

Abstract
Inducible prostaglandin synthase (cyclooxygenase-2, COX-2) is expressed in rheumatoid and osteoarthritic cartilage and produces high amounts of proinflammatory prostanoids in the joint. In the present study we investigated the effects of the inhibitors of mitogen-activated protein kinase (MAPK) pathways Erk1/2, p38, and JNK on COX-2 expression and prostaglandin E2 (PGE2) production in human chondrocytes. Proinflammatory cytokine IL-1β caused a transient activation of Erk1/2, p38, and JNK in immortalized human T/C28a2 chondrocytes and that was followed by enhanced COX-2 expression and PGE2 production. PD98059 (an inhibitor of Erk1/2 pathway) suppressed IL-1-induced COX-2 expression and PGE2 production in a dose-dependent manner, and seemed to have an inhibitory effect on COX-2 activity. SB203580 (an inhibitor of p38 pathway) but not its negative control compound SB202474 inhibited COX-2 protein and mRNA expression and subsequent PGE2 synthesis at micromolar drug concentrations. SP600125 (a recently developed JNK inhibitor) but not its negative control compound N1-methyl-1,9-pyrazolanthrone downregulated COX-2 expression and PGE2 formation in a dose-dependent manner. SP600125 did not downregulate IL-1-induced COX-2 mRNA expression when measured 2 h after addition of IL-1β but suppressed mRNA levels in the later time points suggesting post-transcriptional regulation. Our results suggest that activation of Erk1/2, p38, and JNK pathways belongs to the signaling cascades that mediate the upregulation of COX-2 expression and PGE2 production in human chondrocytes exposed to proinflammatory cytokine IL-1β.
INTRODUCTION
Prostaglandins (PGs) are present in a wide variety of human tissues, where they regulate physiological responses, including vascular tone, blood clotting, kidney function, gastric secretion and reproduction [1]. In arthritis, prostaglandins (especially PGE2) are produced in much higher amounts, and they mediate inflammation, tissue destruction, and inflammatory pain. PGs are synthesized from arachidonic acid by cyclooxygenase (COX) enzymes [2, 3]. Two isoforms of COX have been identified: COX-1 is constitutively expressed and produces low physiological levels of prostanoids, whereas the expression of the inducible isoform, COX-2, is increased in response to proinflammatory cytokines or bacterial products [4]. COX-2 is highly expressed in rheumatoid (RA) and osteoarthritic (OA) cartilage [5, 6]. Interleukin (IL)-1 is a key cytokine involved in the joint destruction in RA and OA and it has been shown to enhance COX-2 expression in articular chondrocytes [5, 7, 8].
Mitogen-activated protein kinases (MAPKs) are a family of serine/threonine kinases, that are part of the signal transduction pathways which connect inflammatory and other extracellular signals to intracellular responses, for example, gene expression. The three better characterized MAPK pathways are extracellular signal-regulated kinase 1 and 2 (Erk1/2), p38, and c-Jun N-terminal kinase (JNK). The growth-factor-induced Erk1/2, and the stress-activated p38 and JNK protein kinases are phosphorylated in response to extracellular stimuli at conserved threonine and tyrosine residues and have regulatory functions in inflammation [9, 10]. Inhibitors of p38 and JNK are under development for treatment of arthritis and they have shown efficacy in experimentally induced arthritis and joint pain [11, 12]. Inhibition of MAPKs is likely to result in suppression of inflammatory mediators, which in turn leads to the desired therapeutic effects. We hypothesized that one of the inflammatory pathways in the cartilage, that might be down-regulated by MAPK inhibitors, is COX-2-PGE2 pathway. The aim of the present study was to investigate if inhibitors of JNK (SP600125), p38 (SB203580), and Erk1/2 (PD98059) MAP-kinase pathways downregulate IL-induced COX-2 expression and PGE2 production in human chondrocytes.
MATERIALS AND METHODS
Materials
Reagents were obtained as follows: SP600125 (anthra[1,9-cd]pyrazol-6(2H)-one), SB203580 (4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-imidazole), and PD98059 (2-(2′-amino-3′-methoxy-phenyl)-oxanaphthalen-4-one) were from Calbiochem (La Jolla, Calif); goat polyclonal human COX-2, rabbit polyclonal JNK, donkey anti-goat polyclonal, and goat anti-rabbit polyclonal antibodies were from Santa Cruz Biotechnology, Inc, (Santa Cruz, Calif); and rabbit polyclonal phospho-JNK, phospho-Erk1/2, Erk1/2, phospho-p38, and p38 antibodies were from Cell Signaling Technology, Inc, (Beverly, Mass). All other reagents were from Sigma Chemical Co (St Louis, Mo).
Cell culture
Immortalized human T/C28a2 chondrocytes [13] were grown in Dulbecco's modified Eagle's medium (Cambrex Bioproducts Europe, Verviers, Belgium) and Ham's F-12 medium (Gibco, Paisley, Scotland) (1:1, v/v). Culture media contained 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, and 250 ng/mL amphotericin B (all from Gibco, Paisley, Scotland). Cells were seeded on 24-well plates for prostaglandin E2 measurements and on 6-well plates for Western blot and RT-PCR. Cell monolayers were grown for 72 h to confluence before the experiments were started and the compounds of interest were added in fresh medium.
Prostaglandin E2 assays
At the indicated time points, the culture medium was collected for prostaglandin E2 (PGE2) measurement. PGE2 concentrations were determined by radioimmunoassay using reagents from the Institute of Isotopes (Budapest, Hungary).
Western blot analysis
At the indicated time points, cells were rapidly washed with ice-cold PBS and solubilized in cold lysis buffer containing 10 mM Tris base, 5 mM EDTA, 50 mM NaCl, 1% Triton X-100, 0.5 mM phenylmethylsulfonyl fluoride, 2 mM sodium orthovanadate, 10 μg/mL leupeptin, 25 μg/mL aprotinin, 1.25 mM NaF, 1 mM sodium pyrophosphate, and 10 mM n-octyl-β-D-glucopyranoside. After incubation for 20 min on ice, lysates were centrifuged (14 500 g for 10 min), and supernatants were mixed in a ratio of 1:4 with SDS loading buffer (62.5 mM Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, 0.025% bromophenol blue, and 5% β-mercaptoethanol) and boiled for 5 min. Protein concentrations in the samples were measured by the Coomassie blue method [14]. After boiling for 5 min, equal aliquots of protein (20 μg) were loaded on a 10% SDS-polyacrylamide electrophoresis gel and electrophoresed for 4 h at 100 V in a buffer containing 95 mM Tris-HCl, 960 mM glycine, and 0.5% SDS. After electrophoresis, the proteins were transferred to Hybond-enhanced chemiluminescence nitrocellulose membrane (Amersham, Buckinghamshire, UK) with semidry blotter at 2.5 mA/cm2 for 60 min. After transfer, the membrane was blocked in TBS/T (20 mM Tris-base pH 7.6, 150 mM NaCl, 0.1% Tween-20) containing 5% nonfat milk for 1 h at room temperature and incubated overnight at 4°C with COX-2, JNK, p38, Erk1/2, phospho-specific JNK, phospho-specific p38, or phospho-specific Erk1/2 antibodies in TBS/T containing 5% nonfat milk. Thereafter the membrane was washed 4 times with TBS/T for 5 min, incubated with secondary antibody coupled to horseradish peroxidase in the blocking solution for 0.5 h at room temperature, and washed four times with TBS/T for 5 min. Bound antibody was detected using SuperSignal West Pico chemiluminescent substrate (Pierce, Cheshire, UK) and FluorChem 8800 imaging system (Alpha Innotech Corp, San Leandro, Calif). The quantitation of the chemiluminescent signal was carried out with the use of FluorChem software version 3.1.
RNA extraction and real-time RT-PCR
At the indicated time points, cell monolayers were rapidly washed with ice-cold PBS, and cells were homogenized using QIAshredder (QIAGEN, Valencia, Calif). RNA extraction was carried out with the use of RNeasy kit for isolation of total RNA (QIAGEN). Total RNA (25 ng) was reverse-transcribed to cDNA using TaqMan reverse transcription reagents and random hexamers (Applied Biosystems, Foster City, Calif). cDNA obtained from the RT reaction (amount corresponding to approximately 1 ng of total RNA) was subjected to PCR using TaqMan Universal PCR Master Mix and ABI PRISM 7000 Sequence detection system (Applied Biosystems). The primer and probe sequences and concentrations were optimized according to manufacturer's guidelines in TaqMan Universal PCR Master Mix Protocol part number 4304449 revision C and they were 5′ - CAACTCTATATTGCTGGAACATGGA - 3′ (human COX-2 forward primer, 300 nM), 5′-TGGAAGCCTGTGATACTTTCTGTACT-3′ (human COX-2 reverse primer, 300 nM), 5′-TCCTACCACCAGCAACCCTGCCA-3′ (human COX-2 probe containing 6-FAM as 5′-reporter dye and TAMRA as 3′-quencher, 150 nM). Human β-actin was obtained from TaqMan Human β-actin Reagents kit (Applied Biosystems), containing VIC as 5′-reporter dye and TAMRA as 3′-quencher. PCR reaction parameters were as follows: incubation at 50°C for 2 min, incubation at 95°C for 10 min, and thereafter 40 cycles of denaturation at 95°C for 15 s and annealing and extension at 60°C for 1 min. Each sample was determined in duplicate.
A standard curve method was used to determine the relative mRNA levels as described in the Applied Biosystems User Bulletin: a standard curve for each gene was created using RNA isolated from IL-1β-stimulated human T/C28a2 chondrocytes. Isolated RNA was reverse-transcribed and dilution series of cDNA ranging from 1 pg to 10 ng were subjected to real-time PCR. The obtained threshold cycle values were plotted against the dilution factor to create a standard curve. Relative mRNA levels in test samples were then calculated from the standard curve.
Statistics
Results are expressed as the mean ± SEM. Statistical significances were calculated by analyses of variance supported by the Dunnett's multiple comparisons test. Differences were considered significant at P < .05.
RESULTS
IL-1β-activated JNK, p38, and Erk1/2 in human T/C28a2 chondrocytes
The ability of IL-1β to activate JNK, p38, and Erk1/2 pathways was studied by Western blot analysis using antibodies directed against Thr-183/Tyr-185, Thr-180/Tyr-182, and Thr-202/Tyr-204 phosphorylated (ie, activated) JNK, p38, and Erk1/2, respectively. JNK activation was seen 20 min after addition of IL-1β. The activation peaked at 1 hour and decreased thereafter. The activation of p38 and Erk1/2 was detected 3–6 min after addition of IL-1β, peaked at 10–20 min, and declined after 1 hour (Figure 1).
Figure 1.

The effects of IL-1β on JNK, p38, and Erk1/2 MAPK activation in human T/C28a2 chondrocytes. The chondrocytes were stimulated with IL-1β (100 pg/mL). Incubations were terminated at the indicated time points. Two parallel immunoblots were run from same cell lysates using antibodies against the Thr-183/Tyr-185, Thr-180/Tyr-182, and Thr-202/Tyr-204 phosphorylated (ie, activated) JNK (p-JNK), p38 (p-p38), and Erk1/2 (p-Erk1/2) and against total JNK, p38, and Erk1/2. The experiment was repeated three times with similar results.
IL-1β-induced COX-2 expression and PGE2 production in human T/C28a2 chondrocytes
IL-1β enhanced COX-2 expression in a concentration-dependent manner, being detectable at 10 pg/mL and increasing up to 1000 pg/lmL (Figure 2a). Radioimmunoassay of prostaglandin E2 (PGE2) in the culture medium was carried out to investigate PGE2 production. IL-1β induced PGE2 production in a concentration-dependent manner. Increased PGE2 production was detected at 10 pg/mL of IL-1β and was maximal at 100 pg/mL remaining elevated up to 1000 pg/mL (Figure 2b).
Figure 2.


The effects of IL-1β on COX-2 protein expression and PGE2 production in human T/C28a2 chondrocytes. (a) Human chondrocytes were incubated for 24 h in the presence of increasing concentrations of IL-1β, and COX-2 protein was measured by Western blot. (b) Human chondrocytes were incubated for 24 h in the presence of increasing concentrations of IL-1β, and PGE2 concentrations in the culture medium were measured by radioimmunoassay. COX inhibitor ibuprofen (10 μM) was used as a control compound. Mean ± SEM, n = 4 − 6. In (a), a representative gel is shown under the bars.
SP600125, SB203580, and PD98059 suppressed IL-1β-induced PGE2 production in human T/C28a2 chondrocytes
Inhibitors of JNK (SP600125), p38 (SB203580), and Erk1/2 (PD98059) reduced IL-1β-induced PGE2 production in T/C28a2 chondrocytes in a concentration-dependent manner (Figures 3a, 3b, 3c). In the further studies, SP600125, SB203580, and PD98059 were added in T/C28a2 chondrocyte cultures at the same time or 6 h after IL-1β. In contrast to the inhibitory effect seen when added at the same time, SP600125 (10 μM) and SB203580 (1 μM) did not inhibit PGE2 production when added 6 h after IL-1β. PD98059 (10 μM) inhibited the production of PGE2 also when added 6 h after IL-1β, but the inhibition was notably less than when added to cells at the same time as the stimulus (Figure 3g).
Figure 3.

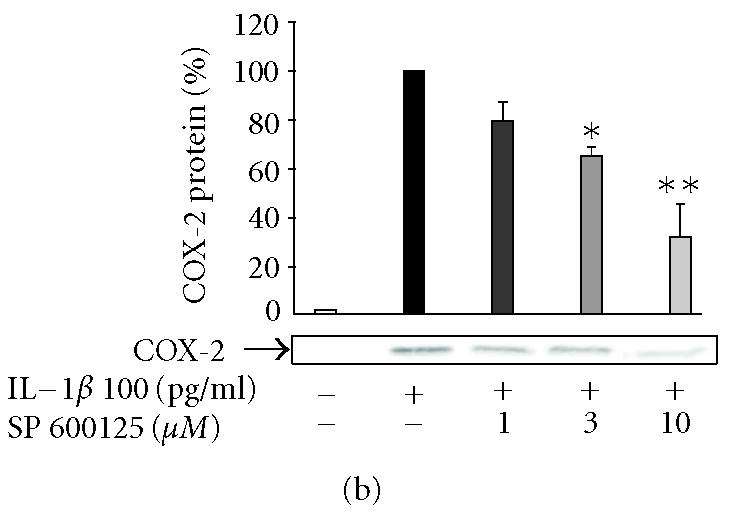
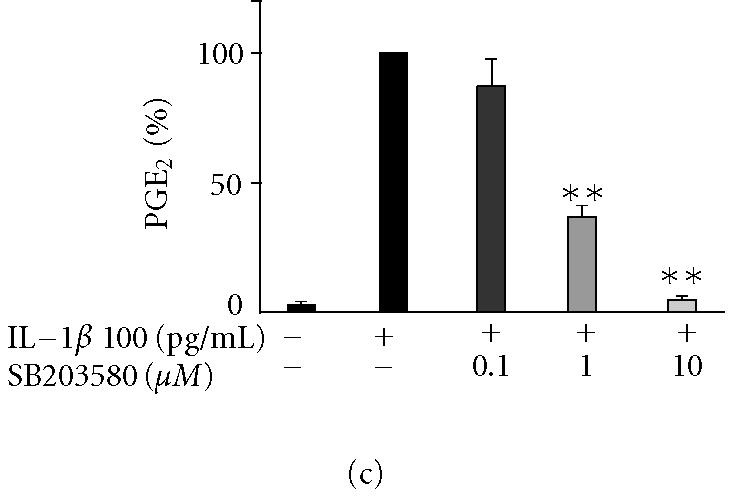
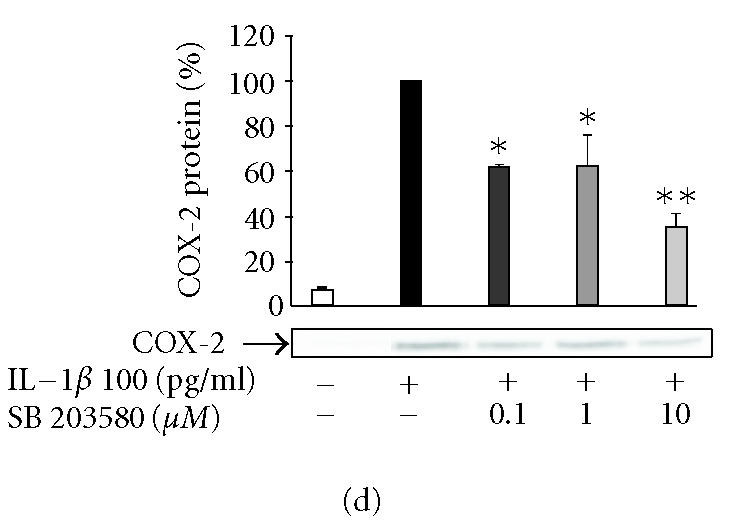

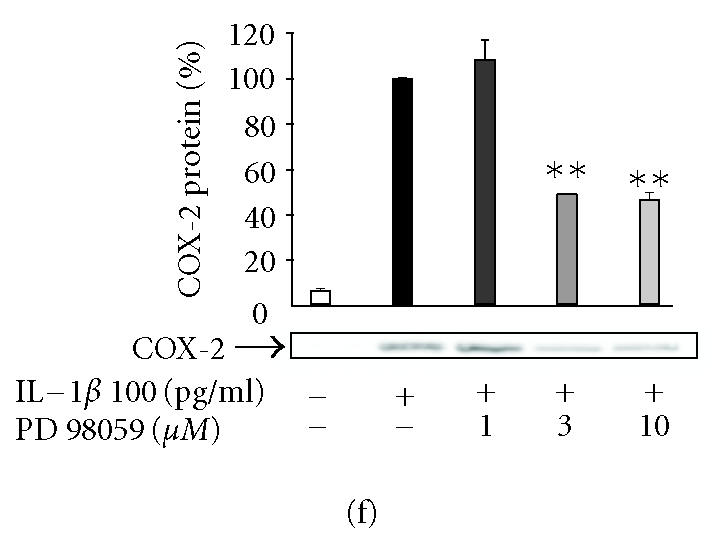

The effects of SP600125, SB203580, and PD98059 on PGE2 production and COX-2 protein expression in IL-1β-stimulated human T/C28a2 chondrocytes. Human chondrocytes were incubated with IL-1β (100 pg/mL) and increasing concentrations of (a), (b) SP600125 (JNK inhibitor), (c), (d) SB203580 (p38 inhibitor), and (e), (f) PD98059 (Erk1/2 inhibitor). After 24 h, incubations were terminated, and PGE2 concentrations in the culture medium were measured by RIA ((a), (c), (e)) and COX-2 protein was measured by Western blot ((b), (d), (f)). (g) SP600125 (10 μM), SB203580 (1 μM), and PD98059 (10 μM) were added to the cell culture at the same time (0 h) or 6 h after IL-1β (6 h). After 24 h, PGE2 concentrations were measured in the culture medium by RIA. Mean ± SEM, n = 4, ∗∗ indicates P < .01 as compared with the respective control. In (b), (d), and (f) a representative gel is shown under the bars.
SP600125, SB203580, and PD98059 inhibited COX-2 expression in human T/C28a2 chondrocytes
In the further studies, we measured the effects of SP600125, SB203580, and PD98059 on IL-1β-induced COX-2 protein expression. Western blots using antibody against COX-2 showed that the three inhibitors caused a concentration-dependent reduction in IL-1β-induced COX-2 protein levels (Figures 3b, 3d, 3f). Negative control compounds were available for SP600125 and SB203580 and their effects on COX-2 expression were also tested. N1-methyl-substituted pyrazolanthrone is structurally related to SP600125 but it is over 100-fold less potent inhibitor of JNK than SP600125 [15]. N1-methyl-1,9-pyrazolanthrone (10 μM) had no effect on COX-2 expression while SP600125 (10 μM) reduced COX-2 expression by 70%. SB202474 is structurally related to SB203580 but does not inhibit p38 [16]. SB202474 (1 μM) did not suppress COX-2 expression while SB203580 (1 μM) inhibited COX-2 expression by 40%.
SP600125, SB203580, and PD98059 inhibited COX-2 mRNA expression in human T/C28a2 chondrocytes
We used real-time RT-PCR to investigate the effects of SP600125, SB203580, and PD98059 on COX-2 mRNA expression. IL-1β induced transient COX-2 expression that peaked 4 h after addition of IL-1. SB203580 and PD98059 reduced IL-1β-induced COX-2 mRNA expression significantly when measured either 2 h or 8 h after IL-1β stimulation. In contrast, SP600125 had no marked effect on IL-1β-induced COX-2 mRNA expression at the 2 h time point, whereas the level of COX-2 mRNA was reduced by about 75% at the 8 h time point (Figure 4).
Figure 4.
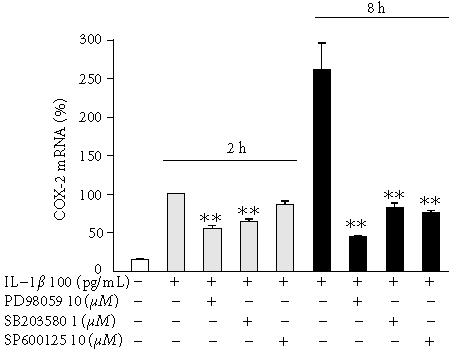
The effects of SP600125, SB203580, and PD98059 on COX-2 mRNA levels in IL-1β-stimulated human chondrocytes. Human chondrocytes were incubated with IL-1β (100 pg/ml) and with or without SP600125, SB203580, and PD98059. Incubations were terminated at the indicated time points, and the extracted total RNA was subjected to real-time RT-PCR. COX-2 mRNA levels were normalized against β-actin mRNA. Mean ± SEM, n = 6, ∗∗ indicates P < .01 as compared with cells treated with IL-1β only.
DISCUSSION
In the present study, we found that inhibitors of JNK, p38, and Erk1/2 pathways downregulate IL-1-induced COX-2 expression and PGE2 production in human chondrocytes.
Consistently with earlier findings using primary articular chondrocytes [17, 18], our results show that IL-1β causes a rapid activation of JNK, p38, and Erk1/2 MAP kinases in immortalized human T/C28a2 chondrocytes. These events were followed by enhanced COX-2 expression and subsequent PGE2 production. Inhibition of JNK activity by SP600125, p38 activity by SB203580, and Erk1/2 activity by PD98059 resulted in a reduction in the amount of PGE2 produced. However, when SP600125 and SB203580 were added 6 h after IL-1β, they did not affect PGE2 production. These findings suggest that SP600125 and SB203580 did not inhibit COX-2 activity, but rather reduced the expression of COX-2. PD98059 inhibited the production of PGE2 also when added 6 h after IL-1β, but the inhibition was notably smaller than in those experiments where PD98059 was added at the same time as IL-1β. This suggests that PD98059 may have also some inhibitory effect on cyclooxygenase activity in activated chondrocytes, as has been earlier reported in arachidonic-acid-stimulated human platelets [19]. Western blot analysis showed that all the three inhibitors (SP600125, SB203580, and PD98059) caused also a concentration-dependent reduction in COX-2 protein levels in IL-1-treated chondrocytes.
In the real-time RT-PCR studies, SP600125 had practically no effect on IL-1β-induced COX-2 mRNA expression in human T/C28a2 chondrocytes when measured 2 h after IL-1β, whereas when measured 8 h after the addition of IL-1β, a significant reduction in the levels of COX-2 mRNA was observed in the presence of SP600125. These results suggest that inhibition of JNK pathway does not affect the early events in IL-1-induced COX-2 expression but it may regulate the process at post-transcriptional level.
SB203580 and PD98059 had a significant effect on COX-2 mRNA expression when measured 2 h after addition of IL-1β. Our results are consistent with previous reports showing that p38 and Erk1/2 MAP kinase pathways are involved in the cellular events leading to upregulation of COX-2 gene transcription in human monocytes and in RAW264 macrophages [20, 21, 22, 23, 24]. In addition, p38 has also reported to stabilize COX-2 mRNA in mammary carcinoma cells, in HeLa-TO cells, and another chondrocyte cell line [25, 26, 27].
We have earlier shown that, in J774 macrophages, SP600125, SB203580, and PD98059 do not have an effect on nuclear translocation and DNA binding activity of NF-κB [28, 29, 30] which plays a role in stimulating COX-2 expression [31]. In addition to NF-κB, the expression of COX-2 is regulated by other factors including NF-IL6 and AP-2 [32, 33]. Further studies are needed to determine the molecular mechanisms which mediate the effects of JNK, p38, and Erk1/2 inhibitors on COX-2 expression.
Inhibitors of p38 and JNK are under development for treatment of arthritis and they have been shown to have antiinflammatory and antierosive effects in experimentally induced arthritis, and to relief inflammatory pain [10, 11]. The inhibition of MAP kinases and subsequent inhibition of the synthesis of a number of important proinflammatory cytokines like IL-1, TNF-α, IL-6, IL-8, and matrix metalloproteases has been identified as a mechanism contributing to the antiinflammatory activity of these compounds [10, 11, 12, 15, 34, 35]. The present results show that inhibitors of JNK, p38, and Erk1/2 MAP kinases also downregulate COX-2 expression and PGE2 production in human chondrocytes which is likely involved in the mechanisms of their therapeutic effects in arthritis and other inflammatory diseases.
ACKNOWLEDGMENT
We thank Mrs Niina Ikonen and Mrs Jaana Tägtström for skillful technical assistance and Mrs Heli Määttä for secretarial help.
References
- 1.Dubois RN, Abramson SB, Crofford L, et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12(12):1063–1073. [PubMed] [Google Scholar]
- 2.Needleman P, Turk J, Jakschik BA, Morrison AR, Lefkowith JB. Arachidonic acid metabolism. Annu Rev Biochem. 1986;55:69–102. doi: 10.1146/annurev.bi.55.070186.000441. [DOI] [PubMed] [Google Scholar]
- 3.Turini ME, DuBois RN. Cyclooxygenase-2: a therapeutic target. Annu Rev Med. 2002;53:35–57. doi: 10.1146/annurev.med.53.082901.103952. [DOI] [PubMed] [Google Scholar]
- 4.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 5.Amin AR, Attur M, Patel RN, et al. Superinduction of cyclooxygenase-2 activity in human osteoarthritis-affected cartilage. Influence of nitric oxide. J Clin Invest. 1997;99(6):1231–1237. doi: 10.1172/JCI119280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pelletier JP, Fernandes JC, Jovanovic DV, Reboul P, Martel-Pelletier J. Chondrocyte death in experimental osteoarthritis is mediated by MEK 1/2 and p38 pathways: role of cyclooxygenase-2 and inducible nitric oxide synthase. J Rheumatol. 2001;28(11):2509–2519. [PubMed] [Google Scholar]
- 7.Berenbaum F, Jacques C, Thomas G, Corvol MT, Bereziat G, Masliah J. Synergistic effect of interleukin-1 beta and tumor necrosis factor alpha on PGE2 production by articular chondrocytes does not involve PLA2 stimulation. Exp Cell Res. 1996;222(2):379–384. doi: 10.1006/excr.1996.0047. [DOI] [PubMed] [Google Scholar]
- 8.Lyons-Giordano B, Pratta MA, Galbraith W, Davis GL, Arner EC. Interleukin-1 differentially modulates chondrocyte expression of cyclooxygenase-2 and phospholipase A2. Exp Cell Res. 1993;206(1):58–62. doi: 10.1006/excr.1993.1120. [DOI] [PubMed] [Google Scholar]
- 9.Su B, Karin M. Mitogen-activated protein kinase cascades and regulation of gene expression. Curr Opin Immunol. 1996;8(3):402–411. doi: 10.1016/s0952-7915(96)80131-2. [DOI] [PubMed] [Google Scholar]
- 10.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 11.Badger AM, Bradbeer JN, Votta B, Lee JC, Adams JL, Griswold DE. Pharmacological profile of SB 203580, a selective inhibitor of cytokine suppressive binding protein/p38 kinase, in animal models of arthritis, bone resorption, endotoxin shock and immune function. J Pharmacol Exp Ther. 1996;279(3):1453–1461. [PubMed] [Google Scholar]
- 12.Han Z, Chang L, Yamanishi Y, Karin M, Firestein GS. Joint damage and inflammation in c-Jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis Rheum. 2002;46(3):818–823. doi: 10.1002/art.10104. [DOI] [PubMed] [Google Scholar]
- 13.Goldring MB, Birkhead JR, Suen LF, et al. Interleukin-1 beta-modulated gene expression in immortalized human chondrocytes. J Clin Invest. 1994;94(6):2307–2316. doi: 10.1172/JCI117595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 15.Bennett BL, Sasaki DT, Murray BW, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98(24):13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372(6508):739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 17.Geng Y, Valbracht J, Lotz M. Selective activation of the mitogen-activated protein kinase subgroups c-Jun NH2 terminal kinase and p38 by IL-1 and TNF in human articular chondrocytes. J Clin Invest. 1996;98(10):2425–2430. doi: 10.1172/JCI119056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scherle PA, Pratta MA, Feeser WS, Tancula EJ, Arner EC. The effects of IL-1 on mitogen-activated protein kinases in rabbit articular chondrocytes. Biochem Biophys Res Commun. 1997;230(3):573–577. doi: 10.1006/bbrc.1996.5985. [DOI] [PubMed] [Google Scholar]
- 19.Borsch-Haubold AG, Pasquet S, Watson SP. Direct inhibition of cyclooxygenase-1 and -2 by the kinase inhibitors SB 203580 and PD 98059. SB 203580 also inhibits thromboxane synthase. J Biol Chem. 1998;273(44):28766–28772. doi: 10.1074/jbc.273.44.28766. [DOI] [PubMed] [Google Scholar]
- 20.Jones MK, Sasaki E, Halter F, et al. HGF triggers activation of the COX-2 gene in rat gastric epithelial cells: action mediated through the ERK2 signaling pathway. FASEB J. 1999;13(15):2186–2194. doi: 10.1096/fasebj.13.15.2186. [DOI] [PubMed] [Google Scholar]
- 21.Subbaramaiah K, Hart JC, Norton L, Dannenberg AJ. Microtubule-interfering agents stimulate the transcription of cyclooxygenase-2. Evidence for involvement of ERK1/2 and p38 mitogen-activated protein kinase pathways. J Biol Chem. 2000;275(20):14838–14845. doi: 10.1074/jbc.275.20.14838. [DOI] [PubMed] [Google Scholar]
- 22.Caivano M, Cohen P. Role of mitogen-activated protein kinase cascades in mediating lipopolysac-charide-stimulated induction of cyclooxygenase-2 and IL-1 beta in RAW264 macrophages. J Immunol. 2000;164(6):3018–3025. doi: 10.4049/jimmunol.164.6.3018. [DOI] [PubMed] [Google Scholar]
- 23.Dean JL, Brook M, Clark AR, Saklatvala J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem. 1999;274(1):264–269. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- 24.Shalom-Barak T, Quach J, Lotz M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J Biol Chem. 1998;273(42):27467–27473. doi: 10.1074/jbc.273.42.27467. [DOI] [PubMed] [Google Scholar]
- 25.Jang BC, Sanchez T, Schaefers HJ, et al. Serum withdrawal-induced post-transcriptional stabilization of cyclooxygenase-2 mRNA in MDA-MB-231 mammary carcinoma cells requires the activity of the p38 stress-activated protein kinase. J Biol Chem. 2000;275(50):39507–39515. doi: 10.1074/jbc.M003224200. [DOI] [PubMed] [Google Scholar]
- 26.Thomas B, Thirion S, Humbert L, et al. Differentiation regulates interleukin-1beta-induced cyclo-oxygenase-2 in human articular chondrocytes: role of p38 mitogen-activated protein kinase. Biochem J. 2002;362(pt 2):367–373. doi: 10.1042/0264-6021:3620367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lasa M, Brook M, Saklatvala J, Clark AR. Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol Cell Biol. 2001;21(3):771–780. doi: 10.1128/MCB.21.3.771-780.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lahti A, Lahde M, Kankaanranta H, Moilanen E. Inhibition of extracellular signal-regulated kinase suppresses endotoxin-induced nitric oxide synthesis in mouse macrophages and in human colon epithelial cells. J Pharmacol Exp Ther. 2000;294(3):1188–1194. [PubMed] [Google Scholar]
- 29.Lahti A, Kankaanranta H, Moilanen E. P38 mitogen-activated protein kinase inhibitor SB203580 has a bi-directional effect on iNOS expression and NO production. Eur J Pharmacol. 2002;454(2-3):115–123. doi: 10.1016/s0014-2999(02)02490-1. [DOI] [PubMed] [Google Scholar]
- 30.Lahti A, Jalonen U, Kankaanranta H, Moilanen E. c-Jun NH2-terminal kinase inhibitor anthra(1,9-cd)pyrazol-6(2H)-one reduces inducible nitric-oxide synthase expression by destabilizing mRNA in activated macrophages. Mol Pharmacol. 2003;64(2):308–315. doi: 10.1124/mol.64.2.308. [DOI] [PubMed] [Google Scholar]
- 31.Crofford LJ. COX-2 in synovial tissues. Osteoarthritis Cartilage. 1999;7(4):406–408. doi: 10.1053/joca.1999.0226. [DOI] [PubMed] [Google Scholar]
- 32.Inoue H, Yokoyama C, Hara S, Tone Y, Tanabe T. Transcriptional regulation of human prostaglandin-endoperoxide synthase-2 gene by lipopolysaccharide and phorbol ester in vascular endothelial cells. Involvement of both nuclear factor for interleukin-6 expression site and cAMP response element. J Biol Chem. 1995;270(42):24965–24971. doi: 10.1074/jbc.270.42.24965. [DOI] [PubMed] [Google Scholar]
- 33.Tanabe T, Tohnai N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 2002;68-69:95–114. doi: 10.1016/s0090-6980(02)00024-2. [DOI] [PubMed] [Google Scholar]
- 34.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9(2):240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 35.Lee JC, Kassis S, Kumar S, Badger A, Adams JL. p38 mitogen-activated protein kinase inhibitors-mechanisms and therapeutic potentials. Pharmacol Ther. 1999;82(2-3):389–397. doi: 10.1016/s0163-7258(99)00008-x. [DOI] [PubMed] [Google Scholar]