

Abstract
During development, neurons are guided to their targets by short- and long-range attractive and repulsive cues. MICAL, a large multidomain protein, is required for the combined action of semaphorins and plexins in axon guidance. Here, we present the structure of the N-terminal region of MICAL (MICALfd) determined by x-ray diffraction to 2.0 Å resolution. The structure shows that MICALfd is an FAD-containing module structurally similar to aromatic hydroxylases and amine oxidases. In addition, we present biochemical data that show that MICALfd is a flavoenzyme that in the presence of NADPH reduces molecular oxygen to H2O2 (Km,NAPDH = 222 μM; kcat = 77 sec-1), a molecule with known signaling properties. We propose that the H2O2 produced by this reaction may be one of the signaling molecules involved in axon guidance by MICAL.
Keywords: hydrogen peroxide, hydroxylase, monooxygenase, x-ray diffraction
During neural development, axons are guided to their targets by a variety of environmental cues. These guidance cues may take the form of long-range signaling by diffusible molecules or short-range interactions by membrane-bound molecules, which either attract or repel the axon. One family of effector molecules, the semaphorins, includes secreted and transmembrane proteins that act as repellents in a variety of axon development processes. Repulsive guidance by semaphorins is mediated through their interaction with plexins, a family of transmembrane proteins that function as their receptors (1). It was shown recently that, in Drosophila, a large multidomain cytosolic protein, MICAL, is required for the repulsive axon guidance mediated by interaction of Semaphorin 1a and Plexin A (2).
Since the MICAL family of proteins was first identified, family members have been implicated in a variety of cellular processes ranging from vesicle trafficking to association with intermediate filaments in mesenchymal cells to involvement in axon pathfinding (2-4). Three MICAL genes (MICAL-1 to -3) have been identified in mammals and one in Drosophila (2). In all cases, MICAL is a >1,000-aa protein that contains a large N-terminal domain, followed by a calponin homology (CH) domain, a LIM domain (double Zn-finger), a proline-rich domain, and a coiled-coil ERM α-like motif (2). This domain architecture, conserved among fly, mouse, and human family members (2), explains the large number of protein-protein associations involving MICAL that have been observed. The proline-rich domain in human MICAL-1 was shown to be a binding site for CasL, a molecule in the β1 integrin signaling cascade (5). (The name MICAL derives from Molecule Interacting with CasL.) The CH and LIM domains (5) have been implicated in signal transduction and cytoskeletal organization (6, 7). Finally, the N-terminal portion of MICAL is an FAD-binding domain that has sequence motifs characteristic of monooxygenases (2).
Yeast two-hybrid assays and immunoprecipitation experiments showed that Drosophila MICAL (D-MICAL) binds to the Plexin A receptor via its C-terminal coiled-coil motif (2). Classical genetic analysis indicated that D-MICAL mediates Sema-1a-induced defasciculation, branching, and general axon pathfinding (2). Genetic analysis of flies carrying a mutation in the FAD-binding domain of D-MICAL indicated that this domain is necessary for Sema-1a signaling. Finally, Terman et al. (2) showed that flavoprotein monooxygenase inhibitors were able to abrogate the repulsive guidance response in rat nerve growth factor-dependent dorsal root ganglion axons. These findings were interpreted as indicating that MICAL's axon guidance activity may be mediated by its putative monooxygenase activity.
Flavoprotein monooxygenases contain a characteristic N-terminal GxGxxG consensus sequence that is required for FAD binding (8). MICAL contains this sequence but lacks an identifiable NADPH/NADH-binding consensus sequence (2), reminiscent of the glutathione reductase (GR2) oxidoreductase family members (8). Because no substrate has been identified for the MICAL FAD-binding domain (MICALfd), various hypotheses linking MICAL's enzymatic activity with cytoskeletal reorganization were proposed (2, 9), including direct oxidation and destabilization of actin filaments, production of reactive oxygen species that affect downstream signaling molecules, and direct oxidation of other signaling molecules (9).
Despite extensive investigation, questions related to substrate identity, mechanism of action, and the interactions of MICALfd with other domains within MICAL remain unanswered. In this article, we report the determination of the structure of the FAD-binding domain of mouse MICAL-1 (MICALfd). In addition, we determined that in the presence of NADPH and molecular oxygen, MICALfd uses the reducing equivalents of NADPH to produce H2O2, a molecule that may be acting as a neuron guidance signal.
Materials and Methods
Cloning and Mutational Surface Engineering. A plasmid containing cDNA coding for residues 1-484 of MICAL1 from Mus musculus (accession no. BC021477) was cloned into the pET28a expression vector that contains an N-terminal 6× His-tag. After extensive unsuccessful crystallization trials, surface-site mutations (K141A and K142A) were made within the same construct to enhance crystallization (10-13). Mutations were introduced with the QuikChange Site-Directed Mutagenesis kit (Stratagene).
Protein Purification and Crystallization. The construct was transformed into Escherichia coli Rosetta (DE3) (pLysS). MICALfd expression was induced by the addition of 1 mM isopropyl-β-d-thiogalactoside (IPTG), and cells were grown for 14 h at 15°C before harvesting by centrifugation. The protein was purified by using Ni-Sepharose affinity chromatography with a gradient of 20 mM Tris·HCl, 500 NaCl, and 5-250 mM imidazole (pH 8). After elution, the protein was dialyzed against 20 mM Hepes, 100 mM NaCl, and 1 mM DTT (pH 7) and digested overnight with thrombin (1 units/ml) at room temperature to remove the His-tag. The cleaved product was purified by using a Source S15 cation exchange column, eluting with a gradient of 20 mM Hepes, 0.1-1 M NaCl, and 1 mM DTT (pH 7). The final yield was ≈10 mg per liter of culture. The purified protein has an absorbance spectrum with peaks at 360 and 460 nm (2). For crystallization, 1 μl of protein solution [4 mg/ml in 20 mM Hepes, 100 mM NaCl, and 1 mM DTT (pH 7)] was combined with 1 μl of reservoir solution [100 mM NaOAc (pH 4.6), 23.5% PEG 2000 MME, and 0.3 M (NH4)2SO4] and equilibrated against 0.5 ml of reservoir solution. Crystals (≈50 × 300 × 300 μm) grew in 24 h at 20°C. MICALfd crystals were frozen in 100 mM NaOAc (pH 4.6), 42% PEG 2000 MME, and 0.3 M (NH4)2SO4. For heavy atom preparation, crystals were soaked in 10 μl of reservoir solution supplemented with heavy atom compounds. MICALfd crystals soaked with 1 mM PCMBS (p-chloromercuribenzene sulfonate) for 2 h were used for phase determination.
Data Collection, Structure Determination, and Refinement. Diffraction data of both native and heavy atom-containing crystals, collected at Brookhaven National Laboratory beamlines X6A and X29, were processed with hkl2000 (14). Isomorphous and anomalous differences of the PCMBS derivative (Table 1) were used to calculate phases with the program solve (15). Phases were improved and extended in the range 30-2.5 Å by density modification with the program resolve (15). After automatic (15) and manual building with o (16-18), the local symmetry and the molecular envelope were identified [Uppsala Software Factory package (16-18)] and used for map improvement by density averaging with the program dm. The structure was refined by using data from 50 to 2.0 Å with the program refmac (19). A subset of 5% of the data was used for cross-validation. After rebuilding and several rounds of positional and atomic B-factor refinement (maximum-likelihood residual), solvent molecules were added with the program arp/warp (20) and manually. A solvent electron density larger than a water molecule present in a highly positively charged pocket was interpreted as a chloride ion. The final rounds of refinement were carried out by using translation, libration, screw-rotation (TLS) refinement (21) as implemented in refmac5. The final model contained 951 amino acids spanning residues 8-146 and 151-484 (monomer A) and residues 1-146, 152-264, and 266-484 (monomer B), two FAD molecules, a chloride ion, and 617 water molecules (Table 1).
Table 1. Data collection and refinement statistics.
| Data set | Native | PCMBS |
|---|---|---|
| Data collection and phasing statistics | ||
| Space group | P21 | P21 |
| Cell dimensions (a, b, c) | 72.97, 87.34, 80.81 | 73.06, 87.72, 80.86 |
| Cell angles (α, β, γ), ° | 90, 111.68, 90 | 90, 112, 90 |
| Resolution, Å | 28-2.0 | 30-2.5 |
| Soaking conditions | ||
| Metal, mM | 1 | |
| Time, h | 2 | |
| Observed reflections | 217,045 | 124,310 |
| Unique reflections | 62,811 | 34,642 |
| Completeness,* % | 98.9 (94.0) | 98.9 (93.8) |
| I/σ† | 15.4 (1.6) | 32.2 (9.1) |
| Rsym‡ | 0.11 (0.59) | 0.08 (0.15) |
| Figure of merit | 0.34 | |
| Refinement | ||
| Rcrystal/Rfree | 19.2/26.6 | |
| Model composition§ | ||
| Amino acids | 951 | |
| Cofactor | FAD | FAD |
| Water molecules | 617 | |
| Ions | Cl− | |
| Total atoms | 8,137 | |
| Stereochemistry¶ | ||
| rms bond length, Å | 0.02 | |
| rms angles, ° | 2.0 | |
| Temp factors, Å2 | ||
| B-factor protein | 35.0 | |
| B-factor cofactor | 25.1 | |
| B-factor water | 43.4 |
Completeness in the highest-resolution shell (in parentheses).
I/σ in the highest-resolution shell (in parentheses).
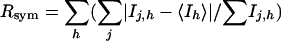 , where h is set of Miller indices and j is set of observations of reflections h.
, where h is set of Miller indices and j is set of observations of reflections h.
Model composition shows two monomers in asymmetric unit.
Over 96% of main chain dihedrals fall within the “most allowed regions” of the Ramachandran plot.
Drawings were prepared with molscript (22), pymol (23),grasp (24), povray (25), espript (26), and vmd (27).
Sedimentation Velocity Analysis. The molar mass and sedimentation coefficient of MICALfd were analyzed by using a Beckman Coulter XL-I analytical ultracentrifuge. Samples contained 4 mg/ml MICALfd in 20 mM Hepes, 100 mM NaCl, and 1 mM BME (pH 7.0). Reference and sample solutions were loaded into double-sector centerpieces and mounted in a Beckman An-50 Ti rotor. Experiments were performed at 4°C and 20°C with a rotor speed of 50,000 rpm. Sample absorbances at 295 and 370 nm were monitored in a continuous mode with no delay and a step size of 0.003 cm without averaging. Multiple scans at different time points were fit to a continuous size distribution by using sedfit (28). The c(s) distribution in sedfit was exported to sedphat, where experimental values were corrected to S20,w. A partial specific volume of 0.7426 was used in all calculations based on the amino acid sequence of the protein. The solvent density and viscosity were calculated by using sednterp (29).
Kinetic Studies. The UV-visible absorption changes in the reactions of MICALfd were monitored at 298 K with a Cary 50 Bio UV-Visible spectrophotometer. Enzyme activity was monitored by the rate of NAD(P)H oxidation (ε340 = 6.2 mM-1·cm-1), and the rate of H2O2 production was measured by using the Amplex Red hydrogen peroxide/peroxidase assay kit (catalog no. A-22188, Molecular Probes; ε560 = 44.3 mM-1·cm-1). Absorbance values were measured over a period of 5 min.
Assays were carried out in 110 μl of a solution containing 20 mM Hepes, 100 mM NaCl (pH 7), and 6.02 nM MICALfd. In assays measuring the Km and kcat for NAD(P)H oxidation, NAD(P)H was added at concentrations ranging from 20 to 500 μM. For the inhibition studies, inhibitor (-)-epigallocatechin gallate (EGCG) was added at concentrations of 30 and 60 μM, and initial rates measured at NADPH concentrations varied from 20 to 200 μM.
Results
Structure Determination. Crystals obtained under several conditions with the wild-type protein were not suitable for x-ray diffraction studies. To favor the formation of other crystal forms, we mutated two surface lysines (K141A and K142A) (12) that were inferred by sequence analysis to be on the surface of the protein. (The structure determination confirmed that the two residues were on the surface of the protein far from the active site.) Well diffracting crystals of the mutated protein were readily obtained. They contain two molecules in the asymmetric unit and have a solvent content of 47%. The structure was determined by using a single isomorphous heavy atom derivative including its anomalous signal (Table 1). The final model was refined with data to 2.0 Å resolution to an R-value of 19.2% (Rfree = 26.6%) with excellent geometry.
Description of the Structure. MICALfd is a mixed α/β protein that contains both parallel and antiparallel β-sheets (Fig. 1A). It is composed of two subdomains of different sizes linked by two β-strands (β9 and β15; residues 227-233 and 367-373) (Fig. 1B). The large subdomain (domain-1; residues 1-226 and 373-484) contains the two FAD sequence motifs (residues 84-114 and 386-416) and a third conserved motif (residues 212-225) (2). The first sequence, which contains the GXGXXG motif (Fig. 2), is part of a Rossmann β-α-β fold (β1-α5-β2 in MICALfd;Fig.1B). This is a sequence commonly found in FAD- and NAD(P)H-dependent oxidoreductases (8). The second motif, which contains the conserved GD sequence in hydroxylases, forms part of a strand and a helix (β17 and α15). In MICALfd, this second conserved sequence (30), makes contacts with the ribose moiety of FAD in a manner similar to that observed in the rubredoxin reductase of Pseudomonas oleovorans (31). The third conserved motif is part of a β-strand (β8) and the following loop.
Fig. 1.

Secondary and tertiary structure of MICALfd.(A) Ribbon diagram of the MICAL FAD-binding domain. In this view, the two subdomains of the structure and the two strands that connect them are clearly visible. In domain-1 (large domain at the top of the figure), the central core is formed by a parallel β-sheet. All of the connecting helices are in one side of the sheet, and a short β-hairpin covers the other side. Domain-2 (small domain at the bottom) contains an antiparallel β-sheet and several short helices. (B) Schematic representation of the MICALfd secondary structure. Strands are represented by arrows, helices are represented by cylinders, and loops are represented by a connecting line. Residue numbers at the beginning and the end of elements of secondary structure are shown. The different structural features are colored for identification. The N-terminal portion, containing the two helical hairpins, is colored blue. Other elements of domain-1 are colored red, maroon, and orange (red, main β-sheet; maroon, helices at one side; orange, strands at the other side). Domain-2 is colored light green, and the two connecting strands are colored pale green.
Fig. 2.

Alignment of the sequences of MICALfd with structural homologs. Single-letter amino acid codes are used. Three sequences are included. (i) MICAL: residues 85-442 of the MICAL-1 of mouse. Residues 1-484 of this protein correspond to MICALfd, but residues 1-84 of MICALfd form a domain that is not present in the other enzymes. (ii) Human monoamine oxidase B (Protein Data Bank entry 1GOS). Only residues 4-95 and 198-456 are shown. (iii) pHBH from Pseudomonas fluorescens residues 1-340 (Protein Data Bank entry 1PBE). The positions of the elements of secondary structure of MICAL, pHBH, and 1GOS are shown at the top of each sequence. The sequences corresponding to the conserved motifs are shown with blue letters. Letters with green background correspond to regions with large insertions and deletions. The two alanine residues with the green background correspond to the mutations introduced to improve the crystals. Residues identical in all three sequences have magenta backgrounds. Residues that contact the FAD cofactor are shown with cyan background. An insertion of 100 residues between α7 and β4, present only in amine oxidase, has been omitted.
In domain-1, residues 1-71 form two perpendicular helical hairpins (helices α1-α4) that connect to the central strand (β1) of the major parallel β-sheet. This sheet, composed of five parallel strands (β1, β2, β5, β8, and β17) and one antiparallel strand (β16), is centrally located within the molecule. The small subdomain (domain-2; residues 238-366) is inserted between strands β8 and β16 of this main sheet. It contains a five-stranded antiparallel β-sheet (β10-β14) and several connecting helices (α10-α14). Two additional strands, one at the beginning (β9) and one at the end (β15) of this domain, form a two-stranded sheet that makes main chain H-bonds with the equivalent two-stranded sheet of another monomer in the crystal asymmetric unit (see below). Several other elements complete the structure of MICALfd, including a C-terminal helical hairpin that ends in an additional short helix (Fig. 1B).
Structural Similarity to Other FAD-Containing Enzymes. The fold of MICALfd is similar to that of other FAD-requiring enzymes. A dali search of known structures revealed that the protein is most similar to aromatic hydroxylases, especially the p-hydroxybenzoate hydroxylase (pHBH) from Pseudomonas fluorescens (rms 1.79 Å for 199 of 484 aligned α-carbons) (32). It is clear from the fraction of aligned residues that, although the two proteins share large portions of their folds, many of the regions connecting the elements of secondary structure diverge between the two proteins (Fig. 2). In particular, the bacterial hydroxylase lacks the two N-terminal helical hairpins of MICALfd. The sequence identity between the two proteins is only 5% for the whole sequence and 11% for the structurally matched residues. A portion of the MICALfd fold is also shared by amine oxidases, the most similar of which is the human monoamine oxidase B (rms 1.85 Å for 116 aligned α-carbons; sequence identity: total 5%, matched residues 21%) (33, 34). The three proteins share core β-sheets and helices (Fig. 2) and diverge in stretches connecting secondary structure elements. The monoamine oxidase differs from the monooxygenases in two significant ways: (i) it contains an additional 100-residue helical domain between α7 and β4 (not shown in Fig. 2); and (ii) the loop connecting strand β17 to helix α15 (residues 390-405) is shorter in monoamine oxidase, allowing the isoalloxazine ring of the FAD to move 3.5 Å closer to the center of molecule. This displacement enlarges the active site cavity above the position of the FAD N5, allowing the covalent attachment of long amine substrates.
MICALfd Is a Monomer. The crystallographic asymmetric unit of the crystals used in this study contains two MICALfd molecules related by a local twofold axis of symmetry. A continuous four-stranded β-sheet (β15-β9-β9′-β15′; primes refer to the second monomer) extends across the dimer interface, with two equivalent β9-strands (residues 228-233), one from each monomer, making six main chain H-bonds to each other and burying 1,601 Å2. This arrangement suggested that the active form of MICAL might be a dimer. However, sedimentation velocity experiments at 4°C and 20°C with an analytical ultracentrifuge showed that in a 80 μM protein solution (4 mg/ml, the concentration used in the crystallization experiments), MICALfd behaves as a monomer with no indication of the presence of dimers (Fig. 3).
Fig. 3.

Analytical ultracentrifugation experiments. The sedimentation of MICALfd was analyzed with a Beckman Coulter XL-I analytical ultracentrifuge at 20°C and 4°C and at 50,000 rpm. Only the experiments at 20°C are shown. (A) Typical sedimentation velocity absorbance trace of MICALfd at 370 nm. (B) Residuals of the experimental fits. (C) Continuous sedimentation coefficient distribution of the protein. (D) Continuous molar mass distribution of the protein showing a single peak with a molecular mass of 52.4 kDa.
FAD Conformation and FAD-Binding Site. FAD binds toward one side of the MICALfd molecule between the two subdomains (Fig. 1A). The isoalloxazine ring is highly planar, and its dimethyl benzene and middle rings stack almost perfectly with the indole moiety of Trp-400 (Fig. 4 A and B), a residue within the region of the second conserved sequence motif. The side chain of Asn-123 makes hydrogen bonds to N5 and O4 of the isoalloxazine, and O4 also makes a hydrogen bond to the OH of Tyr-293. The ribitol moiety extends in a direction almost perpendicular to the isoalloxazine, bending at C5′ before connecting to the diphosphate. O2′ forms an H-bond to the guanidinium of Arg-121 and O3′ to the carboxylate of Asp-393 of the second motif. The ribitol phosphate makes H-bonds to the main chain NHs of Asp-393 and Cys-95 of the first motif. The ribose phosphate makes two H-bonds to the guanidinium of Arg-121. Glu-114 (first motif) makes H-bonds to O2′ and O3′ of the ribose. O2′ also makes an H-bond to the guanidinium group of Arg-116. The adenine ring stacks against the extended aliphatic side chain of Lys-115, with N6 making an H-bond to the main chain NH of Phe-181.
Fig. 4.

Binding of FAD to MICALfd.(A) Electron density of the bound FAD. The SigmaA-weighted map was calculated by using 2mFo-DFc coefficients (45). Some of the protein residues contacting the FAD are shown. (B) Stereoview of the interactions of FAD with MICALfd residues. The following coloring scheme was used for the protein atoms: C, cyan; N, blue; O, red. Water molecules are represented by red spheres. Colors used for the FAD differ only in that carbons are green and phosphates are magenta. (C) Two bound conformations of FAD. The surface of the protein in the cavity around the FAD is shown as an electrostatic surface. The protein atoms surrounding the cavity are shown with carbons colored green, nitrogens colored blue, and oxygens colored orange. The same colors are used for the FAD in the “out” conformation (crystal) with the addition of magenta for phosphorous. The portion of the FAD that has moved in the “in” conformation (model) has yellow carbon atoms.
The edges of the pyrimidine and middle rings of the FAD are exposed to solvent through separate openings at either side of the protein. This exposure is in contrast to other monooxygenases, in which the path for accessing the isoalloxazine ring is from only one side and is usually less open than either opening of MICALfd (32-34). All donors and acceptors of the pyrimidine ring make H-bonds with either protein atoms or water molecules (Fig. 4A).
The conformation of the FAD in the structure reported here is equivalent to the oxidized, “out” conformation in the bacterial pHBH (35-37). In reduced pHBH the FADH2 adopts an “in” conformation that occupies a volume surrounded by the glycine residues of the second conserved motif. This “in” conformation cannot accomodate larger side chains at these positions, explaining the high conservation of this motif. MICALfd has the same four glycines (Fig. 2) and therefore has the same available space adjacent to these residues but in the oxidized form reported here is occupied by waters. However, it is possible for the reduced isoalloxazine to pivot around the C4′-C5′ bond of ribitol and swing into this volume (Fig. 4C), displacing the occupying water molecules.
Identification of an Enzymatic Activity of MICALfd. The strong structural similarity of MICALfd to the P. fluorescens p-hydroxybenzene hydroxylase suggested that the two proteins might have similar enzymatic activities. Because, as isolated, MICALfd contains oxidized FAD, reduction of the cofactor was tested by using either NADH or NADPH. Under the conditions of the experiment, although no net reduction of the FAD was detected (data not shown), a steady, time-dependent oxidation of reduced nicotinamide dinucleotide was observed (Fig. 5A). This observation suggested that enzyme-bound FADH2 was formed but was then reoxidized by oxygen. The resulting production of H2O2 was confirmed by monitoring its formation with horseradish peroxidase in the presence of 10-acetyl-3,7-dihydroxyphenoxazine (Fig. 5A). The rate of reaction was >10 times faster with 200 μM NADPH than with 200 μM NADH (data not shown), suggesting that MICAL is probably an NADPH-dependent enzyme.
Fig. 5.

Kinetic characterization of the MICALfd reaction. (A) Kinetics of NADPH oxidation and H2O2 production. MICALfd (6.02 nM) was incubated in a reaction mixture containing 200 μM NADPH and 3 units of the Amplex Red peroxide/peroxidase assay kit with 1.5 mM 10-acetyl-3,7-dihydroxyphenoxazine. The absorbance peak at 340 nm reflects the concentration of reduced NADPH, and the peak at 560 nm reflects the concentration of H2O2. The experiment ran for 5 min after the addition of the enzyme (time 0). Spectra are shown every 1 min. (Inset) Initial rates as a function the NADPH concentration. The continuous curve was adjusted to the Michaelis-Menten equation by nonlinear least squares with GNUPLOT. (B) pH dependence of the MICAL reaction. (C) Inhibition by EGCG. The reaction rate was measured as a function of the NADPH concentration for concentrations of 0.0, 30.0, and 60.0 μM.
The origin of this activity may correspond to one of three cases. First, that H2O2 is the physiological product of the enzyme and is a component of the avoidance signal. Second, as with other FAD hydroxylases, in the absence of substrate the hydroperoxide form of the enzyme (the MICALfd-FADH-O2H intermediate) decomposes, producing hydrogen peroxide and oxidized enzyme-bound FAD (Scheme 1). Third, the enzyme may actually be an amine oxidase in which the FADH2 is reoxidized by molecular oxygen, and the reduction by NADPH is a fortuitous, nonspecific reaction. Although this last case is unlikely, discrimination among these possibilities requires further experimentation.
Scheme 1.

Kinetics and Inhibition of MICALfd. Kinetic characterization of MICALfd was carried out by measuring the rate of oxidation of NAD(P)H for concentrations between 20 and 500 μM. Analysis of the data showed that the kcat for H2O2 production is 77 sec-1 and the Km of NADPH is 222 μM (Fig. 5 A Inset). The optimum pH of this activity is ≈7 (Fig. 5B). A monooxygenase inhibitor, EGCG, has been shown to inhibit the axon guidance activity of MICAL (2). In the presence of NADPH, EGCG inhibits H2O2 production by MICALfd with an inhibition constant (Ki) of 2 μM. The inhibition by EGCG is noncompetitive with respect to NADPH (Fig. 5C), indicating that EGCG does not bind to the NADPH site.
Binding of NADPH to MICALfd. Mechanistically, NADPH must bind to the oxidized form of the enzyme. In this form, there is a cavity at the exposed end of the pyrimidine ring of the FAD that is lined with positively charged residues (arginines 136, 158, 356, and 116, and lysine 118; Fig. 6). A Cl- ion found in this region of the structure may occupy the position that the NADPH 2′-phosphate occupies in the complex.
Fig. 6.
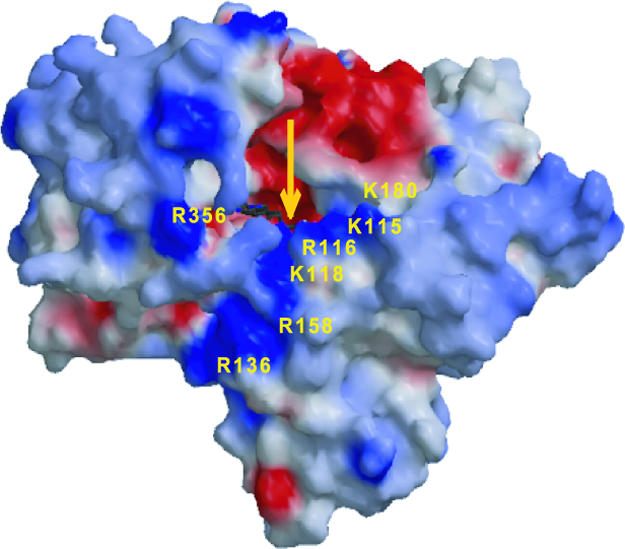
Charge distribution in the MICALfd surface. Positive areas are shown in blue, negative areas are shown in red, and neutral areas are shown in gray. The residues surrounding a strong positive charge feature assumed to be involved in NADPH binding are indicated. The yellow arrow indicates to the location of the FAD isoalloxazine ring.
Discussion
In Drosophila, the multidomain cytosolic protein MICAL is expressed in axons, where it interacts with the neuronal Plexin A receptor (2). Expression of MICAL is required for Semaphorin 1a/Plexin A-mediated repulsive axon guidance. It was shown that MICALfd is required for MICAL's physiological function and that inhibitors of monooxygenase activity such as EGCG abate the action of MICAL (2). We have demonstrated that MICALfd can reduce molecular oxygen to H2O2 using NADPH as the source of reducing equivalents, and that this activity is inhibited by EGCG.
The synthesis of a specific metabolite and the production of reactive oxygen species have been proposed previously as possible mechanisms of MICAL signaling (9). We showed that the FAD-binding domain of MICAL can generate at least the second kind of signals: MICAL reduces molecular oxygen using NADPH to produce H2O2, a possible signaling molecule in processes involving protein phosphorylation (38, 39) and direct actin modification (40-42). Oxidation of actin has been shown to play a major role in actin remodeling. For example, G-actin oxidized by air (41) results in disulfide-bonded dimers that may be incorporated into F-actin filaments during assembly and allow for cross-linking between filaments. In vitro studies of exposure of G-actin to H2O2 showed that several methionine residues may also be oxidized (43, 44). Furthermore, oxidation of methionine and cysteine residues produces conformational changes within the actin molecule that may affect the integrity of the assembled F-actin. Thus, the action of hydrogen peroxide in axon guidance may be related to its ability to modify the biophysical characteristics of the cytoskeleton.
Independently of the mechanism, our observations relate the enzymatic activity of MICAL to its physiological function by identifying a small-molecule candidate that can act as a diffusible effector in axon guidance. In addition, hydrogen peroxide may react with endogenous small molecules and change their properties so they can act as additional signaling molecules. In addition to molecular oxygen, other endogenous molecules or their products of reaction with H2O2 may also be substrates of MICAL resulting in additional signaling molecules. Thus, the results presented here offer a clear path for the identification of additional signaling small molecules active in axon guidance by MICAL.
Acknowledgments
We thank A. Kolodkin (The Johns Hopkins University School of Medicine) for the gift of the DNA containing the MICALfd coding sequence and D. Leahy and J. Lorsch for reading the manuscript. The data were collected on beamlines X6A and X29A of the National Synchrotron Light Source at the Brookhaven National Laboratory. We thank M. Allaire and H. Robinson for help during data collection at the National Synchrotron Light Source. This work was supported by National Institute of General Medical Sciences Grant GM45540.
Author contributions: M.N., M.A.B., S.B.G., J.B., and L.M.A. designed research; M.N., M.A.B., S.B.G., and J.B. performed research; M.N., M.A.B., S.B.G., J.B., and L.M.A. analyzed data; and M.N. and L.M.A. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: EGCG, (-)-epigallocatechin gallate; pHBH, p-hydroxybenzoate hydroxylase.
Data deposition: The atomic coordinates of MICALfd have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2BRA).
References
- 1.Winberg, M. L., Noordermeer, J. N., Tamagnone, L., Comoglio, P. M., Spriggs, M. K., Tessier-Lavigne, M. & Goodman, C. S. (1998) Cell 95, 903-916. [DOI] [PubMed] [Google Scholar]
- 2.Terman, J. R., Mao, T., Pasterkamp, R. J., Yu, H. H. & Kolodkin, A. L. (2002) Cell 109, 887-900. [DOI] [PubMed] [Google Scholar]
- 3.Fischer, J., Weide, T. & Barnekow, A. (2005) Biochem. Biophys. Res. Commun. 328, 415-423. [DOI] [PubMed] [Google Scholar]
- 4.Weide, T., Teuber, J., Bayer, M. & Barnekow, A. (2003) Biochem. Biophys. Res. Commun. 306, 79-86. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki, T., Nakamoto, T., Ogawa, S., Seo, S., Matsumura, T., Tachibana, K., Morimoto, C. & Hirai, H. (2002) J. Biol. Chem. 277, 14933-14941. [DOI] [PubMed] [Google Scholar]
- 6.Gimona, M., Djinovic-Carugo, K., Kranewitter, W. J. & Winder, S. J. (2002) FEBS Lett. 513, 98-106. [DOI] [PubMed] [Google Scholar]
- 7.Bach, I. (2000) Mech. Dev. 91, 5-17. [DOI] [PubMed] [Google Scholar]
- 8.Dym, O. & Eisenberg, D. (2001) Protein Sci. 10, 1712-1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ventura, A. & Pelicci, P. G. (2002) Sci STKE 2002, PE44. [DOI] [PubMed] [Google Scholar]
- 10.Chou, K. C. (2000) Anal. Biochem. 286, 1-16. [DOI] [PubMed] [Google Scholar]
- 11.Zhang, C.-T. & Chou, K. C. (1997) Biopolymers 41, 673-702. [Google Scholar]
- 12.Derewenda, Z. S. (2004) Structure (Cambridge, Mass.) 12, 529-535. [Google Scholar]
- 13.Shepherd, A. J., Gorse, D. & Thornton, J. M. (1999) Protein Sci. 8, 1045-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 276, 307-326. [DOI] [PubMed] [Google Scholar]
- 15.Terwilliger, T. (2004) J. Synchrotron Radiat. 11, 49-52. [DOI] [PubMed] [Google Scholar]
- 16.Kleywegt, G. J. & Jones, T. A. (1996) Acta Crystallogr. D 52, 826-828. [DOI] [PubMed] [Google Scholar]
- 17.Kleywegt, G. & Jones, T. (1994) From First Map to Final Model (Science and Engineering Research Council, Daresbury Laboratory, Warrington, U.K.).
- 18.Jones, T., Zou, J., Cowan, S. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110-119. [DOI] [PubMed] [Google Scholar]
- 19.Collaborative Computational Project, Number 4 (1994) Acta Crystallogr. D 50, 760-763.15299374 [Google Scholar]
- 20.Perrakis, A., Sixma, T. K., Wilson, K. S. & Lamzin, V. S. (1997) Acta Crystallogr. D 53, 448-455. [DOI] [PubMed] [Google Scholar]
- 21.Winn, M. D., Isupov, M. N. & Murshudov, G. N. (2001) Acta Crystallogr. D 57, 122-133. [DOI] [PubMed] [Google Scholar]
- 22.Kraulis, P. (1991) J. Appl. Crystallogr. 24, 946-950. [Google Scholar]
- 23.Delano, W. (2002) The PyMOL User's Manual (Delano Scientific, San Carlos, CA).
- 24.Nicholls, A., Sharp, K. A. & Honig, B. (1991) Proteins 11, 281-296. [DOI] [PubMed] [Google Scholar]
- 25.Fenn, T. D., Ringe, D. & Petsko, G. A. (2003) J. Appl. Crystallogr. 36, 944-947. [Google Scholar]
- 26.Gouet, P., Courcelle, E., Stuart, D. I. & Metoz, F. (1999) Bioinformatics 15, 305-308. [DOI] [PubMed] [Google Scholar]
- 27.Humphrey, W., Dalke, A. & Schulten, K. (1996) J. Mol. Graphics 14, 33-38. [DOI] [PubMed] [Google Scholar]
- 28.Schuck, P. (2000) Biophys. J. 78, 1606-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Philo, J. S. (2005) Biophys. J. 72, 435-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eppink, M. H., Schreuder, H. A. & Van Berkel, W. J. (1997) Protein Sci. 6, 2454-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eggink, G., Engel, H., Vriend, G. & Terpstra, P. (1990) J. Mol. Biol. 212, 135-142. [DOI] [PubMed] [Google Scholar]
- 32.Wierenga, R. K., de Jong, R. J., Kalk, K. H., Hol, W. G. & Drenth, J. (1979) J. Mol. Biol. 131, 55-73. [DOI] [PubMed] [Google Scholar]
- 33.Binda, C., Newton-Vinson, P., Hubalek, F., Edmondson, D. E. & Mattevi, A. (2002) Nat. Struct. Biol. 9, 22-26. [DOI] [PubMed] [Google Scholar]
- 34.Binda, C., Hubalek, F., Li, M., Herzig, Y., Sterling, J., Edmondson, D. E. & Mattevi, A. (2004) J. Med. Chem. 47, 1767-1774. [DOI] [PubMed] [Google Scholar]
- 35.Wang, J., Ortiz-Maldonado, M., Entsch, B., Massey, V., Ballou, D. & Gatti, D. L. (2002) Proc. Natl. Acad. Sci. USA 99, 608-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Enroth, C., Neujahr, H., Schneider, G. & Lindqvist, Y. (1998) Structure 6, 605-617. [DOI] [PubMed] [Google Scholar]
- 37.Gatti, D. L., Palfey, B. A., Lah, M. S., Entsch, B., Massey, V., Ballou, D. P. & Ludwig, M. L. (1994) Science 266, 110-114. [DOI] [PubMed] [Google Scholar]
- 38.Rhee, S. G., Bae, Y. S., Lee, S. R. & Kwon, J. (2000) Sci STKE 2000, PE1. [DOI] [PubMed] [Google Scholar]
- 39.Rhee, S. G. (1999) Exp. Mol. Med. 31, 53-59. [DOI] [PubMed] [Google Scholar]
- 40.Milzani, A., Dalledonne, I. & Colombo, R. (1997) Arch. Biochem. Biophys. 339, 267-274. [DOI] [PubMed] [Google Scholar]
- 41.Tang, J. X., Janmey, P. A., Stossel, T. P. & Tadanao, I. (1999) Biophys. J. 76, 2208-2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dalledonne, I., Milzani, A. & Colombo, R. (1995) Biophys. J. 69, 2710-2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Milzani, A., Rossi, R., Di Simplicio, P., Giustarini, D., Colombo, R. & Dalledonne, I. (2000) Protein Sci. 9, 1774-1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dalledonne, I., Rossi, R., Giustarini, D., Gagliano, N., Di Simplicio, P., Colombo, R. & Milzani, A. (2002) Free Radical Biol. Med. 32, 927-937. [DOI] [PubMed] [Google Scholar]
- 45.Read, R. J. (1986) Acta Crystallogr. A 42, 140-149. [Google Scholar]