

Abstract
Biogenesis of phagolysosomes is a very rapid event in neutrophils which takes place with nascent unclosed phagosomes, leading to the release of lysosomal enzymes such as β-glucuronidase in the extracellular medium. We have previously shown that, under nonopsonic conditions, both pathogenic and nonpathogenic mycobacteria uncouple phagocytosis from fusion of azurophil granules (specialized secretory lysosomes) with phagosomes. In the present study we questioned whether they actively act on neutrophils to block this process or use phagocytic receptors that negatively control the biogenesis of phagolysosomes. As for live unicellular Mycobacterium smegmatis, we observed that nonopsonic phagocytosis of heat-killed mycobacteria did not induce the release of β-glucuronidase, indicating that M. smegmatis does not actively act on the fusion process in neutrophils. In contrast, phagocytosis of unicellular M. smegmatis opsonized in immune serum or that of small nonopsonized mycobacterial aggregates restored the biogenesis of phagolysosomes. Aggregates were internalized in a CR3- and cholesterol-dependent manner as unicellular mycobacteria. However, aggregates but not unicellular bacteria triggered F-actin and Hck recruitment at the phagosomes, events that have been associated with lysosome fusion. Thus, we propose that M. smegmatis does not actively control the fusion of azurophil granules at early time points postinfection and that mycobacterial aggregates recruit large clusters of receptors at the neutrophil surface which could trap proteins implicated in the biogenesis of phagolysosomes.
Neutrophils constitute the first line of defense against infectious agents that penetrate the body's physical barriers. They are the first cells to be recruited to sites of infection or injury. Their major role is to internalize and destroy infectious agents by their microbicidal mechanisms. Phagocytosis is triggered by the binding of serum-opsonized microorganisms through opsonin receptors or by the binding of nonopsonized microorganisms mostly through lectin-sugar recognition (23). Two microbicidal processes are activated concomitantly with phagocytosis to create a highly toxic environment inside phagosomes: (i) NADPH oxidase-dependent production of O2−, a precursor of other reactive oxygen species, and (ii) degranulation, which corresponds to the release of azurophil granule content (specialized secretory lysosomes) and other granule types into phagosomes (30). In neutrophils, fusion of azurophil granules with phagosomes occurs very rapidly and takes place with nascent, unclosed phagosomes (33, 34), leading to the release of azurophil granule enzymes in the extracellular medium (4). Therefore, when neutrophils ingest particles, it is easy to follow the biogenesis of phagolysosomes by simply measuring the release of these enzymes in the cell supernatant (7, 22). When compared to neutrophils, fusion of lysosomes with phagosomes in macrophages is delayed by several minutes (31) and inhibited by mycobacteria (1). Since neutrophils have the particularity to very rapidly fuse their azurophil granules with phagosomes, we have recently addressed the question of whether mycobacteria can inhibit this very rapid process (22). We have found that when human neutrophils ingest pathogenic (Mycobacterium avium, M. kansasii) or nonpathogenic (M. smegmatis, M. phlei) mycobacteria under nonopsonic conditions, fusion of azurophil granules with phagosomes does not take place (22). We have recently reported that M. kansasii used the complement receptor 3 (CR3) (also known as CD11b/CD18) associated with glycosylphosphatidylinositol (GPI)-anchored proteins in cholesterol-enriched domains to enter neutrophils (26). Two hypotheses could explain this phenomenon: (i) mycobacteria have the ability to actively and very rapidly control the fusion of azurophil granules, or (ii) mycobacteria are internalized in neutrophils through receptors which trigger phagocytosis but do not initiate intracellular signals, leading to fusion of azurophil granules with phagosomes. To distinguish between these two hypotheses, the release of the lysosomal enzyme β-glucuronidase was monitored during phagocytosis of live or heat-killed M. smegmatis, a representative mycobacterial species which inhibits the biogenesis of phagolysosomes in neutrophils (22).
To study whether it is possible to restore the biogenesis of phagolysosomes by redirecting mycobacteria to other phagocytic receptors, two approaches were used. One approach was to grow bacilli in nutrient-starved medium to induce physiological adaptation of the bacteria, expecting a change in the composition of the mycobacterial surface molecules (29). The second approach was to opsonize mycobacteria in immune serum to direct the bacteria to opsonic receptors. We were able to restore the fusion of azurophil granules during phagocytosis of IgG-opsonized unicellular mycobacteria or mycobacterial clumps. Taken together, these results indicate that mycobacteria do not actively control the behavior of their phagosomes at early time points postinfection (p.i.).
MATERIALS AND METHODS
Reagents.
Ficoll and Dextran T500 were purchased from Amersham-Pharmacia-Biotech (Courtaboeuf, France). Phosphate-buffered saline (PBS), minimum essential medium (MEM), and HEPES were purchased from Life Technology (Cergy Pontoise, France). Fluorescein isothiocyanate (FITC) and rhodamine-conjugated phalloidin were from Molecular Probes (Leiden, The Netherlands). Methyl β-cyclodextrin, fluorescein diacetate, zymosan A, and tetramethyl rhodamine isocyanate (TRITC)-conjugated goat anti-rabbit immunoglobulin G (IgG) were purchased from Sigma (Saint Quentin Fallavier, France). Mouse anti-human CR3 antibodies (clone 2LPM19C) were from Dako (Trappes, France).
Human neutrophils.
Neutrophils were isolated from the blood of healthy donors, separated by the dextran-Ficoll method as previously described (18), resuspended in MEM containing 10 mM HEPES (pH 7.4), and maintained for 20 min at 37°C before stimulation. The final cell suspension contained >95% neutrophils.
Cell viability.
The viability of cells was estimated by measuring the release of the cytosolic enzyme lactate dehydrogenase using the colorimetric assay kit from Boehringer as previously described (7).
Mycobacteria.
M. smegmatis strain ATCC 607 (from the American Type Culture Collection, Manassas, Va.) was used for all experiments. A preculture was prepared by inoculating M. smegmatis from Jensen stock cultures kept at 4°C (Lowenstein-Jensen medium; Institut Pasteur, Paris, France) into 250-ml flasks containing 100 ml of Sauton broth medium. This first culture was grown at 37°C as a surface pellicle for 4 days. The second culture was inoculated from the preculture and grown under the same conditions for 3 days. The pellicle was either used to inoculate new pellicle cultures or disrupted by gentle shaking with glass beads (4-mm diameter) for 30 s (25) and resuspended at an optical density at 650 nm of 0.2 in PBS, pH 7.4, for inoculation of shaken cultures. Bacteria were grown at 37°C in a shaking incubator (250 rpm). Cultures were centrifuged at 10,000 × g for 10 min. Pellicles from day 3 or day 6 cultures or pellets from shaken cultures were disrupted by gentle shaking with glass beads for 30 s and resuspended in PBS, pH 7.4. To remove the larger clumps, the bacterial suspensions were sedimented for 15 min; the supernatants were collected and centrifuged for 10 min at 200 × g (22). The resulting supernatants contained unicellular mycobacteria, whereas pellets contained small mycobacterial aggregates. Unicellular mycobacteria and resuspended small aggregates were counted under a microscope in a Thoma chamber and stored at −80°C in 10% glycerol.
The viability of mycobacteria was assessed by labeling both live and dead bacteria diluted to approximately 107/ml in PBS with 5 μl of propidium iodide (0.1 mg/ml in water) and 5 μl of fluorescein diacetate (1 mg/ml in dimethyl sulfoxide) for 30 min in the dark at room temperature. Bacteria were washed twice with PBS, pH 7.4, and visualized by fluorescence microscopy. The percentage of live (fluorescein-positive [green]) or dead (propidium iodide-positive [red]) bacteria was determined by counting at least 100 bacteria.
In some experiments, M. smegmatis in suspension was heat killed at 80°C for 30 min, and the killing efficiency was ascertained by both the absence of CFU on Sauton medium agar and the lack of fluorescein diacetate labeling.
The surface-exposed components of the mycobacterial cell envelopes were prepared and analyzed as previously described (24, 25).
Opsonisation of zymosan and mycobacteria.
Zymosan particles were incubated in pooled human sera for 20 min at 37°C; washed twice with PBS, pH 7.4; and resuspended in the same buffer containing 1 mM CaCl2 (26) and 0.5 mM MgCl2. Using the above protocol, mycobacteria were opsonized in the serum of the rabbit Camelia immunized against mycobacteria (17), a serum that immunoreacted more with mycobacteria than did human serum from healthy donors (22).
Exocytosis of the lysosomal enzyme β-glucuronidase.
Neutrophils in suspension (5 × 106/ml) were infected for the indicated period of time with mycobacteria or zymosan and pelleted. The supernatants were centrifuged (10,000 × g for 10 min) to eliminate bacteria. The β-glucuronidase activity was measured at 405 nm in both the cell pellets lysed in Triton X-100 and the cell supernatants as described previously (12). Comparable results were obtained with mycobacteria stained or not by FITC.
Phagocytosis measurement.
FITC staining of mycobacteria (109 bacteria) was performed in 1 ml of 0.01% FITC in 0.2 M Na2CO3-Na2HCO3 buffer, pH 9.2, for 10 min and washed twice with PBS, pH 7.4.
Phagocytosis experiments were performed on neutrophils adhering on glass coverslips (7 × 105 cells/ml in a 24-well tissue culture plate) as previously described (22). Briefly, phagocytosis of FITC-stained bacteria (50:1) was carried out for 30 min at 37°C in MEM-HEPES, pH 7.4. Then, neutrophils were washed to remove most of the extracellular bacteria and fixed with 3.7% paraformaldehyde in PBS containing 15 mM sucrose, pH 7.4, at room temperature. After fixation, aldehyde groups were neutralized with 50 mM NH4Cl, and cells were washed in PBS. Extracellular mycobacteria were stained with antimycobacterial antibodies from the rabbit Camelia (17), revealed by TRITC-conjugated anti-rabbit antibodies. Cells which contained green but not red mycobacteria (intracellular bacteria) were counted by fluorescence microscopy. For each condition, duplicate experiments were performed, and at least 100 cells were counted per slide.
We have verified that FITC staining of M. smegmatis did not interfere with phagocytosis. Cells were fixed and extracellular bacteria stained by Camelia antibodies revealed by TRITC-conjugated secondary antibodies, and then cells were permeabilized with 0.1% Triton X-100 and intracellular and extracellular bacteria were revealed by Camelia antibodies and FITC-conjugated secondary antibodies. Extracellular bacteria were yellow (red plus green); intracellular bacteria were green.
Polystyrene microspheres 1 or 3 μm in diameter (2 × 109) were coated with bovine serum albumin as described previously (3). Extracellular latex beads were revealed using anti-bovine serum albumin antibodies and TRITC-conjugated secondary antibodies.
In some experiments, neutrophils were exposed to methyl-β-cyclodextrin (5 mM) for 15 min at 37°C or incubated for 15 min at 37°C with antibodies directed against the I domain of CR3 (2LPM [2 μg/ml]) before infection with FITC-stained bacteria, as described previously (26).
For immunolocalization of Hck, adherent neutrophils were fixed and permeabilized in methanol at −20°C for 6 min, washed in PBS containing 0.1% Tween 20, and then exposed to affinity-purified anti-Hck antibodies (Santa Cruz Biotechnologies, Santa Cruz, Calif.) revealed by rhodamine-conjugated secondary antibodies as previously described (21).
Filamentous actin staining.
Synchronized phagocytosis experiments were performed as previously described (7). Adherent neutrophils were incubated at 4°C with FITC-stained mycobacteria for 20 min and washed to remove nonadherent bacteria, and phagocytosis was initiated by adding the incubation medium at 37°C. Cells were incubated for 5, 15, and 30 min at 37°C and fixed for 6 min at −20°C with acetone. Cells were washed and incubated with rhodamine-conjugated phalloidin for 20 min at room temperature, washed, and visualized by direct fluorescence microscopy. At least 100 cells having ingested mycobacteria were counted per slide.
Statistical analysis.
Data are presented as the mean ± standard deviation (SD) of the indicated number of experiments performed in duplicate or triplicate. Data were analyzed by two-way analysis of variance followed by post hoc multiple comparisons made with Newman-Keuls or Dunnett's tests. The level of significance was set at P < 0.05. Statistical analysis was performed using PRISM 2.0 (GraphPad software).
RESULTS
Mycobacterium smegmatis does not actively control the fusion capacity of phagosomes with azurophil granules.
We have previously shown that when neutrophils ingest nonpathogenic or pathogenic mycobacteria the biogenesis of phagolysosomes does not take place, as measured by the absence of both β-glucuronidase and myeloperoxidase in the extracellular medium and by the lack of lysosomal enzyme translocation to phagosomes by immunoelectron and immunofluorescence microscopy (7, 22). In the present study, M. smegmatis, one of the species previously shown to impair the biogenesis of phagolysosomes in neutrophils (22), was used, and the release of β-glucuronidase was measured as an index of phagolysosome biogenesis (33, 34). Neutrophils were incubated with unicellular bacteria collected during the exponential phase of pellicle growth. As previously reported, live M. smegmatis did not trigger the release of the lysosomal enzyme β-glucuronidase (22). In the presence of heat-killed (30 min at 80°C) mycobacteria, no release of β-glucuronidase was obtained (Fig. 1A). When M. smegmatis was opsonized in the immune serum of the rabbit Camelia, previously reported to contain mycobacterium-reactive IgG (26), the release of β-glucuronidase was triggered (Fig. 1A). In contrast, opsonization of M. smegmatis in serum devoid of immunoreactive IgG did not induce the release of β-glucuronidase (data not shown), indicating that binding of immunoglobulins to the mycobacterial surface is necessary to stimulate this process. Since both dead and live M. smegmatis organisms reside in phagosomes that do not fuse with azurophil granules, and opsonization in immune serum triggers the fusion of azurophil granules with phagosomes, this implies that these bacteria do not actively control the biogenesis of phagolysosomes.
FIG. 1.

Fusion of azurophil granules with phagosomes in neutrophils is triggered by IgG opsonization of M. smegmatis. (A) Neutrophils were incubated with immune serum-opsonized zymosan (OZ) (20 particles per cell) for 20 min or with live, heat-killed or immune serum-opsonized M. smegmatis for 40 min (50 particle per cell). The release of the lysosomal enzyme β-glucuronidase was measured. Results are expressed as means + SDs (error bars) of three experiments. ∗∗∗, P < 0.001 versus control; ○○○, P < 0.001 versus M. smegmatis. (B) M. smegmatis was grown for different periods of time (in days [D]), and the growth was monitored by measuring the optical density at 650 nm and the cell wet weight (insert). Neutrophils were incubated with M. smegmatis collected at different growth stages, and the release of β-glucuronidase was measured. OZ was used to show the level of β-glucuronidase released when lysosomal degranulation is triggered. Results are expressed as means + SDs (error bars) of three experiments. ∗, P < 0.05 versus control.
Infection of human neutrophils with either exponential or stationary phase M. smegmatis does not trigger exocytosis of β-glucuronidase.
Mycobacteria have developed a program toadapt to the limitation of nutrients, and morphological changes have been noticed when they enter into the stationary phase (29). This may lead to the absence or the nonexposure of a putative mycobacterial surface ligand recognized by the couple CR3-GPI receptor shown to be used by mycobacteria to enter neutrophils (26). This absence, in turn, may redirect mycobacteria to other receptors which could activate other transducing signals able to reconstitute the biogenesis of phagolysosomes. To test this hypothesis, neutrophils were incubated with M. smegmatis grown for various periods of time (0 to 15 days).
To compare our data to those already published on stationary-phase adaptation of mycobacteria, we used cultures of M. smegmatis performed in a shaking incubator as previously described (29). The bacilli multiplied actively for the first 3 days, and after the late exponential phase, they entered the stationary phase at day 7 (Fig. 1B, inset). The viability of M. smegmatis was not affected until day 11; during the following days, when the bacilli progressed in the late stationary phase, a rapid increase in the mortality was observed as previously reported (29). Consequently, the subsequent experiments were carried out with bacteria cultured for less than 12 days. No difference was found in the biochemical composition of surface-exposed material when M. smegmatis was collected at exponential or stationary growth phases (data not shown).
When freshly isolated neutrophils were infected with unicellular M. smegmatis grown for different time periods, no exocytosis of azurophil granule lysosomal enzymes was observed (Fig. 1B) and phagocytosis of mycobacteria remained constant (data not shown), indicating that the biogenesis of phagolysosomes was independent of the growth stage of mycobacteria.
Clumps of M. smegmatis trigger the release of β-glucuronidase in human neutrophils.
Mycobacteria are known to form aggregates in culture, and much caution has been taken to infect cells with unicellular bacteria. For this purpose, mycobacterial clumps are gently disrupted with glass beads (24); large aggregates are sedimented, whereas small aggregates and unicellular bacteria remain in the supernatant. A low-speed centrifugation step is necessary to pellet the small bacterial aggregates (22). Unicellular bacteria in the supernatant represent 5 to 10% of the initial culture. To test the possibility that the biogenesis of phagolysosomes may be influenced by the size of engulfed particles, neutrophils were infected with the small bacterial aggregates obtained in the pellets obtained by low-speed centrifugation, which are made of two to five bacteria, as observed by electron microscopy (data not shown). Under these conditions, an extensive release of β-glucuronidase was obtained (Fig. 2A); this did not result from a cytotoxic effect, since no release of the cytosolic enzyme lactate dehydrogenase, a marker of cell death, was observed in these experiments (data not shown). To investigate whether it was possible to reverse the biogenesis of phagolysosomes, the small clumps used as described above were disrupted with glass beads to obtain unicellular mycobacteria which were used to infect neutrophils. The degranulation effect was totally abolished by the declumping of aggregates since no significant release of β-glucuronidase was observed under these conditions (Fig. 2A). Finally, clumps prepared from 3-, 7-, 8-, and 11-day-old cultures were identical in the efficiency with which they triggered the exocytosis of azurophil granules (data not shown).
FIG. 2.

Release of β-glucuronidase is triggered by aggregates of M. smegmatis. (A) Neutrophils were incubated with 6-day-old unicellular mycobacteria (bar 1), small aggregates of mycobacteria (bar 2), or unicellular mycobacteria obtained by disaggregating small aggregates with glass beads (bar 3). The release of β-glucuronidase was measured, and the results are expressed as means + SDs (error bars) of three experiments. Stimulation of β-glucuronidase exocytosis triggered by OZ is also shown. (B) Neutrophils were incubated with 6-day-old unicellular mycobacteria (bars with oblique lines) or small aggregates of mycobacteria (bars with vertical lines) at the indicated multiplicity of infection (MOI). The release of β-glucuronidase was measured, and the results are expressed as means + SDs (error bars) of two experiments performed in triplicate. Statistically significant difference versus control: ∗, P < 0.05; ∗∗∗, P < 0.001.
To determine whether the effect observed with the small aggregates was not the result of an increased bacterium-to-cell ratio, neutrophils were infected with unicellular mycobacteria or aggregates at different multiplicities of infection. As shown in Fig. 2B, unicellular mycobacteria at a ratio as high as 250 did not trigger the exocytosis of β-glucuronidase, while aggregates triggered the exocytosis of the lysosomal enzyme at a 10:1 ratio. These results indicate that only aggregates have the ability to induce the biogenesis of phagolysosomes. In addition, the percentage of neutrophils that internalized aggregates is lower than the percentage that internalized unicellular bacteria (16.7 versus 28.6%), indicating that the release of β-glucuronidase is not dependent on the phagocytic rate but on the type of particle ingested as previously reported (22).
Translocation of the Src family tyrosine kinase Hck onto phagosomes has been shown to reflect the biogenesis of phagolysosomes (35). The presence of Hck on phagosomes was examined 30 min p.i. While Hck is not present on phagosomes containing nonopsonized mycobacteria (22) or heat-killed M. smegmatis (Fig. 3), the kinase was found associated to phagosomes containing nonopsonized M. smegmatis aggregates (Fig. 3).
FIG. 3.
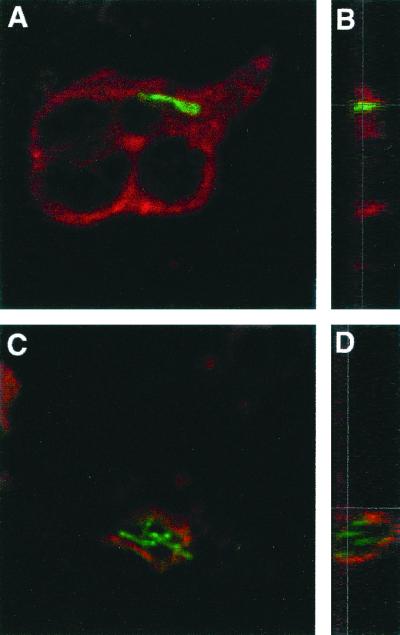
Hck is present on phagosomes containing clumps of M. smegmatis. Neutrophils were coincubated with FITC-stained M. smegmatis aggregates or heat-killed unicellular mycobacteria for 30 min at 37°C. Cells were fixed and permeabilized with methanol at −20°C, and the tyrosine kinase Hck was revealed by TRITC immunostaining. Merged confocal images of phagosomes are shown in horizontal (A and C) or in vertical (B and D) planes. As shown here, phagosomes containing a single bacterium are most of the time in the plane of the cell (A), while phagosomes containing aggregated bacteria are above the plane of the cell (C). Phagosomes surrounding aggregates of M. smegmatis are Hck positive all over their periphery (D). In contrast, Hck staining is diffuse around heat-killed unicellular mycobacteria representing Hck in the cytoplasm and absent at the apex of phagosomes (B), indicating that the kinase is not associated with the phagosomal membrane. One experiment representative of four is shown.
The size of phagocytic particles does not influence the biogenesis of phagolysosomes in human neutrophils.
A previous report on IgG-coated latex beads has shown that macrophage phagosomes containing beads of 1 μm or larger have no lag for delivery to lysosomes, in contrast to smaller beads (16). To further gain insight into the mechanisms by which clumps induce fusion of azurophil granules while unicellular mycobacteria fail to do so, we investigated the effect of the size of the particles ingested by neutrophils on the biogenesis of phagolysosomes. The exocytosis of β-glucuronidase was therefore measured in response to latex beads with diameters of 1 or 3 μm, which approximately correspond to the size of unicellular and clumped bacteria, respectively. The phagocytic rates of 1- and 3-μm-diameter beads were 31.6 and 24.8%, respectively, which were comparable to the phagocytic rates of unicellular and aggregated mycobacteria (Fig. 4), respectively. However, no release of β-glucuronidase was triggered by phagocytosis of latex beads (data not shown). These data indicate that the size of the phagocytic particle and therefore the size of the phagosomes is not critical for azurophil granule exocytosis.
FIG. 4.

Phagocytosis of unicellular or aggregated M. smegmatis is cholesterol and CR3 dependent. Adherent neutrophils were incubated at 37°C for 10 min with 5 mM methyl-β-cyclodextrin or anti-CR3 antibody (2LPM) (2 μg/ml), and unicellular or aggregated FITC-stained mycobacteria were added (50:1). Cells were washed and fixed in paraformaldehyde, and the remaining extracellular bacteria were revealed by rabbit antimycobacterial antiserum and TRITC-conjugated anti-rabbit IgG. The number of cells having ingested at least one particle was determined and expressed as the percentage of the number of phagocytic cells. Results are means + SDs (error bars) of five experiments. Statistically significant difference versus control: ∗, P < 0.05; ∗∗, P < 0.01; ∗∗∗, P < 0.001.
Unicellular or aggregated M. smegmatis bacteria are internalized through the same phagocytic receptors, but only the aggregates recruit F-actin on phagosomes.
We have previously reported that phagocytosis of unicellular M. kansasii by human neutrophils is dependent on cholesterol, CR3, and GPI-anchored receptors (26). Based on the observed differential behavior of unicellular M. smegmatis versus aggregates towards the biogenesis of phagolysosomes, we investigated the possibility that clumped bacteria could be internalized through receptors other than those used by unicellular mycobacteria (26). When neutrophils were incubated with methyl-β-cyclodextrin, a chelator of sterols used to remove cholesterol from the plasma membrane (10, 15, 28, 32) which does not affect phagocytosis of zymosan or opsonized zymosan (26), internalization of unicellular or clumped bacteria was strongly inhibited (Fig. 4). Similarly, preincubation of neutrophils with anti-CR3 antibodies directed against CD11b affected not only phagocytosis of unicellular mycobacteria as previously reported (26) but also that of clumped bacteria (Fig. 4). These results demonstrate that clumped and unicellular M. smegmatis bacteria are internalized through CR3 in a cholesterol-dependent manner as described for unicellular M. kansasii (26).
Receptor clustering has been shown to be required for phagocytosis and actin rearrangements necessary for internalization of particles (11). In addition, rapid delivery of lysosomal markers to phagosomes has been correlated with the presence of actin around phagosomes at early time points (16). Our next attempt was to determine whether clumped mycobacteria, which probably recruit more phagocytic receptors than unicellular mycobacteria, also recruited polymerized actin on their phagosomes. Neutrophils incubated with unicellular or aggregated mycobacteria at different time points were fixed, and F-actin was stained with rhodamine-conjugated phalloidin. As shown in Fig. 5, phagocytic cups of F-actin were observed very early after the contact of neutrophils with aggregated M. smegmatis but were never seen with unicellular bacteria (data not shown). Similarly, 30 min after infection, about 60% of phagosomes containing mycobacterial clumps were F-actin positive, versus 0% of phagosomes containing unicellular M. smegmatis.
FIG. 5.

Phagocytosis of mycobacterial aggregates induces actin polymerization. Adherent neutrophils were incubated in the presence of unicellular FITC-stained mycobacteria (50:1) for 30 min (A and B) or aggregated FITC-stained mycobacteria (C to F) for 5 min (C and D) or 30 min (E and F). Cells were fixed and permeabilized in acetone at −20°C, and F-actin was stained with rhodamine-conjugated phalloidin (A, C, and E) visualized by fluorescence microscopy. At 5 min, phagocytic cups of F-actin (C) (black arrowhead) are seen in front of a mycobacterial clump (D) (white arrowhead). At 30 min p.i., phagosomes containing mycobacterial aggregates are surrounded by F-actin (E and F), whereas this is not seen around phagosomes containing unicellular mycobacteria (A and B). One representative experiment out of three is shown.
DISCUSSION
We have previously shown that at early time points p.i. of human neutrophils with pathogenic or nonpathogenic mycobacteria, the fusion of azurophil granules with phagosomes is inhibited (22). We have also reported that nonopsonic phagocytosis of M. kansasii by human neutrophils takes place in rafts through CR3 associated with GPI-anchored proteins (26). In the present study, we first investigated whether this inhibition process is actively controlled by mycobacteria; the nonpathogenic strain M. smegmatis was used to avoid the potential influence of virulence factors. We showed that heat-killed M. smegmatis resides in phagosomes which do not fuse with azurophil granules, indicating that by 1 h p.i., mycobacteria do not actively release inhibitory molecules or actively act on the phagosomal membrane to modify its fusion competence with azurophil granules. However, additional events may operate at later stages since it has been shown that heat-killed pathogenic mycobacteria reside in phagolysosomes 1 day p.i. (5). Consequently, we propose that, at early time points, mycobacteria do not actively act on their phagosomes but rather the receptor of entry plays a crucial role. In contrast, at late time points, such as hours or days p.i., the receptors of entry may no longer control the phagosomes, and at this stage, mycobacteria may need to be alive to control the fusion capacity of their phagosomes. This hypothesis is consistent with the observation that, at early time points p.i., nonpathogenic as well as pathogenic mycobacteria are found in phagosomes unable to fuse with azurophil granules in both human neutrophils (22) and macrophages (8), whereas it is well known that a few days after infection, only pathogenic mycobacteria reside in phagosomes not fusing with lysosomes. This suggests that after a certain time, virulent mycobacterial factors are expressed to control the behavior of the phagosomes. In addition, we showed that opsonization of mycobacteria in immune serum is able to restore the fusion capacity of their phagosomes with azurophil granules. Similarly, at early time points p.i., human macrophage phagosomes containing mycobacteria do not fuse with Hck-positive lysosomes, while they fuse when they contain immune-opsonized M. kansasii (2). Taken together, these results support the hypothesis that nonopsonic phagocytosis of mycobacteria involves receptors that do not trigger the biogenesis of phagolysosomes. We have reported that, in human macrophages, phagosomes containing particles internalized through the mannose receptor do not fuse with Hck-positive lysosomes (2, 3).
We have recently been able to develop an in vitro assay for fusion between neutrophil phagosomes and azurophil granules (27). This fusion assay requires the presence of cytosol isolated from neutrophils which have been activated to trigger the fusion of azurophil granules. When the activated cytosol is diluted by a cytosol collected from neutrophils infected by live but not heat-killed M. smegmatis (27) the fusion assay is inhibited, suggesting the presence of a factor able to counteract the activated fusion assay. In the present study, we worked in a different context using intact resting neutrophils. When they are infected by dead or live M. smegmatis, no release of β-glucuronidase is observed, suggesting that the inhibitory factor could specifically operate when the biogenesis of phagolysosomes is activated. Thus, mycobacteria probably use multiple mechanisms to control cells of the host.
To examine whether it is possible to restore the fusion of azurophil granules with phagosomes in mycobacterium-infected neutrophils, we searched to redirect M. smegmatis towards other nonopsonic phagocytic receptors. To this aim, mycobacteria were collected at different stages of growth and, consequently, under different physiologic states, expecting that they would differentially interact with neutrophils. We report here that, independently of their state of growth, unicellular M. smegmatis organisms always reside in phagosomes unable to fuse with azurophil granules. In patients with chronic tuberculosis, bacteria are found in different metabolic states, ranging from actively growing bacteria to mycobacteria in the stationary phase (9, 20). It would be interesting to examine on isolated alveolar macrophages whether these bacteria also remain in phagosomes not fusing with lysosomes. We observed that the conditions which permit the restoration of phagolysosome biogenesis under nonopsonic conditions are those involving ingestion of small aggregates of mycobacteria by neutrophils. Similarly, macrophages that ingest aggregates of M. tuberculosis fuse their lysosomes with phagosomes (5). When aggregates of M. smegmatis were declumped with glass beads, no fusion of azurophil granules was obtained. It is therefore clear that only aggregated mycobacteria possess the capacity to trigger the biogenesis of phagolysosomes and that this occurs independently of whether they are dead or alive.
Because we demonstrated that aggregates, as well as unicellular M. smegmatis, were internalized in a cholesterol- and CR3-dependent manner (26), the question of how aggregates can restore fusion was addressed. We showed that the size of the particles by itself was not critical, since phagosomes containing latex beads of 3 μm, a size similar to the average size of the bacterial clumps, did not fuse with azurophil granules. This could be due to the clustering of phagocytic receptors, which is certainly more important in the presence of bacterial clumps than in the presence of a single bacterium. This hypothesis is further supported by a recent work showing that CR3 can trigger distinct signaling pathways in response to its activation and its clustering (36). Actin filaments have been shown to facilitate fusion events at late stages of the endocytic pathway and between phagosomes and endocytic vesicles (6, 14). Recruitment of F-actin at early time points of the phagocytic process correlates with the biogenesis of phagolysosomes (16), and interestingly, this phenomenon was observed during phagocytosis of mycobacterial aggregates by neutrophils but not during phagocytosis of unicellular mycobacteria (this report). Similar observations have recently been reported in mouse macrophages infected with M. avium (13). Moreover, phagocytosis of clumped but not unicellular mycobacteria has been shown to induce an increase in the intracellular calcium concentration (19), an event also known to trigger the exocytosis of azurophil granules in neutrophils (4). Therefore, we propose that clustering of receptors could trap or recruit proteins involved in the signaling towards azurophil granules.
In conclusion, it is possible to restore the fusion of azurophil granules with phagosomes containing M. smegmatis when bacteria are either opsonized in immune serum or ingested as small clumps but not when they are heat killed. This indicates that, at early time points p.i., unicellular mycobacteria do not actively act on the fusogenic capacity of their phagosomes but rather use nonopsonic receptors of entry, which do not trigger the biogenesis of phagolysosomes. Although mycobacterial clumps are internalized through the same receptors, we propose that during their phagocytosis, large clusters of these receptors are produced, leading to the initiation of fusogenic signals for azurophil granules.
Acknowledgments
We gratefully acknowledge A. Labrousse for helpful advice on confocal microscopy, M.-A. Dupont for electron microscopy experiments on mycobacteria, P. Peyron and S. Angot for immunofluorescence experiments, C. Bordier for expert technical assistance, F. Viala for iconography, and C. Gouardères for statistical analysis.
This work was supported in part by Sidaction and European Community grant QLK2CT1999-01093.
REFERENCES
- 1.Armstrong, J. A., and P. D. Hart. 1975. Phagosome-lysosome interactions in cultured macrophages infected with virulent tubercle bacilli. Reversal of the usual nonfusion pattern and observations on bacterial survival. J. Exp. Med. 142:1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Astarie-Dequeker, C., S. Carreno, C. Cougoule, and I. Maridonneau-Parini. The protein tyrosine kinase Hck is located on lysosomal vesicles physically and functionally distinct from CD63-positive lysosomes in human macrophages. J. Cell Sci., in press. [DOI] [PubMed]
- 3.Astarie-Dequeker, C., E. N. N'Diaye, V. Le Cabec, M. G. Rittig, J. Prandi, and I. Maridonneau-Parini. 1999. The mannose receptor mediates uptake of pathogenic and nonpathogenic mycobacteria and bypasses bactericidal responses in human macrophages. Infect. Immun. 67:469-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borregaard, N., and J. Cowland. 1997. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 89:3503-3521. [PubMed] [Google Scholar]
- 5.Clemens, D., and M. Horwitz. 1995. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J. Exp. Med. 181:257-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durrbach, A., D. Louvard, and E. Coudrier. 1996. Actin filaments facilitate two steps of endocytosis. J. Cell Sci. 109:457-465. [DOI] [PubMed] [Google Scholar]
- 7.Francois, M., V. Le Cabec, M. A. Dupont, P. J. Sansonetti, and I. Maridonneau-Parini. 2000. Induction of necrosis in human neutrophils by Shigella flexneri requires type III secretion, IpaB and IpaC invasins, and actin polymerization. Infect. Immun. 68:1289-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frehel, C., C. de Chastellier, T. Lang, and N. Rastogi. 1986. Evidence for inhibition of fusion of lysosomal and prelysosomal compartments with phagosomes in macrophages infected with pathogenic Mycobacterium avium. Infect. Immun. 52:252-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Girling, D. 1989. The chemotherapy of tuberculosis, p. 285-323. In C. Ratledge, J. Stanford, and J. M. Grange (ed.), The biology of the mycobacteria, vol. 3. Academic Press, New York, N.Y.
- 10.Green, J. M., A. Zhelesnyak, J. Chung, F. P. Lindberg, M. Sarfati, W. A. Frazier, and E. J. Brown. 1999. Role of cholesterol in formation and function of a signaling complex involving αvβ3, integrin-associated protein (CD47), and heterotrimeric G proteins. J. Cell Biol. 146:673-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenberg, S. 1999. Modular components of phagocytosis. J. Leukoc. Biol. 66:712-717. [DOI] [PubMed] [Google Scholar]
- 12.Gregoire, C., H. Welch, C. Astarie-Dequeker, and I. Maridonneau-Parini. 1998. Expression of azurophil and specific granule proteins during differentiation of NB4 cells in neutrophils. J. Cell. Physiol. 175:203-210. [DOI] [PubMed] [Google Scholar]
- 13.Guerin, I., and C. de Chastellier. 2000. Disruption of the actin filament network affects delivery of endocytic contents marker to phagosomes with early endosome characteristics: the case of phagosomes with pathogenic mycobacteria. Eur. J. Cell Biol. 79:735-749. [DOI] [PubMed] [Google Scholar]
- 14.Jahraus, A., M. Egeberg, B. Hinner, A. Habermann, E. Sackman, A. Pralle, H. Faulstich, V. Rybin, H. Defacque, and G. Griffiths. 2001. ATP-dependent membrane assembly of F-actin facilitates membrane fusion. Mol. Biol. Cell 12:155-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keller, P., and K. Simons. 1998. Cholesterol is required for surface transport of influenza virus hemagglutinin. J. Cell Biol. 140:1357-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koval, M., K. Preiter, C. Adles, P. D. Stahl, and T. H. Steinberg. 1998. Size of IgG-opsonized particles determines macrophage response during internalization. Exp. Cell Res. 242:265-273. [DOI] [PubMed] [Google Scholar]
- 17.Le Cabec, V., C. Cols, and I. Maridonneau-Parini. 2000. Nonopsonic phagocytosis of zymosan and Mycobacterium kansasii by CR3 (CD11b/CD18) involves distinct molecular determinants and is or is not coupled with NADPH oxidase activation. Infect. Immun. 68:4736-4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Cabec, V., and I. Maridonneau-Parini. 1994. Annexin 3 is associated with cytoplasmic granules in neutrophils and monocytes and translocates to the plasma membrane in activated cells. Biochem. J. 303:481-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majeed, M., N. Perskvist, J. D. Ernst, K. Orselius, and O. Stendahl. 1998. Roles of calcium and annexins in phagocytosis and elimination of an attenuated strain of Mycobacterium tuberculosis in human neutrophils. Microb. Pathog. 24:309-320. [DOI] [PubMed] [Google Scholar]
- 20.Mitchison, D. 1980. Treatment of tuberculosis. The Mitchel lecture 1979. J. R. Coll. Physicians Lond. 14:98-99. [PMC free article] [PubMed] [Google Scholar]
- 21.Mohn, H., V. Le Cabec, S. Fischer, and I. Maridonneau-Parini. 1995. The src-family protein-tyrosine kinase p59hck is located on the secretory granules in human neutrophils and translocates towards the phagosome during cell activation. Biochem. J. 309:657-665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.N'Diaye, E. N., X. Darzacq, C. Astarie-Dequeker, M. Daffe, J. Calafat, and I. Maridonneau-Parini. 1998. Fusion of azurophil granules with phagosomes and activation of the tyrosine kinase HcK are specifically inhibited during phagocytosis of mycobacteria by human neutrophils. J. Immunol. 161:4983-4991. [PubMed] [Google Scholar]
- 23.Ofek, I., J. Goldhar, Y. Keisari, and N. Sharon. 1995. Nonopsonic phagocytosis of microorganisms. Annu. Rev. Microbiol. 49:239-276. [DOI] [PubMed] [Google Scholar]
- 24.Ortalo-Magne, A., M. A. Dupont, A. Lemassu, A. B. Andersen, P. Gounon, and M. Daffe. 1995. Molecular composition of the outermost capsular material of the tubercle bacillus. Microbiology 141:1609-1620. [DOI] [PubMed] [Google Scholar]
- 25.Ortalo-Magne, A., A. Lemassu, M. A. Laneelle, F. Bardou, G. Silve, P. Gounon, G. Marchal, and M. Daffe. 1996. Identification of the surface-exposed lipids on the cell envelopes of Mycobacterium tuberculosis and other mycobacterial species. J. Bacteriol. 178:456-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peyron, P., C. Bordier, E. N. N’Diaye, and I. Maridonneau-Parini. 2000. Nonopsonic phagocytosis of Mycobacterium kansasii by human neutrophils depends on cholesterol and is mediated by CR3 associated with glycosylphosphatidylinositol-anchored proteins. J. Immunol. 165:5186-5191. [DOI] [PubMed] [Google Scholar]
- 27.Peyron, P., I. Maridonneau-Parini, and T. Stegmann. 2001. Fusion of human neutrophil phagosomes with lysosomes in vitro. Involvement of tyrosine kinases of the src family and inhibition by mycobacteria. J. Biol. Chem. 276:35512-35517. [DOI] [PubMed] [Google Scholar]
- 28.Rothberg, K. G., Y. S. Ying, B. A. Kamen, and R. G. Anderson. 1990. Cholesterol controls the clustering of the glycophospholipid-anchored membrane receptor for 5-methyltetrahydrofolate. J. Cell Biol. 111:2931-2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smeulders, M. J., J. Keer, R. A. Speight, and H. D. Williams. 1999. Adaptation of Mycobacterium smegmatis to stationary phase. J. Bacteriol. 181:270-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith, J. 1994. Neutrophils, host defense, and inflammation: a double-edged sword. J. Leukoc. Biol. 56:672-686. [DOI] [PubMed] [Google Scholar]
- 31.Storrie, B., and M. Desjardins. 1996. The biogenesis of lysosomes: is it a kiss and run, continuous fusion and fission process? Bioessays 18:895-903. [DOI] [PubMed] [Google Scholar]
- 32.Subtil, A., I. Gaidarov, K. Kobylarz, M. A. Lampson, J. H. Keen, and T. E. McGraw. 1999. Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc. Natl. Acad. Sci. USA 96:6775-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzaki, E., H. Kobayashi, Y. Kodama, T. Masujima, and S. Terakawa. 1997. Video-rate dynamics of exocytotic events associated with phagocytosis in neutrophils. Cell. Motil. Cytoskeleton 38:215-228. [DOI] [PubMed] [Google Scholar]
- 34.Tapper, H., and S. Grinstein. 1997. Fc receptor-triggered insertion of secretory granules into the plasma membrane of human neutrophils: selective retrieval during phagocytosis. J. Immunol. 159:409-418. [PubMed] [Google Scholar]
- 35.Welch, H., and I. Maridonneau-Parini. 1997. Hck is activated by opsonized zymosan and A23187 in distinct subcellular fractions of human granulocytes J. Biol. Chem. 272:102-109. [DOI] [PubMed] [Google Scholar]
- 36.Whitlock, B. B., S. Gardai, V. Fadok, D. Bratton, and P. M. Henson. 2000. Differential roles for alpha(M)beta(2) integrin clustering or activation in the control of apoptosis via regulation of akt and ERK survival mechanisms. J. Cell Biol. 151:1305-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]