

Abstract
The phosphate carrier (PiC) catalyses the import of phosphate into mitochondria where it is needed for ATP synthesis. We have analysed the 5′-flanking region of the human PiC gene and found that it has a single transcriptional initiation site and lacks a TATA box. Through deletion analysis of the −1213/−25 nt region, we identified an activation domain (−223/−25) and an inhibition domain (−1017/−814). The most effective promoter activity in transfected HeLa cells corresponded to the region containing putative binding sites for Sp1 (−163/−142; where Sp1 stands for stimulating protein-1) and CREB (−138/−116; where CREB stands for cAMP-response-element-binding protein). These DNA sequences were active in gel-shift assays in the presence of HeLa cell nuclear extracts or recombinant Sp1 and CREB respectively. Forskolin increased PiC promoter activity via the CREB site. Both footprinting and transfection of deletion constructs of the inhibition region (−1017/−814) showed that PiC silencer activity extends over 25 nt (−943/−919), which specifically binds two proteins present in HeLa cell nuclear extracts. These transcription factors were purified by DNA affinity, analysed by MS and identified as p54nrb/NonO (nuclear RNA binding protein) and PSF (protein-associated splicing factor). The PiC silencer region cloned in front of the ferritin promoter conferred a strong inhibition to the heterologous promoter. These findings may provide insight into control of PiC gene expression in different cell types and under different growth conditions. To our knowledge, this is the first study to analyse the regulation of the PiC gene expression in any cell.
Keywords: gene expression, mitochondria, phosphate carrier, promoter, silencer, transcription factor
Abbreviations: CAT, chloramphenicol acetyltransferase; CREB, cAMP-response-element-binding protein; EMSA, electrophoretic mobility-shift assay; PiC, phosphate carrier; PSF, protein-associated splicing factor; RACE, rapid amplification of cDNA ends; RT, reverse transcriptase; Sp1, stimulating protein-1
INTRODUCTION
The phosphate carrier (PiC) is a nuclear-encoded protein, which is an essential component of inner mitochondrial membranes. Its physiological role is to catalyse the transport of inorganic phosphate into the mitochondrial matrix, as proton-compensated phosphate symport, at the expense of the pH gradient across the membrane. Uptake of phosphate into mitochondria is essential for the oxidative phosphorylation of ADP to ATP. Inside the mitochondria, phosphate fulfils additional functions by facilitating the uptake of other metabolites via exchange mechanisms catalysed by other mitochondrial transporters and then recycling on its own carrier. The PiC belongs to the mitochondrial carrier protein family [1]; it is made up of three tandemly related domains of approx. 100 amino acids in length [2]; it spans the membrane six times with both the N- and C-termini protruding towards the cytosol [3]; and it functions as a homodimer by a sequential mechanism in reconstituted liposomes [4]. Among the family members, the mammalian, but not the yeast, PiC is exceptional in having a processed N-terminal sequence that helps to target the protein into mitochondria [5]. Only one gene for the PiC has been detected in human and cow [6]. The human gene (named SLC25A3) has been localized to chromosome 12q23 [7]. In both human and cow, two closely related exons named IIIA and IIIB appear to be alternatively spliced [6]. The presence of the two PiC isoforms (A and B) has been demonstrated both at transcript and protein levels [8]. PiC-A is present only in heart and skeletal muscles, whereas PiC-B is ubiquitous. The recombinant, reconstituted isoforms A and B both exhibit similar substrate specificity and inhibitor sensitivity, but differ in their kinetic parameters [8]. The yeast PiC has been the target of a number of mutations involving charged residues and hydroxy residues. The data generated by these mutations pointed out to a coupled proton–phosphate transport pathway within the PiC protein and shed light on possible subunit–subunit contact sites [9]. In addition, the PiC has been shown to be a controlling step in ATP synthesis and O2 consumption [10].
Although PiC plays an important role in mitochondrial metabolism, nothing is known about the transcriptional control of its gene in any cell. In order to investigate the transcriptional regulation of the PiC promoter, we used the human PiC gene 5′-flanking region (from −1213 to +95 nt) and its deletion constructs in CAT (chloramphenicol acetyltransferase) reporter gene studies, footprinting and EMSA (electrophoretic mobility-shift assay) experiments. We identified an activating domain (−223/−25) and a silencer domain (−943/−919). Within the activating domain, the sequences −163/−142 and −138/−116 bound specifically recombinant CREB (cAMP-response-element-binding protein) and Sp1 (stimulating protein-1) respectively. Mutations in the CREB element prevented binding of recombinant CREB, decreased transcription in transfected cells and abolished the increase in expression by forskolin. The silencer element was active in gel shift assays in the presence of HeLa cell nuclear extracts, whereas silencer mutants were not, and were devoid of silencing activity. Two HeLa cell extract proteins of apparent molecular masses of 100 and 55 kDa, which bind the silencer region, were identified by Southwestern-blot analysis. These proteins were purified and identified as p54nrb/NonO (nuclear RNA binding protein) and PSF (protein-associated splicing factor). To our knowledge, this is the first study to analyse the PiC gene promoter in any cell.
EXPERIMENTAL
Plasmids and constructs
To analyse the promoter of the human PiC gene, reporter plasmid constructs were prepared. Progressive deletion fragments of the regions from −1213 to −25 nt and from −1017 to −837 nt of the PiC gene promoter were obtained by PCR and cloned into the pEMBL8-CAT upstream of the CAT gene [11]. For heterologous promoter expression, the minimal promoter region of the human ferritin gene was inserted in the pEMBL8-CAT vector in front of the CAT gene [12]; then heterologous promoter constructs were prepared by cloning the PiC silencer region (−943/−919), in both directions and repeated twice, into the pEMBL8-CAT vector upstream of the ferritin gene promoter. The sequences of the inserts were verified.
Primer extension and RACE (rapid amplification of cDNA ends)
The primer extension analysis was carried out as described previously [13]. The oligonucleotide primer (15 ng), complementary to nt +51/+75 of the human PiC-A cDNA [6], was labelled at the 5′-end using T4 polynucleotide kinase (Roche, Lewes, East Sussex, U.K.) and [γ-32P]ATP (Amersham Biosciences) at 37 °C for 30 min, and then incubated at 85 °C for 10 min with 20 μg of total RNA extracted from HeLa or Hep3B cells. Hybridization between the mRNA and the labelled oligonucleotide was accomplished overnight at 42 °C. RT (reverse transcriptase)–PCR was initiated by adding 200 units of RT Superscript II (Gibco BRL, Paisley, Renfrewshire, Scotland, U.K.) to the mRNA–oligonucleotide mixture. The size of the primer extension products was determined by comparison with a DNA sequencing ladder. RACE products were generated by touchdown PCR performed on Smart-RACE cDNA (ClonTech Laboratories, Basingstoke, U.K.), using two reverse nested primers complementary to nt +51/+75 and +76/+98 of the human PiC-A cDNA and two forward adaptor primers AP-1 and AP-2 (ClonTech Laboratories). A single band of approx. 60 nt was obtained, cloned into pCR II vector (Invitrogen) and sequenced.
Cell culture and transfection
HeLa cells from 4C-Biotech (Seneffe, Belgium) were grown in Dulbecco's modified Eagle's medium containing 10% (v/v) heat-inactivated fetal calf serum supplemented with 2 mM L-glutamine, 100 units of penicillin and 100 μg/ml streptomycin at 37 °C in 5% CO2. When cells were 60–70% confluent, they were co-transfected with 3 μg of pBlue-RS TOPO-lacZ and 3 μg of the pEMBL8-CAT plasmid either empty or containing the PiC gene 5′-flanking region (−1213/+95). In some cases, deletion fragments of the 5′-flanking region were inserted replacing the full-length region. In others the ferritin promoter was inserted downstream of deletion fragments. Transfection was performed in the presence of metafectene (Biontex Laboratories, Munich, Germany) following the manufacturer's instructions. Transfected cells were collected after 48 h and assayed for CAT activity [14]. Where indicated, transfected cells were supplemented with 10 μg/ml forskolin after 24 h, and assayed after an additional 24 h. The extent of transfection was normalized by β-galactosidase activity [15].
EMSA
EMSA experiments were performed as described by Sambrook et al. [15] using the labelled DNA probes and the proteins indicated in the legends to Figures 5, 7 and 8. The double-stranded oligonucleotide probes were 5′-end labelled using T4 polynucleotide kinase and [γ-32P]ATP at 37 °C for 30 min. Where indicated, the unlabelled DNA probe, the mutated probe or an unrelated oligonucleotide was added. The gels were dried and the images acquired by phosphoimager (Bio-Rad Laboratories, Hemel Hempstead, Herts., U.K.).
Figure 5. Specificity of protein binding to the PiC gene silencer.

(A) Binding of the PiC gene silencer corresponding to nt −943/−919 of the PiC gene promoter to 5 μg of protein of HeLa cell nuclear extracts in the presence and absence of unlabelled specific (WT) or unspecific (unsp) probe in 100-fold molar excess. The sequence of the unspecific competitor was TTATTTCGCTCACCATATGGATGGT. (B) Binding of the PiC gene silencer (WT) and of its mutated forms (M1–M3) to 5 μg of protein of HeLa cell nuclear extracts in the presence or absence of unlabelled specific competitor. The sequences of WT, M1, M2 and M3 are GACAGCAGAGCATTAAATGAAGTGT, GACAGCAGAGCATTAAATGCCGTGT, ACAGCCTCTCATTAAATGAAGTGT and GACAGCAGAGCAGGAAATGAAGTGT respectively, where the mutated nucleotides are underlined. (C) The DNA sequence corresponding to nt −943/+95 of the PiC gene promoter and three mutated forms of this sequence were cloned into the pEMBL8-CAT plasmid and tested for CAT expression activity in transfected HeLa cells. WT indicates the wild-type sequence; M1, M2 and M3 indicate the mutated sequences (from −943 to −919) reported in (B). The values were set relative to the wild-type fragment. The data represent the means±S.D. for three duplicate independent experiments.
Figure 7. Identification and purification of the silencer-binding proteins.

(A) Southwestern-blot analysis. Increasing amounts of HeLa cell nuclear extracts were separated by SDS/PAGE and blotted on to a PVDF membrane, which was incubated with a 5′-end labelled DNA probe corresponding to nt −943/−919 of the PiC gene promoter (i.e. the silencer region). Specific signals are indicated by arrows. (B) Affinity purification of the silencer-binding proteins. A concatamer of the DNA sequence corresponding to nt −943/−919 of the PiC gene promoter (i.e. the silencer region) was ligated to streptavidin magnetic particles. These particles were incubated with HeLa cell nuclear extracts. The supernatant was removed with a magnetic separator and the bound proteins were separated with four washing steps and then with a buffer containing the indicated KCl concentrations. The protein fractions (30 μl) were analysed by SDS/PAGE and stained with Coomassie Blue. Lane U, supernatant; lane W, last washing step; lanes 1.5, 1, 0.8, 0.6 and 0.4, fractions eluted with 1.5, 1, 0.8, 0.6 and 0.4 M KCl. (C) Binding of the 5′-end labelled PiC gene silencer corresponding to nt −943/−919 of the PiC gene promoter to the protein fractions (30 μl) shown in (B). Lane P, labelled DNA probe only; lane C1, binding of the labelled DNA probe to 5 μg of protein of HeLa nuclear extracts; and lane C2, as in lane C1 in the presence of a 100-fold molar excess of unlabelled probe.
Figure 8. Specificity of protein binding to the PiC activator elements.

(A) Binding of the potential Sp1 and CREB sites within the PiC gene promoter to HeLa cell nuclear extracts. The 5′-end labelled DNA probe corresponding to nt −163/−142 or to nt −138/−116 of the PiC gene promoter was incubated with 5 μg of protein of HeLa cell nuclear extracts in the presence or absence of a 100-fold molar excess of unlabelled probe (competitor). (B) Binding of recombinant CREB to the DNA sequence from nt −138 to −116 of the PiC gene promoter. The DNA probe (−138/−116) labelled at the 5′-end was incubated with 1 μg of recombinant CREB alone and in the presence of 100- or 200-fold molar excess of the unlabelled probe (WT1 and WT2 respectively) or mutated probe (M1 and M2 respectively). The sequence of the mutated probe was GGAGAACGCGATGCATCTACGGG. (C) Binding of recombinant Sp1 to the DNA sequence from nt −163 to −142 of the PiC gene promoter. The 5′-end labelled DNA probe (−163/−142) was incubated with 2 μg of recombinant Sp1 alone and in the presence of 100- or 200-fold molar excess of the unlabelled probe (WT1 and WT2 respectively) or mutated probe (M1 and M2 respectively). The sequence of the mutated probe was GGCATTGGCTTGAATAAGGTTG.
DNase I protection assay
DNase I protection assay was performed as described by Lichtsteiner et al. [16], except that the labelled DNA probe was partially digested for 1 min at 25 °C using RNase-free DNase I (Roche).
Southwestern-blot analysis
Southwestern-blot analysis was performed as reported by Vinson et al. [17]. HeLa cell nuclear extracts were separated by SDS/PAGE (10% polyacrylamide) and electroblotted on to a PVDF membrane (Amersham Biosciences). The membrane was incubated with the 5′-end labelled DNA probe for 2 h at room temperature (25 °C). The image was acquired by phosphoimager.
Protein purification
Protein purification was carried out using the DNA-binding protein purification kit (Roche), following the manufacturer's instructions. A long concatamer of the DNA sequence corresponding to the −943/−919 nt of the PiC gene promoter was obtained by self-primed PCR technique [18]; it was phosphorylated at its 5′-end and ligated to streptavidin magnetic particles. The particles were mixed with HeLa cell nuclear extracts (70 μg) and incubated with occasional swirling for 1 h at 4 °C. DNA-binding proteins were eluted with decreasing KCl concentrations from 1.5 to 0.4 M and loaded on to SDS/PAGE (10% polyacrylamide). All eluted fractions were tested by EMSA experiments using the radioactively labelled −943/−919 oligonucleotide as the probe.
Mass analysis and protein identification
Digestion of the protein bands, excised from the gel, was performed with 20 ng/μl modified trypsin (Promega, Chilworth, Southampton, U.K.) in 50 mM NH4HCO3, at 37 °C overnight in a shaking incubator, as described by Castegna et al. [19]. A Bruker Autoflex matrix-assisted laser-desorption ionization–time-of-flight mass spectrometer operated in the reflectron mode was used to generate peptide mass ‘fingerprints’. External calibration of the mass axis was used for acquisition, whereas internal calibration, using either trypsin autolysis ions or matrix clusters, was applied post-acquisition for accurate mass determination. The spectra of the tryptic fragments were searched against the NCBI protein databases using the MASCOT search engine (http://www.matrixscience.com). Peptide mass ‘fingerprinting’ was obtained with the assumption that peptides are monoisotopic, oxidized at methionine residues and carbamidomethylated at cysteine residues. Up to one missed trypsin cleavage was allowed, although most matches did not contain any missed cleavage. Mass tolerance of 150 p.p.m. was the window of error allowed for matching the peptide mass values.
RT–PCR and Western blotting
HeLa cells (1×106) were seeded in 60-mm culture dishes. The next day, 10 μg/ml forskolin (Sigma) was added where indicated. For RT–PCR, after 4 h, the cells were collected and total RNA was extracted. RT–PCR assay was performed with GeneAmp RNA PCR core kit (Applied Biosystems, Warrington, Cheshire, U.K.). The forward and reverse primers corresponded to nt +438/+462 and +795/+816 respectively of the PiC-A cDNA [6]. The level of β-actin was used as an internal standard. For Western blotting, after 24 h, the cells were lysed in 50 mM Tris/HCl (pH 7.8), 150 mM NaCl and 1% Nonidet P40 and then freeze–thawed. The proteins were subjected to SDS/PAGE and electroblotted on to nitrocellulose (Bio-Rad Laboratories). The nitrocellulose membranes were treated with an antibody (1:2000) raised against the peptide 1−10 of the human PiC. The immunoreaction was detected by the ECL® system (Amersham Biosciences).
RESULTS
Identification of the transcription start site
The transcription start site of the human PiC cDNA was investigated first by primer extension analysis carried out using total RNA from HeLa or Hep3B cells and then using a DNA fragment from nt +51 to nt +75 of the human PiC-A cDNA as the probe. This probe corresponds to the genomic sequence from nt +288 to nt +313 (see Figure 1). With this analysis, a single fragment of approx. 60 nt was obtained (Figure 2). The transcription starting nucleotide was mapped precisely using 5′-RACE analysis. The sequences of 12 clones out of 15 began at nt −55 of the PiC-A cDNA, which corresponds to nt +1 of the genome sequence (Figure 1), 289 nt upstream of the ATG translation start codon.
Figure 1. Sequence of the 5′-flanking region of the human PiC gene.

The nucleotides preceding the transcription start site (boxed G at position +1) are indicated by negative numbers. The Sp1 and CREB binding sites and the silencer region (WT) are represented in italics and underlined. Boxed nucleotide sequences indicate the forward (F1–F7) and reverse (REV) primers employed to generate a series of deletion fragments. The translational start codon ATG is also underlined.
Figure 2. Primer extension analysis of PiC transcript from HeLa and Hep3B cells.
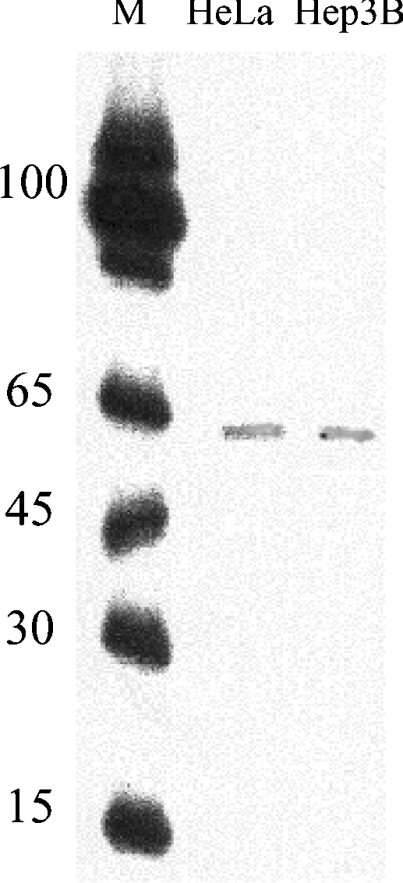
The antisense oligonucleotide corresponding to nt +51/+75 of the human PiC-A cDNA was incubated with 20 μg of total RNA from HeLa or Hep3B cells (second and third lanes respectively), as described in the Experimental section. Lane M, 5′-end labelled oligonucleotide markers of different lengths.
Identification of the regulatory domains within the promoter of the PiC gene
To investigate the promoter activity, deletion mutants of PiC promoter-driven CAT reporter gene constructs and control vector pEMBL8-CAT were transfected into HeLa cells, and the relative CAT expression activity of each reporter gene construct was measured (Figure 3). Our serial promoter deletions from −1213 to −25 nt showed different promoter activities. Removal of the nucleotides from region −1213 to −1018 (C2 in Figure 3) led to a drastic reduction in CAT expression. In contrast, progressive deletions from −1017 to −224 (C3–C6 in Figure 3) restored the PiC gene promoter activity, and construct C6 showed a 6.1-fold increase in promoter activity with respect to the activity of construct C2.
Figure 3. Deletion analysis of the 5′-flanking region of the human PiC gene.

The deletion mutants named C1–C8 were cloned into the pEMBL8-CAT plasmid and tested for CAT expression activity in transfected HeLa cells. CAT indicates the pEMBL8-CAT vector alone. Numbering indicates the extent of fragments, whereas dark bars indicate CAT activity. The values were set relative to the C1 construct. Means±S.D. for five duplicate independent experiments are shown.
These results led us to identify two domains in the 5′-flanking regulatory region of the PiC gene: an inhibitory domain between −1017 and −814 nt and an activation domain between −223 and −25 nt.
Characterization of the inhibitory domain of the PiC gene promoter
The presence of a negative regulatory region from −1017 to −814 nt of the PiC gene was confirmed by the results obtained with construct C8. This construct was made by skipping the region from −1017 to −814 of the PiC carrier gene (i.e. linking nt −1018 to nt −813). By using C8 to measure the CAT activity in transfected HeLa cells, a promoter activity much higher than that of C2 and similar to those of C1 and C3 was obtained (Figure 3). Preliminary EMSA experiments were performed using HeLa cell nuclear extracts and a labelled DNA fragment corresponding to nt from −1017 to −814 as the probe showed that this oligonucleotide formed a specific DNA–protein complex (results not shown), indicating that HeLa cell nuclear extracts contain one or more protein factors that bind to the inhibitory domain −1017/−814 of the PiC gene.
The protein-binding site on the negative regulatory domain was identified by a DNase footprinting assay performed using the labelled DNA fragment corresponding to nt −1017/−814 and nuclear extracts from HeLa cells. A protected region of 25 nt from −943 to −919 (corresponding to the sequence 5′-GACAGCAGAGCATTAAATGAAGTGT-3′, see Figure 1) was observed (Figure 4A). Furthermore, a strong DNase hypersensitive site was evident next to the protected region. The fact that the −943/−919 region acts as a silencer is apparent from observations of various deletion mutants of the −1017/−837 PiC sequence-driven CAT reporter gene constructs. Only the one that starts with the sequence from −943 to −919 exhibited a very strong reduction in CAT activity (Figure 4B).
Figure 4. DNase footprinting analysis and deletion analysis of the PiC gene promoter inhibitory domain.

(A) The labelled DNA probe, corresponding to −1017/−814 nt of the PiC gene promoter, was partially digested by 100 or 400 μg/ml DNase I in the presence of 30 μg of HeLa cell nuclear extracts (NE). In the left-hand lane, the DNA probe was digested by 40 μg/ml DNase I in the absence of nuclear extracts. The protected region is indicated by a bracket and an arrow; it extends between −943 and −919 nt. (B) The indicated deletion mutants of the region from −1017 to −837 nt of the PiC gene promoter were cloned into the pEMBL8-CAT plasmid and tested for CAT expression activity in transfected HeLa cells. Numbering indicates the extent of fragments, whereas dark bars indicate CAT activity. The values were set relative to the construct −1017/+95. Means±S.D. for four duplicate independent experiments are shown.
The binding activity of the PiC gene silencer was investigated in detail by EMSA experiments on HeLa cell nuclear extracts. Figure 5(A) shows that the labelled probe encompassing the silencer region (−943/−919) binds one or more proteins of the nuclear extracts. The specificity of this binding activity was demonstrated by competition showing that binding was abolished by a 100-fold excess of the unlabelled probe. Moreover probe binding was unaffected by an unrelated DNA fragment (Figure 5A). No protein binding was observed with two oligonucleotides from regions immediately upstream (from −990 to −964) and downstream (from −918 to −887) of the sequence of the probe used in Figure 5(A) (results not shown). Further evidence for the specificity of the PiC gene silencer was obtained using mutants of the silencer (see Figure 5B). No binding was observed when the dinucleotide AA at positions −924/−923 was mutated into CC and the tetranucleotide AGAG at positions −937/−934 was changed to CTCT. In contrast, the oligonucleotide M3, in which TT at positions −931/−930 were mutated into GG, retained protein binding activity (Figure 5B). Interestingly, the silencer mutants M1 and M2 with no binding activity increased the expression of CAT in transfected HeLa cells approx. 2-fold, as compared with the wild-type sequence (Figure 5C). In contrast, M3 that has binding activity did not cause any increase in CAT expression. These findings indicate that specific nucleotides of the silencer are indispensable for binding one or more proteins of HeLa nuclear extracts and for the silencing activity of the PiC gene promoter.
Conferring silencer activity to a heterologous promoter
In order to investigate whether the silencing can be conferred on a heterologous promoter, the PiC gene fragment from −943 to −919 was ligated in both directions into the pEMBL8-CAT vector upstream of the ferritin gene promoter. These constructs as well as the one containing the ferritin gene promoter alone were transfected into HeLa cells and assayed for CAT activity. The construct with the correct orientation showed a strong reduction of the ferritin promoter activity (by ∼73%) with respect to the control consisting of the ferritin gene promoter alone (Figure 6). In contrast, the opposite orientation of the fragment (−919/−943) showed a much lower silencing activity. These findings demonstrate that the silencing associated with the −943/−919 region of the PiC gene can be conferred to other gene promoters. They also suggest that the silencer sequence of the PiC gene belongs to a family of transcriptional negative elements that recognize factors acting as repressors on different gene promoters.
Figure 6. The PiC gene silencer confers inhibition to a heterologous promoter.

The silencer of the PiC gene corresponding to nt −943/−919 was cloned in both directions into the pEMBL8-CAT plasmid upstream of the ferritin promoter and tested for CAT expression activity in transfected HeLa cells. Numbering indicates the direction of the DNA sequence inserted in the vector. The values are expressed as percentage of the activity of the pEMBL8-CAT plasmid containing the ferritin promoter (pFerritin) alone. Means±S.D. for three duplicate independent experiments are shown.
Purification and identification of the silencer-binding proteins
A computer search of vertebrate transfactor databases (http://www.cbrc.jp/research/db/TFSEARCH.html and http://www.genomatix/bin/matinspector/pl) with the −943/−919 region of the PiC gene failed to reveal a transfactor that recognizes the silencer DNA sequence of the PiC gene. To estimate the size of the protein(s) that binds to this DNA sequence, a Southwestern-blot analysis was performed. Using HeLa nuclear extracts and the wild-type −943/−919 oligonucleotide as the probes, two polypeptides with apparent molecular masses of approx. 100 and 55 kDa were found to hybridize with the probes (Figure 7A), suggesting that at least two proteins are involved in binding to the DNA sequence of the PiC gene silencer.
The proteins that bind to the silencer were purified from HeLa nuclear extracts by DNA affinity using a long concatameric oligonucleotide of the region from −943 to −919 of the PiC gene as the affinity ligand. The purified fraction eluted at 1 M KCl contained three major protein bands with apparent molecular masses of approx. 100, 74 and 55 kDa (Figure 7B). These bands were also present in the fraction eluted at 1.5 M KCl but together with many other protein bands. MS analysis of the tryptic digests obtained from the three proteins excised from the gel, together with database search, identified the protein bands at 100 and 74 kDa as PSF and the protein band at 55 kDa as p54nrb/NonO [20], as shown in Table 1. It should be noted that the molecular mass of PSF, previously known as 100 kDa-associated splicing factor, was determined to be approx. 75 kDa by DNA sequencing [21]. Our results, showing a strongly stained band at 100 kDa and a weakly stained band at 74 kDa, both identified as a single protein, confirm the previous findings [21].
Table 1. Identification of transcription factors that bind to the PiC gene silencer.
A total of 39 tryptic peptide masses corresponding to the protein bands at 74 and 100 kDa and 36 tryptic peptide masses corresponding to the protein band at 55 kDa were searched against the NCBI database. The pI, molecular mass and gi accession number of the proteins, as well as the number of matched peptides and the percentage sequence coverage, are listed here. The probability-based Mowse score, associated with each result, displays a high level of significance for the identified proteins.
| Excised protein band (kDa) | Identified protein | pI | Molecular mass (kDa) | gi number | Matched peptides (no.) | Sequence coverage (%) | Mowse score | P value |
|---|---|---|---|---|---|---|---|---|
| 100 | Human PSF | 9.50 | 76.255 | 29881667 | 16 | 21 | 109 | ≪0.05 |
| 74 | Human PSF | 9.50 | 76.255 | 29881667 | 16 | 21 | 109 | ≪0.05 |
| 55 | Human p54nrb/NonO | 9.01 | 54.311 | 13124797 | 20 | 28 | 140 | ≪0.05 |
The functional integrity of the DNA affinity-purified proteins was verified by EMSA. Figure 7(C) demonstrates that the fractions eluted at 1.5 and 1.0 M KCl showed strongly shifted bands (lanes 1.5 and 1), which were similar to that obtained with HeLa cell nuclear extracts (lane C1). In contrast, no binding activity was exhibited by the proteins that had not been bound by the DNA ligand (lane U). The other two very faint hybridizing bands present in lane 1 of Figure 7(C) most likely result from unspecific binding. In fact, in competition experiments with the unlabelled DNA sequence (lane C2), the strongly shifted band was markedly decreased, whereas the other two bands were not.
Characterization of the activation domain of the PiC gene promoter
Computer analysis by the TFSEARCH program of the region from −223 to −25 nt, showing the maximal promoter activity (see C6 and C7 in Figure 3), revealed the absence of TATA box, but the presence of CCAAT from −98 to −94 and potential binding sequences for Sp1 from −159 to −152 and for CREB from −128 to −120 (Figure 2). In fact, the sequences −159/−152 and −128/−120 are 88 and 86% identical with the Sp1 and CREB consensus sequences respectively.
As shown in Figure 8(A), HeLa cell nuclear extracts contained transcription factors that were bound by labelled probes corresponding to the DNA regions from −163 to −142 and from −138 to −116 respectively. This binding activity was completely eliminated by a 100-fold molar excess of the unlabelled probes (Figure 8A). To verify whether the putative elements for Sp1 and CREB are capable of binding these transcription factors, EMSA experiments were performed using the same probes as in Figure 8(A) and the recombinant Sp1 and CREB proteins. The binding is highly specific between these transcription factors and their putative elements in the PiC promoter. This is shown by the competition experiments carried out with unlabelled probes and mutated probes (Figures 8B and 8C). Whereas the unlabelled probes abolished the binding between Sp1 and the −159/−152 DNA sequence and between CREB and the −128/−120 DNA sequence, the mutated probes did not affect the binding activity. It should be noted that the labelled DNA sequences −163/−142 and −138/−116, containing mutations in the core of the Sp1 and CREB elements (as reported in the legends of Figures 8B and 8C), when incubated with HeLa cell nuclear extracts exhibited no binding activity (results not shown). This suggests that these regions of the PiC promoter bind mainly Sp1 and CREB respectively, and that no new protein-binding sites are created by the mutations.
Forskolin increases PiC promoter activity and PiC content both at the transcript and protein levels
Since CREB is activated via cAMP, we investigated the possibility that forskolin, which increases endogenous cAMP by activating adenylate cyclase [22], also increases the PiC promoter activity. For this reason, HeLa cells were transfected with the CAT reporter construct containing the activation domain (−223/+95) of the PiC gene promoter and treated with forskolin. This cAMP activator increased CAT activity by approx. 2-fold when compared with untreated cells (Figure 9A). Mutation of the CREB site completely abolished the effect of forskolin on promoter activity (Figure 9A). These results indicate that forskolin increased the transcription of the PiC gene via the CREB binding site. Similarly, forskolin treatment increased the endogenous PiC mRNA level of HeLa cells (Figure 9B) as well as their PiC protein content (Figure 9C). Therefore the CREB site in the PiC promoter plays an important role in both basal and induced expression of the PiC gene.
Figure 9. Effect of forskolin on PiC promoter activity and on PiC content both at the transcript and protein levels.

(A) Effect of forskolin on PiC promoter activity conferred by the DNA fragment −223/+95 with or without mutation in the CREB binding site. Transfected HeLa cells treated or untreated with 10 μg/ml forskolin were assayed for CAT expression activity. Grey bars indicate HeLa cells transfected with the wild-type (W) DNA fragment −223/+95 and the DNA fragment mutated in the CREB site (M). The black bars indicate forskolin-treated HeLa cells transfected with the constructs W and M. Means±S.D. for three duplicate independent experiments are shown. (B) Effect of forskolin on the PiC mRNA level. RT–PCR was performed with specific primers for the cDNA of PiC or β-actin and with total RNA from HeLa cells treated or untreated with forskolin. (C) Effect of forskolin on the PiC protein content. The PiC protein of HeLa cells treated or untreated with forskolin was immunodecorated with an anti-N-terminal PiC antibody. The reactivity of the antibody to recombinant human PiC-B was also tested.
DISCUSSION
In the present study, we have, for the first time, functionally characterized the 5′-flanking regulatory region of the human PiC gene and have identified an activation and an inhibition domain. Two distinct DNA stretches of the activation domain from −163 to −142 nt and from −138 to −116 nt bind two known transcriptional factors, Sp1 and CREB respectively, with high specificity. Consistently, mutations introduced into the core of the Sp1 and CREB elements of the PiC promoter prevented binding of HeLa nuclear extract proteins and recombinant Sp1 and CREB as shown in mobility shift experiments. Although the involvement of additional factors cannot be entirely eliminated, competition shift experiments demonstrate that Sp1 and CREB are the major, if not only, nuclear factors of HeLa cells that bind the reported DNA sequences of the activation domain. It is likely that Sp1 is responsible, at least in part, for the PiC gene basal expression since Sp1 is a ubiquitously expressed factor required for the expression of a large number of constitutive and regulated genes [23]. It is noteworthy that the PiC gene promoter does not contain adjacent Sp1-binding sites that have been shown to synergize the Sp1-dependent activation of transcription [23,24]. It should also be noted that the Sp1 element within the PiC gene promoter is sufficiently distant from the transcriptional start to make it unlikely to have a negative influence on transcription initiation efficiency as reported by Li et al. [23]. CREB is a family of proteins that in their phosphorylated form [25] activates transcription of target genes in response to a diverse array of stimuli such as peptide hormones, growth factors and neuronal activity. These proteins are essential for a variety of cellular processes, such as cell proliferation, differentiation, adaptative responses and control of metabolic pathways [26,27]. The data reported in the present paper indicate that the CREB element in the PiC promoter is not only important for basal expression but also for induced expression through the cAMP–protein kinase pathway. In fact, upon stimulation with forskolin, an activator of adenylate cyclase [22], we have observed a clear increase in the level of both PiC mRNA and PiC protein and a 2-fold activation of CAT expression.
A well-defined region of the PiC gene promoter acts as a silencer. The results presented above show that it consists of 25 nt (from −943 to −919) and exhibits a pronounced repression of transcription, as it strongly decreases CAT expression and ferritin promoter activity in transfected cells. The identification of the PiC promoter silencer has allowed us to purify two DNA-binding proteins, identified as p54nrb/NonO and PSF by MS. These proteins are multifunctional nuclear proteins [28] that are involved in RNA processing [29,30] and in gene transcription [31–33]. p54nrb/NonO and PSF have been shown to cause transcription inhibition of type II nuclear hormone receptor and retinoid X receptor genes [31], of the CYP17 gene [33] and of the P450scc gene [32]. In the human CYP17 gene, p54nrb/NonO and PSF are bound to a DNA sequence containing the oligonucleotide TGAAGA, and in the porcine P450scc gene PSF binds to a DNA sequence containing the oligonucleotide CTGAGTC. Interestingly, both these oligonucleotides are similar to the motifs TGAAGT and CAGAGC, which are present in the PiC gene promoter silencer and have been proven to be essential for the binding activity of this region. p54nrb/NonO and PSF may function separately, as heterodimers or as docking sites for other factors in the formation of transcription complexes [28]. In the hCYP17 gene, the repression complex contains not only PSF and p54nrb/NonO but also SF-1 (steroidogenic factor-1) and Sin3A [33]. Sin3A is also involved in the negative regulation of the nuclear hormone receptor genes together with PSF and p54nrb/NonO [31]. It should be noted that, although only PSF and p54nrb/NonO have been identified in the staining profile of Figure 7(B), lane 1, we cannot exclude the presence of minor components in the complex that mediates repression of the PiC gene expression. It should also be noted that in the case of the PiC gene, neither p54nrb/NonO nor PSF functions only as an ancillary protein of the other, i.e. it is associated with the other but does not bind the DNA directly, because both proteins have been shown by the Southwestern-blot analysis of Figure 7(A) to bind to the silencer region from −943 to −919 nt of the PiC promoter. It may be that the fragment starting at −990 nt (Figure 4B) does not decrease CAT expression, because it contains an enhancer element that suppresses the activity of the silencer region.
The activities of many transport systems are tightly regulated in different ways [34]. In a previous study, we suggested that PiC-A and PiC-B, due to their different Km and Vmax values, can be used to modulate the rate of oxidative ATP production for tissue-specific energetic needs [8]. Several observations point to the need of effective regulation of PiC gene expression in different tissues and under different physiological conditions, metabolic and energetic states, and growth conditions. The PiC has a very high catalytic-centre activity but is present in very minute amounts in the mitochondrial membrane [35,36]; it is rate-limiting for oxidative phosphorylation [10]; and it is essential in maintaining cell metabolism and growth in eukaryotic cells since it makes up the mitochondrial ATP-synthesizing machinery together with the FoF1-ATPase complex and the ADP/ATP carrier that exchanges cytosolic ADP for mitochondrial ATP. Furthermore, the PiC transcripts and PiC protein levels vary in the different tissues [8,37]. We have recently found that PiC mRNA, measured as the sum of PiC-A and PiC-B mRNAs, is significantly higher in HeLa cells when they are stimulated by the addition of serum (V. Iacobazzi, V. Infantino and F. Palmieri, unpublished work). The functional analysis of the PiC gene promoter reported here has helped to identify mechanisms regulating the expression of this gene in HeLa cells. However, the biological relevance of these regulatory mechanisms in different cells and growth conditions remains to be delineated.
Acknowledgments
This work was supported by grants from Ministero dell'Università e della Ricerca Scientifica e Tecnologica (MIUR), University's Local Funds, the CNR, CNR-MIUR project ‘Functional genomics’, the Centro di Eccellenza Geni in campo Biosanitario e Agroalimentare (CEGBA), EC contract LSHM-CT-2004-503116 and by the European Social Fund.
References
- 1.Palmieri F. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch. 2004;447:689–709. doi: 10.1007/s00424-003-1099-7. [DOI] [PubMed] [Google Scholar]
- 2.Runswick M. J., Powell S. J., Nyren P., Walker J. E. Sequence of the bovine mitochondrial phosphate carrier protein: structural relationship to ADP/ATP translocase and the brown fat mitochondria uncoupling protein. EMBO J. 1987;6:1367–1373. doi: 10.1002/j.1460-2075.1987.tb02377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Capobianco L., Brandolin G., Palmieri F. Transmembrane topography of the mitochondrial phosphate carrier explored by peptide-specific antibodies and enzymatic digestion. Biochemistry. 1991;30:4963–4969. doi: 10.1021/bi00234a018. [DOI] [PubMed] [Google Scholar]
- 4.Schroers A., Burkovski A., Wohlrab H., Krämer R. The phosphate carrier from yeast mitochondria. Dimerization is a prerequisite for function. J. Biol. Chem. 1998;273:14269–14276. doi: 10.1074/jbc.273.23.14269. [DOI] [PubMed] [Google Scholar]
- 5.Zara V., Palmieri F., Mahlke K., Pfanner N. The cleavable presequence is not essential for import and assembly of the phosphate carrier of mammalian mitochondria, but enhances the specificity and efficiency of import. J. Biol. Chem. 1992;267:12077–12081. [PubMed] [Google Scholar]
- 6.Dolce V., Iacobazzi V., Palmieri F., Walker J. E. The sequence of human and bovine genes of the phosphate carrier from mitochondria contain evidence of alternatively spliced forms. J. Biol. Chem. 1994;269:10451–10460. [PubMed] [Google Scholar]
- 7.Marsh S., Carter N. P., Dolce V., Iacobazzi V., Palmieri F. Chromosomal localization of the mitochondrial phosphate carrier gene PHC to 12q23. Genomics. 1995;29:814–815. doi: 10.1006/geno.1995.9924. [DOI] [PubMed] [Google Scholar]
- 8.Fiermonte G., Dolce V., Palmieri F. Expression in Escherichia coli, functional characterization, and tissue distribution of isoforms A and B of the phosphate carrier from bovine mitochondria. J. Biol. Chem. 1998;273:22782–22787. doi: 10.1074/jbc.273.35.22782. [DOI] [PubMed] [Google Scholar]
- 9.Wohlrab H. Novel inter- and intrasubunit contacts between transport-relevant residues of the homodimeric mitochondrial phosphate transport protein. Biochem. Biophys. Res. Commun. 2004;320:685–688. doi: 10.1016/j.bbrc.2004.05.211. [DOI] [PubMed] [Google Scholar]
- 10.Mazat J. P., Jean-Bart E., Rigoulet M., Guèrin B. Control of oxidative phosphorylations in yeast mitochondria. Role of the phosphate carrier. Biochim. Biophys. Acta. 1986;849:7–15. [Google Scholar]
- 11.Dente L., Cesareni G., Cortese R. pEMBL: a new family of single stranded plasmids. Nucleic Acids Res. 1983;11:1645–1655. doi: 10.1093/nar/11.6.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bevilacqua M. A., Giordano M., D'Agostino P., Santoro C., Cimino F., Costanzo F. Promoter for the human ferritin heavy chain-encoding gene (FERH): structural and functional characterization. Gene. 1992;111:255–260. doi: 10.1016/0378-1119(92)90696-m. [DOI] [PubMed] [Google Scholar]
- 13.Boorstein W. R., Craig E. A. Primer extension analysis of RNA. Methods Enzymol. 1989;180:347–369. doi: 10.1016/0076-6879(89)80111-9. [DOI] [PubMed] [Google Scholar]
- 14.Gorman C. M., Moffat L. F., Howard B. H. Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol. Cell. Biol. 1982;2:1044–1051. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sambrook J., Fritsch E. F., Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd edn. Plainview, NY: Cold Spring Harbour Laboratory Press; 1989. [Google Scholar]
- 16.Lichtsteiner S., Wuarin J., Schibler U. The interplay of DNA-binding proteins on the promoter of the mouse albumin gene. Cell (Cambridge, Mass.) 1987;51:963–973. doi: 10.1016/0092-8674(87)90583-6. [DOI] [PubMed] [Google Scholar]
- 17.Vinson C. R., LaMarco K. L., Johnson P. F., Landschulz W. H., McKnight S. L. In situ detection of sequence-specific DNA binding activity specified by a recombinant bacteriophage. Genes Dev. 1988;2:801–806. doi: 10.1101/gad.2.7.801. [DOI] [PubMed] [Google Scholar]
- 18.Hemat F., McEntee K. A rapid and efficient PCR-based method for synthesizing high-molecular-weight multimers of oligonucleotides. Biochem. Biophys. Res. Commun. 1994;205:475–481. doi: 10.1006/bbrc.1994.2690. [DOI] [PubMed] [Google Scholar]
- 19.Castegna A., Thongboonkerd V., Klein J. B., Lynn B., Markesbery W. R., Butterfield D. A. Proteomic identification of nitrated proteins in Alzheimer's disease brain. J. Neurochem. 2003;85:1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x. [DOI] [PubMed] [Google Scholar]
- 20.Dong B., Horowitz D. S., Kobayashi R., Krainer A. R. Purification and cDNA cloning of HeLa cell p54nrb, a nuclear protein with two RNA recognition motifs and extensive homology to human splicing factor PSF and Drosophila NONA/BJ6. Nucleic Acids Res. 1993;21:4085–4092. doi: 10.1093/nar/21.17.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patton J. G., Porro E. B., Galceran J., Tempst P., Nadal-Ginard B. Cloning and characterization of PSF, a novel pre-mRNA splicing factor. Genes Dev. 1993;7:393–406. doi: 10.1101/gad.7.3.393. [DOI] [PubMed] [Google Scholar]
- 22.Seamon K. B., Padgett W., Daly J. W. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc. Natl. Acad. Sci. U.S.A. 1981;78:3363–3367. doi: 10.1073/pnas.78.6.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li R., Hodny Z., Luciakova K., Barath P., Nelson B. D. Sp1 activates and inhibits transcription from separate elements in the proximal promoter of human adenine nucleotide translocase 2 (ANT2) gene. J. Biol. Chem. 1996;271:18925–18930. doi: 10.1074/jbc.271.31.18925. [DOI] [PubMed] [Google Scholar]
- 24.Pascal E., Tjian R. Different activation domains of Sp1 govern formation of multimers and mediate transcriptional synergism. Genes Dev. 1991;5:1646–1656. doi: 10.1101/gad.5.9.1646. [DOI] [PubMed] [Google Scholar]
- 25.Quinn P. G. Mechanisms of basal and kinase-inducible transcription activation by CREB. Prog. Nucleic Acid Res. Mol. Biol. 2002;72:269–305. doi: 10.1016/s0079-6603(02)72072-2. [DOI] [PubMed] [Google Scholar]
- 26.Salton S. R., Ferri G. L., Hahm S., Snyder S. E., Wilson A. J., Possenti R., Levi A. VGF: a novel role for this neuronal and neuroendocrine polypeptide in the regulation of energy balance. Front. Neuroendocrinol. 2000;21:199–219. doi: 10.1006/frne.2000.0199. [DOI] [PubMed] [Google Scholar]
- 27.Lonze B. E., Ginty D. D. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 28.Shav-Tal Y., Zipori D. PSF and p54nrb/NonO – multi-functional nuclear proteins. FEBS Lett. 2002;531:109–114. doi: 10.1016/s0014-5793(02)03447-6. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Z., Carmichael G. G. The fate of dsRNA in the nucleus: a p54nrb-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell (Cambridge, Mass.) 2001;106:465–475. doi: 10.1016/s0092-8674(01)00466-4. [DOI] [PubMed] [Google Scholar]
- 30.Teigelkamp S., Mundt C., Achsel T., Will C. L., Luhrmann R. The human U5 snRNP-specific 100-kD protein is an RS domain-containing, putative RNA helicase with significant homology to the yeast splicing factor Prp28p. RNA. 1997;3:1313–1326. [PMC free article] [PubMed] [Google Scholar]
- 31.Mathur M., Tucker P. W., Samuels H. H. PSF is a novel corepressor that mediates its effect through Sin3A and the DNA binding domain of nuclear hormone receptors. Mol. Cell. Biol. 2001;21:2298–2311. doi: 10.1128/MCB.21.7.2298-2311.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Urban R. J., Bodenburg Y., Kurosky A., Wood T. G., Gasic S. Polypyrimidine tract-binding protein-associated splicing factor is a negative regulator of transcriptional activity of the porcine P450scc insulin-like growth factor response element. Mol. Endocrinol. 2000;14:774–782. doi: 10.1210/mend.14.6.0485. [DOI] [PubMed] [Google Scholar]
- 33.Sewer M. B., Nguyen V. Q., Huang C. J., Tucker P. W., Kagawa N., Waterman M. R. Transcriptional activation of human CYP17 in H295R adrenocortical cells depends on complex formation among p54nrb/NonO, protein-associated splicing factor, and SF-1, a complex that also participates in repression of transcription. Endocrinology. 2002;143:1280–1290. doi: 10.1210/endo.143.4.8748. [DOI] [PubMed] [Google Scholar]
- 34.Krämer R. Analysis and modeling of substrate uptake and product release by prokaryotic and eukaryotic cells. Adv. Biochem. Eng. Biotechnol. 1996;54:31–74. doi: 10.1007/BFb0102332. [DOI] [PubMed] [Google Scholar]
- 35.Ligeti E., Brandolin G., Dupont Y., Vignais P. V. Kinetics of Pi-Pi exchange in rat liver mitochondria. Rapid filtration experiments in the millisecond time range. Biochemistry. 1985;24:4423–4428. doi: 10.1021/bi00337a025. [DOI] [PubMed] [Google Scholar]
- 36.Palmieri F. Mitochondrial carrier proteins. FEBS Lett. 1994;346:48–54. doi: 10.1016/0014-5793(94)00329-7. [DOI] [PubMed] [Google Scholar]
- 37.Dolce V., Fiermonte G., Palmieri F. Tissue-specific expression of the two isoforms of the mitochondrial phosphate carrier in bovine tissues. FEBS Lett. 1996;399:95–98. doi: 10.1016/s0014-5793(96)01294-x. [DOI] [PubMed] [Google Scholar]