 3 h. The transglycosylation ratio was defined as the percentage of maltose that undergoes a transferase reaction rather than a hydrolysis, and was calculated on the basis of the molarities of glucose and maltotriose. As shown in Supplemental Table S2 (http://www.BiochemJ.org/bj/391/bj3910269add.htm), the values were very high (>70%). Interestingly, the transglycosylation ratio was not affected in the course of the reaction (which is normally the case when the concentration of maltose decreases). A selective production of high yields of maltotriose (up to 300 g/l) in the reaction at pH 3.0 with αGluFa suggests that F. acidiphilum α-glucosidase has restrictive binding properties. In this regard, Mala et al. [15] suggested that an enzyme having a low capacity to synthesize a tetrasaccharide will also have a low capacity to hydrolyse it, since the conditions to form enzyme–substrate complexes should be the same for both reaction directions. In the case of αGluFa, its hydrolytic activity decreased markedly from maltose to maltotriose and from maltotriose to maltotetraose (Table 1), which suggests limitations for binding of longer oligosaccharides, which in turn enable the selective synthesis of trisaccharides (i.e. maltotriose) even at low or moderate maltose concentrations. This fact highlights the transglycosylation capability of αGluFa even at low or moderate maltose concentrations.
Figure 3. HPLC chromatogram of the reaction mixture in the transglycosylation assay.

Conditions: 600 g/l maltose in 0.2 M sodium citrate buffer (pH 3.0) and 5 μg of purified αGluFa/ml, at 40 °C.
Inactivation by active-site inhibitors and chemical modification
To probe the residues essential for catalysis, αGluFa was incubated with various inhibitors and its hydrolytic activity was determined (Figure 4). We initially studied chemical modification of αGluFa by EDC/nucleophile modification. Since carbodi-imide reacts with the carboxylic groups of aspartic and glutamic residues and the phenolic groups of tyrosine residues, it leads to severe inactivation in glycosidases, α-amylases and cyclodextrin glucanotransferases [24]. Despite the fact that we employed a large excess of EDC compared with enzyme (3000:1) to achieve a high level of modification [24], no inactivation of αGluFa was observed (kcat/Km=285±41 and 293±44 s−1·mM−1 for the treated and untreated enzymes respectively). Nitration of αGluFa with the tyrosine-specific reagent TNM also had no effect on α-glucosidase activity (Figure 4A). These results suggest that αGluFa-mediated hydrolysis does not involve CO2H groups of Asp, Glu or Tyr residues.
Figure 4. Inhibition of αGluFa.

Purified α-glucosidase was incubated with increasing concentrations of (A) TNM (○) and (B) N-ethylmaleimide (■), iodoacetate (□), p-chloromercuribenzoate (▲), PCMPS (△), tosylphenylalanylchloromethane (▼), diethyl pyrocarbonate (▽), iPr2P-F (●), PMSF (○) and 5FαGlcF (◇). Upon incubation, the aliquots were withdrawn and analysed using the standard α-glucosidase assay (see the Materials and methods section). Residual activity was expressed as a percentage of the control value obtained without the addition of inhibitory chemicals.
αGluFa was also resistant to cysteine-specific reagents, such as N-ethylmaleimide, iodoacetate, p-chloromercuribenzoate and PCMPS (Figure 4B), and sequence analysis revealed that αGluFa does not possess any cysteine residue. αGluFa was also not affected by serine hydrolase inhibitors, such as iPr2P-F and PMSF. On the other hand, the enzyme was strongly inhibited by histidine-specific inhibitors, such as tosylphenylalanylchloromethane (‘TPCK’) and diethyl pyrocarbonate (Figure 4B), which caused a marked concentration-dependent decrease of enzyme activity with Ki=4.2±0.12 and 2.9±0.10 μM respectively at pH 3.0; this result strongly suggested that histidine residues are involved in the catalytic function of the enzyme [31–33]. αGluFa was also strongly inhibited by the reversible inhibitor 5FαGlcF, which inhibited immediately upon addition and maintained the same level of inhibition over time. A similar observation has also been reported for an α-glucosidase from A. niger [25]. An apparent Ki value of 3.1±0.10 μM was determined in a second set of experiments that addressed 5FαGlcF as a competitive inhibitor [25]. The presence of glucose and maltose, the end-products of the hydrolysis of sucrose, maltose and maltotriose, did not affect the hydrolytic activity of the enzyme (at least at concentrations ≤0.2 M).
Glu and Asp residues are not catalytic residues in αGluFa
αGluFa contains eight Glu and 17 Asp residues, one of which might in principle mediate sugar hydrolysis. To confirm that none of them is involved in catalysis, as suggested by the carbodi-imide modification, single mutants were constructed in which each of the glutamic and aspartic residues were separately replaced by Gln (E/Q and D/Q respectively). The mutated proteins were expressed in E. coli DH5α, purified, and the kinetic constants kcat/Km of these enzymes with sucrose as substrate were determined. As shown in Supplemental Figure S1 (http://www.BiochemJ.org/bj/391/bj3910269add.htm), similar hydrolytic patterns were observed for the wild-type and αGluFa E/Q and D/Q variants (kcat/Km values from 229±37 to 305±50 s−1·mM−1). We also assayed by HPLC the apparent specific activities of the mutant variants with maltose as substrate. As shown in Supplemental Figure S1 (http://www.BiochemJ.org/bj/391/bj3910269add.htm), the differences among variants and wild-type enzymes were more pronounced with maltose as substrate compared with sucrose as substrate, although none of the mutated proteins was inactive [minimum specific activity: 129 units/mg for D391Q (Asp391→Gln) mutant]. This suggests that neither glutamic nor aspartic residues are catalytic residues in αGluFa and, therefore, that its activesite structure differs substantially from known glycosyl hydrolases (proof awaits three-dimensional structure determination, which is currently in progress), whose sugar-binding sites involve the carboxylic side chains of glutamic and aspartic residues [11].
Identification of Thr212 as a catalytic nucleophile
To identify the catalytic nucleophile, 80 μg of αGluFa was incubated with the inhibitor 5FαGlcF. A sample of α-glucosidase either untreated (Figure 5A) or treated for 5 min with 5FαGlcF (1.6 mM; Figure 5B) was digested with pepsin at pH 2.0 and loaded on to a C18 HPLC column connected to an ESI mass spectrometer, and comparative mass spectra were obtained. ESI-MS analysis revealed the presence of a peptide of m/z 756.62 in the labelled α-glucosidase. The putative labelled peptide was isolated by reversed-phase HPLC and subjected to ESI-MS/MS fragmentation analysis (Figure 5C), which revealed a fragment of 575.6 Da, corresponding to the mass difference between the mass of the peptide, 756.62 Da, and the mass of the 5FαGlcF label, 181 Da. Additionally, eight prominent peaks of m/z 159.1, 273.9, 286.1, 387.2, 501.3, 554.9, 558.6 and 568.5 were detected (Figure 5C). Interpretation of the spectrum indicated that the labelled peptide corresponded to peptide sequence WVTNG, which corresponds to amino acids 210–214 of the protein. Of these amino acids, only threonine and asparagine residues are putative nucleophilic candidates. To identify the nucleophilic residue unambiguously, site-directed mutagenesis was used to replace Thr212 and Asn213 by Gln. Replacement of Thr212 by Gln (T212Q) resulted in a 600-fold reduction in activity (kcat/Km values decreased from 293 to 0.5 s−1·mM−1). Activity could be partially restored by the addition of an external nucleophile, i.e. formate (98 mM formate restored up to 90% original activity). No inactivation of αGluFa occurred through replacement of Asn213 by Gln (kcat/Km=293±44 and 270±41 s−1·mM−1 for the native and modified enzymes respectively). These results suggest that Thr212 is essential for α-glucosidase activity and is the nucleophile of the enzyme. Moreover, the far-UV spectra of the T212Q (Supplemental Figure S3 at http://www.BiochemJ.org/bj/391/bj3910269add.htm) and N213Q (results not shown) variants were very similar to that of the wild-type enzyme, suggesting that the mutations did not introduce significant changes in secondary structure in the enzyme.
Figure 5. Identification of catalytic residues by labelling with 5FαGlcF and MS/MS analysis.

Comparative mass spectra of (A) unlabelled and (B) labelled F. acidiphilum α-glucosidase. The arrow indicates the peptide unique to the peptic digest of labelled enzyme. Only the fragments of m/z ranging from 640 to 810 are shown. No differences were observed above m/z 810 and below m/z 640. (C) MS/MS spectrum of the 5FαGlcF labelled peptide (m/z 755.620) and an interpretation of the spectrum.
His390 is essential for iron binding
Treatment of αGluFa with histidine-specific reagents resulted in a substantial reduction of α-glucosidase activity. To determine which amino acid(s) is(are) important for α-glucosidase activity, we replaced all eight histidine residues in the protein, His9, His41, His47, His243, His275, His325, His377 and His390, by an alanine residue, through site-directed mutagenesis, and analysed the α-glucosidase activity, iron content and secondary structures of the variants produced (Table 2). Complete loss of activity was observed in the variant enzyme with His390 replaced by Ala. Mutations at His47, His243 and His325 only slightly affected the hydrolytic activity, whereas mutations at His9, His41, His275 and His377 had no effect. The iron content of the His390 variant was significantly lower than that of the wild-type enzyme, and mutations at His243 and His47 caused a partial loss of the iron. The lower iron content directly correlated with decreased α-glucosidase activity. Furthermore, the secondary structure of the His9, His41, His275 and His377 mutant proteins was very similar (according to their molar ellipticity) to that of the wild-type enzyme, whereas secondary structure of H390A (see also Supplemental Figure S3 at http://www.BiochemJ.org/bj/391/bj3910269add.htm) and, to a lesser extent, that of the H47A and H243A variants were significantly different.
Table 2. Catalytic specificity (kcat/Km), iron content and molar ellipticity of wild-type α-glucosidase and His→Ala mutants from F. acidiphilum.
| Mutant | kcat/Km (s−1·mM−1)* | Iron content (mol/mol) | Molar ellipticity at 210 nm (%) |
|---|---|---|---|
| Wild-type | 293.0±44.0 | 1.04±0.08 | 100 |
| H9A | 302.0±38.0 | 1.00±0.09 | 100 |
| H41A | 312.0±33.0 | 0.96±0.08 | 100 |
| H47A | 220.0±31.0 | 0.86±0.08 | 88 |
| H243A | 227.0±32.0 | 0.55±0.07 | 65 |
| H275A | 297.0±35.0 | 1.04±0.08 | 100 |
| H325A | 296.0±37.0 | 1.00±0.08 | 100 |
| H277A | 291.0±27.0 | 0.99±0.06 | 102 |
| H390A | 5.4±0.4 | 0.05±10−3 | 2 |
* Measured at 50 °C in 100 mM sodium citrate buffer (pH 3.0) using sucrose as substrate.
We further compared the secondary-structure content of single mutants T212G and H390A and double mutants T212G/H390A (all of them inactive) with that of the wild-type protein. As shown in Supplemental Figure S3 (http://www.BiochemJ.org/bj/391/bj3910269add.htm), only proteins containing mutations at His390 exhibited significant alterations in secondary structure. This demonstrated conclusively that iron elimination in His→Ala variants, rather than the amino acid mutation itself, is responsible for structural perturbation and loss of activity, whereas Thr212 is directly implicated in catalysis, being essential for α-glucosidase activity.
Taken together, these studies indicate that iron plays an essential role in the structural integrity of αGluFa, that His390 is the ligand for iron binding/co-ordination in the enzyme and that Thr212 is the nucleophile in its catalytic activity. This, in turn, suggests that αGluFa is a novel α-glucosidase with a novel catalytic mechanism for sugar glycosylation and transglycosylation in which the formation of a glycosyl-enzyme intermediate is initiated by nucleophilic attack on the carbonyl carbon of the sugar by the hydroxy group of the catalytic threonine residue, rather than by the general acid/base mechanism, mediated by glutamic and/or aspartic residue (as in all glycosidases) or tyrosine residue (in sialidases) [27,34]. The nucleophile water, or probably the iron-interacting water molecules at His390, then probably attack the glycosyl-enzyme intermediate to form the product, analogous to the zinc–water molecule interaction in metalloproteases [35,36]. Although the formation of such intermediates has been proposed previously for hen's-egg white lysozyme, several α-amylases, cyclodextrin glucanotransferases and B. subtilis xylanase, in all these cases the nucleophile and the acid/base catalysts were the carboxylic chains of glutamic and aspartic residues [37]. Moreover, although threonine has been found to be the active-site nucleophile in other hydrolases, such as glycosylasparaginase, dipeptidyl peptidase IV, γ-glutamyltranspeptidase and threonine phosphatases [38–44], in these systems the reactive nucleophile threonine requires activation via deprotonation by histidine or interaction with another threonine residue. Clearly, structural analysis of F. acidiphilum α-glucosidase is needed to confirm these conclusions, although, so far, we have not been able to obtain crystals that are stable within the pH range 2.4–3.5. Nevertheless, the present study opens up new perspectives for the investigation of structural adaptation of proteins to extreme conditions and novel structural features in the glycoside hydrolase superfamily.
Online data
Acknowledgments
M.F. thanks the European Commission for a Marie Curie postdoctoral fellowship and the Spanish Ministerio de Ciencia y Tecnología. This research was supported by European Community projects EVK3-2002-00077 ‘COMMODE’ and MERG-CT-2004-505242, Spanish CICYT (Comisión Interministerial de Ciencia y Tecnología) project BIO2004-03773-C00-01 and BMBF ‘GenoMik’ initiative. K.N.T. gratefully acknowledges the generous support provided by the Fonds der Chemischen Industrie. We also thank R. Getzlaff (GBF – German Research Centre for Biotechnology, Braunschweig, Germany) for protein sequence analyses and A. Ballesteros [Department of Biocatalysis, Institute of Catalysis, CSIC (Consejo Superior de Investigaciones Científicas), Madrid, Spain] for support and encouragement.
References
- 1.Eichler J. Facing extremes: archaeal surface-layer (glyco)proteins. Microbiology. 2003;149:3347–3351. doi: 10.1099/mic.0.26591-0. [DOI] [PubMed] [Google Scholar]
- 2.Johnson D. B., Hallberg K. B. The microbiology of acidic mine waters. Res. Microbiol. 2003;154:466–473. doi: 10.1016/S0923-2508(03)00114-1. [DOI] [PubMed] [Google Scholar]
- 3.Cavicchioli R., Siddiqui K. S., Andrews D., Sowers K. R. Low-temperature extremophiles and their applications. Curr. Opin. Biotechnol. 2002;13:253–261. doi: 10.1016/s0958-1669(02)00317-8. [DOI] [PubMed] [Google Scholar]
- 4.Ciaramella M., Pisani F. M., Rossi M. Molecular biology of extremophiles: recent progress on the hyperthermophilic archaeon Sulfolobus. Antonie Van Leeuwenhoek. 2002;81:85–97. doi: 10.1023/a:1020577510469. [DOI] [PubMed] [Google Scholar]
- 5.Thomas D. N., Dieckmann G. S. Antarctic Sea ice – a habitat for extremophiles. Science. 2002;295:641–644. doi: 10.1126/science.1063391. [DOI] [PubMed] [Google Scholar]
- 6.Rothschild L. J., Mancinelli R. L. Life in extreme environments. Nature (London) 2001;409:1092–1101. doi: 10.1038/35059215. [DOI] [PubMed] [Google Scholar]
- 7.Edwards K. J., Bond P. L., Gihring T. M., Banfield J. F. An archaeal iron-oxidizing extreme acidophile important in acid mine drainage. Science. 2000;287:1796–1799. doi: 10.1126/science.287.5459.1796. [DOI] [PubMed] [Google Scholar]
- 8.Demirijan D. C., Moris-Varas F., Cassidy C. S. Enzymes from extremophiles. Curr. Opin. Chem. Biol. 2001;5:144–151. doi: 10.1016/s1367-5931(00)00183-6. [DOI] [PubMed] [Google Scholar]
- 9.Van der Burg B. Extremophiles as a source for novel enzymes. Curr. Opin. Microbiol. 2003;6:213–218. doi: 10.1016/s1369-5274(03)00060-2. [DOI] [PubMed] [Google Scholar]
- 10.Golyshina O. V., Pivovarova T. A., Karavaiko G. I., Kondrateva T. F., Moore E. R., Abraham W.-R., Lunsdorf H., Timmis K. N., Yakimov M. M., Golyshin P. N. Ferroplasma acidiphilum gen. nov., sp. nov., an acidophilic, autotrophic, ferrous-iron-oxidizing, cell-wall-lacking, mesophilic member of the Ferroplasmaceae fam. nov., comprising a distinct lineage of the Archaea. Int. J. Syst. Evol. Microbiol. 2000;50:997–1006. doi: 10.1099/00207713-50-3-997. [DOI] [PubMed] [Google Scholar]
- 11.Sinnott M. L. Catalytic mechanisms of enzymic glycosyl transfer. Chem. Rev. 1990;96:1171–1202. [Google Scholar]
- 12.Kato N., Suyama S., Shirokane M., Kato M., Kobayashi T., Tsukagoshi N. Novel alpha-glucosidase from Aspergillus nidulans with strong transglycosylation activity. Appl. Environ. Microbiol. 2002;68:1250–1256. doi: 10.1128/AEM.68.3.1250-1256.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiba S. The Amylase Research Society of Japan. Handbook of Amylases and Related Enzymes. Oxford, U.K.: Pergamon Press; 1988. α-Glucosidases; pp. 104–105. [Google Scholar]
- 14.Duan K. J., Sheu D. C., Lin C. T. Transglucosylation of a fungal alpha-glucosidase. The enzyme properties and correlation of isomaltooligosaccharide production. Ann. N.Y. Acad. Sci. 1995;750:325–328. doi: 10.1111/j.1749-6632.1995.tb19974.x. [DOI] [PubMed] [Google Scholar]
- 15.Mala S., Dvorakova H., Hrabal R., Kralova B. Towards regioselective synthesis of oligosaccharides by use of alpha-glucosidases with different substrate specificity. Carbohydr. Res. 1999;322:209–218. doi: 10.1016/s0008-6215(99)00222-0. [DOI] [PubMed] [Google Scholar]
- 16.Buchholz K., Seibel J. Isomaltooligosaccharides. In: Eggleston G., Cote G. L., editors. Oligosaccharides in Food and Agriculture, ACS Symposium Series, vol. 849. Washington, DC: The American Chemical Society; 2003. pp. 63–74. [Google Scholar]
- 17.Crittenden R. G., Playne M. J. Production, properties and applications of food-grade oligosaccharides. Trends Food Sci. Technol. 1996;7:353–361. [Google Scholar]
- 18.Takenaka F., Uchiyama H. Synthesis of alpha-D-glucosylglycerol by alpha-glucosidase and some of its characteristics. Biosci. Biotechnol. Biochem. 2000;64:1821–1826. doi: 10.1271/bbb.64.1821. [DOI] [PubMed] [Google Scholar]
- 19.Sambrook J., Fritsch E. F., Maniatis T. 2nd edn. Plainview, NY: Cold Spring Harbor Laboratory Press; 1989. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 20.McCarter J. D., Withers S. G. 5-Fluoro glycosides: a new class of mechanism-based inhibitors of both α- and β-glucosidases. J. Am. Chem. Soc. 1996;118:241–242. [Google Scholar]
- 21.Sumner J. B., Howell S. F. A method for determination of invertase activity. J. Biol. Chem. 1935;108:51–54. [Google Scholar]
- 22.Villette J. R., Helbecque N., Aibani J. R., Sicard P. J., Bouquelet S. J.-L. Cyclomaltodextrin glucanotransferase from Bacillus circulans E 192: nitration with tetranitromethane. Biotechnol. Appl. Biochem. 1993;17:205–216. [PubMed] [Google Scholar]
- 23.Tenno M., Toba S., Kézdy F. J., Elhammet A. P., Kurosaka A. Identification of two cysteine residues involved in the binding of UDP-GalNAc to UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase 1 (GalNAc-T1) Eur. J. Biochem. 2002;269:4308–4316. doi: 10.1046/j.1432-1033.2002.03123.x. [DOI] [PubMed] [Google Scholar]
- 24.Alcalde M., Plou F. J., Pérez-Boada M., García-Arellano H., Valdés I., Méndez E., Ballesteros A. Chemical modification of carboxylic residues in a cyclodextrin glucanotransferase and its implication in the hydrolysis/transglycosylation ratio of the α-amylase family. J. Mol. Catal. B. 2003;26:57–67. [Google Scholar]
- 25.Lee S. S., He S., Withers S. G. Identification of the catalytic nucleophile of the family 31 alpha-glucosidase from Aspergillus niger via trapping of a 5-fluoroglycosyl-enzyme intermediate. Biochem. J. 2001;359:381–386. doi: 10.1042/0264-6021:3590381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coutinho P. M., Henrissat B. Carbohydrate-active enzymes: an integrated database approach. In: Gilbert H. J., Davies G., Henrissat B., Svensson B., editors. Recent Advances in Carbohydrate Bioengineering. Cambridge, U.K.: The Royal Society of Chemistry; 1999. pp. 3–12. [Google Scholar]
- 27.Liu W., Madsen N. B., Braun C., Withers S. G. Reassessment of the catalytic mechanism of glycogen debranching enzyme. Biochemistry. 1991;30:1419–1424. doi: 10.1021/bi00219a036. [DOI] [PubMed] [Google Scholar]
- 28.Plou F. J., Martín M. T., Gómez de Segura A., Alcalde M., Ballesteros A. Glucosyltransferases acting on starch or sucrose for the synthesis of oligosaccharides. Can. J. Chem. 2002;80:743–752. [Google Scholar]
- 29.Monsan P., Paul F. Enzymatic synthesis of oligosaccharides. FEMS Microbiol. Rev. 1995;16:187–192. [Google Scholar]
- 30.Okada M., Nakayama T., Noguchi A., Yano M., Hemmi H., Nishino T., Ueda T. Site-specific mutagenesis at positions 272 and 273 of the Bacillus sp. SAM1606 α-glucosidase to screen mutants with altered specificity for oligosaccharide production by transglucosylation. J. Mol. Catal. B Enzym. 2002;16:265–274. [Google Scholar]
- 31.Shaw E. Selective chemical modification of proteins. Physiol. Rev. 1970;50:244–296. doi: 10.1152/physrev.1970.50.2.244. [DOI] [PubMed] [Google Scholar]
- 32.Miles E. Modification of histidyl residues in proteins by diethylpyrocarbonate. Methods Enzymol. 1977;47:431–442. doi: 10.1016/0076-6879(77)47043-5. [DOI] [PubMed] [Google Scholar]
- 33.Powers J. Reaction of serine proteases with halomethyl ketones. Methods Enzymol. 1997;46:197–208. doi: 10.1016/s0076-6879(77)46020-8. [DOI] [PubMed] [Google Scholar]
- 34.Chavas L. M., Tringali C., Fusi P., Venerando B., Tettamanti G., Kato R., Monti E., Wakatsuki S. Crystal structure of the human cytosolic sialidase Neu2. Evidence for the dynamic nature of substrate recognition. J. Biol. Chem. 2005;280:469–475. doi: 10.1074/jbc.M411506200. [DOI] [PubMed] [Google Scholar]
- 35.Odintsov S. G., Sabala I., Marcyjaniak M., Bochtler M. Latent LytM at 1.3A resolution. J. Mol. Biol. 2004;335:775–785. doi: 10.1016/j.jmb.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 36.Barth A., Frost K., Wahab M., Brandt W., Schädler H. D., Franke R. Classification of serine proteases derived from steric comparisons of their active sites, part II: ‘Ser, His, Asp arrangements in proteolytic and nonproteolytic proteins’. Drug Des. Discov. 1994;12:89–111. [PubMed] [Google Scholar]
- 37.Withers S. G., Aebersold R. Approaches to labeling and identification of active site residues in glycosidases. Protein Sci. 1995;4:361–372. doi: 10.1002/pro.5560040302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guan C., Liu Y., Shao Y., Cui T., Liao W., Ewel A., Whitaker R., Paulus H. Characterization and functional analysis of the cis-autoproteolysis active center of glycosylasparaginase. J. Biol. Chem. 1998;273:9695–9702. doi: 10.1074/jbc.273.16.9695. [DOI] [PubMed] [Google Scholar]
- 39.Brannigan J. A., Dogson G., Duggledy H. J., Moody P. C. E., Smith J. L., Tomchick D. R., Murzin A. G. Protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature (London) 1995;378:416–419. doi: 10.1038/378416a0. [DOI] [PubMed] [Google Scholar]
- 40.Bjelke J. R., Christensen J., Branner S., Wagtmann N., Olsen C., Kranstrup A. B., Rasmussen H. B. Tyrosine 547 constitutes an essential part of the catalytic mechanism of dipeptidyl peptidase IV. J. Biol. Chem. 2004;279:34691–34697. doi: 10.1074/jbc.M405400200. [DOI] [PubMed] [Google Scholar]
- 41.Inoue M., Hiratake J., Suzuki H., Kumagai H., Sakata K. Identification of catalytic nucleophile of Escherichia coli gamma-glutamyltranspeptidase by gamma-monofluorophosphono derivative of glutamic acid: N-terminal Thr-391 in small subunit is the nucleophile. Biochemistry. 2000;39:7764–7771. doi: 10.1021/bi000220p. [DOI] [PubMed] [Google Scholar]
- 42.Das A. K., Helps N. R., Cohen P. T., Barford D. Crystal structure of the protein serine/threonine phosphatase 2C at 2.0 A resolution. EMBO J. 1996;15:6798–6809. [PMC free article] [PubMed] [Google Scholar]
- 43.Palm G. L., Lubkowsky J., Derst C., Schleper S., Rohm K. H., Wlodawer A. A covalently bound catalytic intermediate in Escherichia coli asparaginase: crystal structure of a Thr-89-Val mutant. FEBS Lett. 1996;390:211–216. doi: 10.1016/0014-5793(96)00660-6. [DOI] [PubMed] [Google Scholar]
- 44.Borek D., Michalska K., Brzezinski K., Kisiel A., Podkowinski J., Bonthron D. T., Krowarsch D., Otlewski J., Jaskolski M. Expression, purification and catalytic activity of Lupinus luteus asparagine beta-amidohydrolase and its Escherichia coli homolog. Eur. J. Biochem. 2004;271:3215–3226. doi: 10.1111/j.1432-1033.2004.04254.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.