

ABSTRACT
Purpose
Von Hippel–Lindau (VHL) disease is a hereditary cancer syndrome expressed in multiple organs caused by germline alterations of the VHL gene. We have shown VHL deletion in the “stromal” cells of retinal angiomas. The VHL protein–associated complex is a primary ubiquitin ligase for the ubiquitination of hypoxia-inducible factor (HIF). This study examines VHL and ubiquitin expression in optic nerve hemangiomas and juxtapapillary angiomas.
Methods
Using microdissection and polymerase chain reaction, four optic nerve hemangiomas (one also had juxtapapillary angioma) associated with VHL disease were analyzed for loss of heterozygosity in the VHL gene. In addition, expression of HIF and ubiquitin was evaluated in these tumors by immunohistochemistry.
Results
All informative optic nerve and juxtapapillary lesions showed loss of heterozygosity in the VHL gene detected in vacuolated “stromal” cells. Both HIF and ubiquitin were highly expressed in the hemangiomas of all four VHL cases.
Conclusions
Like retinal angiomas and other VHL tumor lesions, VHL gene deletion is found in optic nerve hemangiomas and juxtapapillary angiomas. These tumor cells also express HIF and ubiquitin, the protein responsible for the negative regulation of HIF that results in the hypervascularization characteristic of VHL disease.
INTRODUCTION
Von Hippel–Lindau (VHL) disease occurs in roughly 1 in 36,000 live births and is inherited as a highly penetrant autosomal dominant trait (ie, with a high individual risk of manifesting the disease). Von Hippel–Lindau disease is caused by germline alterations of the VHL gene, which has been cloned and identified as a tumor suppressor gene on the short arm of chromosome 3, 3p25.5.1,2 The major lesions in VHL disease include hemangioblastomas in the central nervous system, retinal angiomas, clear cell renal cell carcinomas, pheochromocytomas, pancreatic tumors, epididymal cystadenomas, endolymphatic sac tumors, carcinoid tumors, and multiple cysts of the kidney, pancreas, and epididymis. Retinal angioma and cerebellar hemangioblastoma are the most frequent and earliest manifestations of VHL disease. Retinal angioma has been reported in nearly 60% of patients with VHL disease.3 VHL gene deletion and alteration are reported in VHL tumors in various systemic organs. In the eye, VHL gene deletion is found in the vacuolated clear, or “stromal,” cells of VHL retinal angiomas.4
Histologic features of VHL tumors are characterized by their high degree of vascularization and the presence of a clear cell component.5–7 Hypervascularization, angiogenesis, or both are induced by overexpression of vascular endothelial growth factor (VEGF). Since the principal function of VHL protein is the negative regulation of hypoxia-inducible mRNAs, including VEGF mRNA, inactivation of VHL gene plays a critical role in the angiogenesis of VHL tumors. We have detected expression of VEGF messenger and protein in retinal angiomas associated with VHL disease.4 The VHL protein–associated complex is a primary ubiquitin ligase for ubiquitination of the α subunits of the hypoxia-inducible factor (HIF).8 Loss of VHL protein function in VHL disease leads to the accumulation of ubiquitin and HIF-α during normoxic condition, which in turn causes constitutive induction of HIF-responsive genes, including VEGF.9
Optic nerve hemangiomas and juxtapapillary angiomas are relatively rare, and the prognosis is often poor.10 The present study examines VHL gene and ubiquitin expression in optic nerve hemangiomas obtained from patients with VHL disease.
METHODS
Three pathological specimens with intracranial optic nerve hemangiomas and one specimen with juxtapapillary hemangiomas were collected from four patients with family histories and clinical diagnoses of VHL disease (Figure 1). All patients were examined clinically by one of the authors (E.Y.C.). This study has been approved by the National Cancer Institute and National Eye Institute institutional review boards for human subjects.
Figure 1.
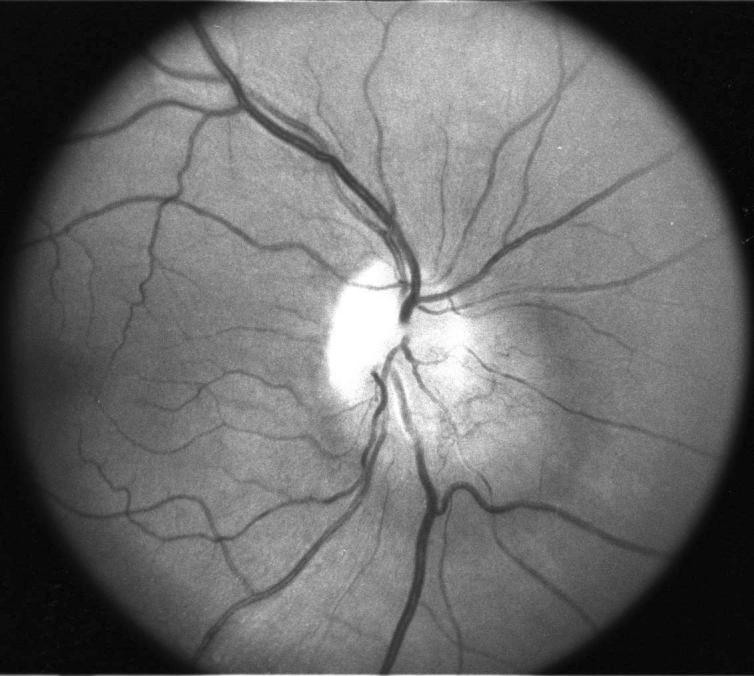
Optic nerve hemangioblastoma involving the optic nerve head in a patient with von Hippel–Lindau disease.
All pathological specimens were embedded in paraffin after fixation in 10% formalin. Sections were cut and thoroughly reviewed to identify each angiomatous lesion in the optic nerve. Manual microdissection was performed to obtain hemangiomas and normal cells separately. In each case, tumor cells were procured from areas with predominantly “stromal” cells, and normal cells were procured from areas without tumor cells and with normal morphology.
Microdissected cells were immediately placed in DNA extraction buffer containing proteinase K and subjected to polymerase chain reaction as described previously.4 Briefly, all samples were examined for loss of heterozygosity (LOH) using the microsatellite markers D3S1038, D3S1110, and D3S2452 flanking the VHL gene (Research Genetics, Huntsville, Alabama). A case was considered informative for a polymorphic marker if normal tissue DNA showed two different alleles (heterozygosity). The criterion for LOH was a complete or near complete absence of one allele in the tumor DNA as defined by direct visualization.
Deparaffinized sections were then subjected to immunohistochemical analysis utilizing the avidin-biotin-peroxidase complex technique. The primary antibodies were mouse anti-HIF monoclonal antibody (Novus Biologicals, Littleton, Colorado) and rabbit anti-ubiquitin proteosome polyclonal antibody (Chemicon International, Temecula, California). The secondary antibodies were biotin conjugated horse anti-mouse IgG or goat anti-rabbit IgG, respectively.
RESULTS
Two specimens were optic nerve only, one specimen was orbital tissue with optic nerve, and one was an enucleated eye with optic nerve. The two optic nerve lesions were surgically removed intracranially from two female patients (15 and 36 years of age). These patients were asymptomatic, and these lesions were detected upon screening (Figure 1). The orbital tissue was obtained from a 44-year-old man who had had an enucleation and radiation treatment for the affected orbit 22 years previously. Magnetic resonance imaging showed that he had a progressive intracranial lesion that was extending from the optic chiasm along the affected optic nerve to the orbit. He had gradual proptosis in the affected orbit that precluded proper fitting of his eyeglasses, which prompted the surgical removal of the orbital tissue. The enucleated eye came from a 12-year-old girl who had exudative and tractional retinal detachment from the retinal angiomas found in the retinal periphery as well as on the optic nerve. With the exception of the youngest patient, who had a normal fellow eye, all remaining patients had retinal angiomas with good vision in their fellow eyes.
Histopathologically, all four specimens contained classic VHL hemangiomas that were either juxtapapillary angioma (one specimen) or optic nerve hemangioma (four specimens) in the neural tissues, which matched clinical observations (Figure 2). The hemangiomas were composed mainly of densely packed, small capillary-like vascular channels and small cells with prominent dark nuclei and little cytoplasm, intermixed with vacuolated “stromal” cells and glial cells (Figure 2A). Scatter hemorrhages were often present inside the VHL lesions.
Figure 2.
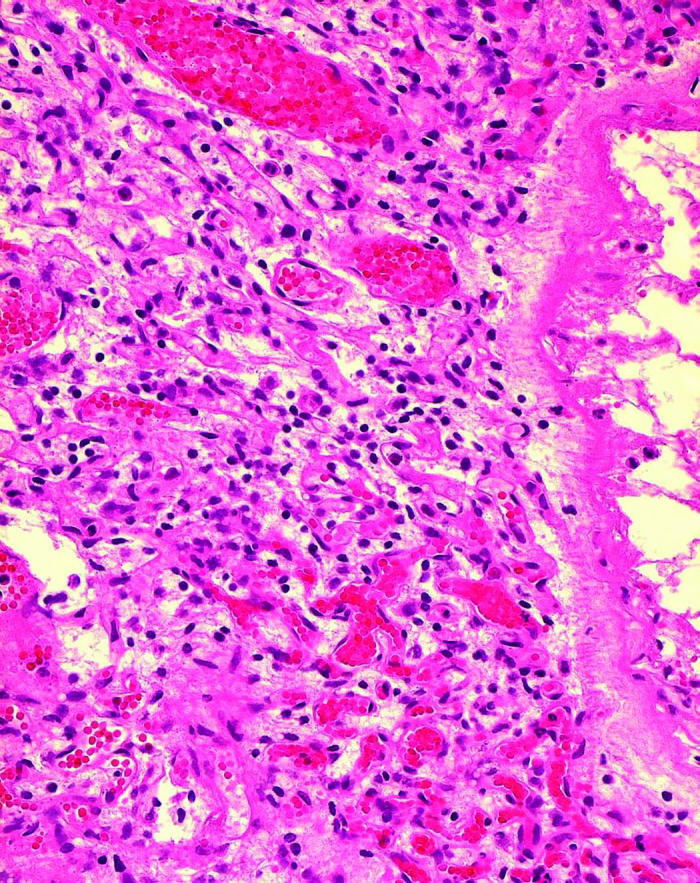
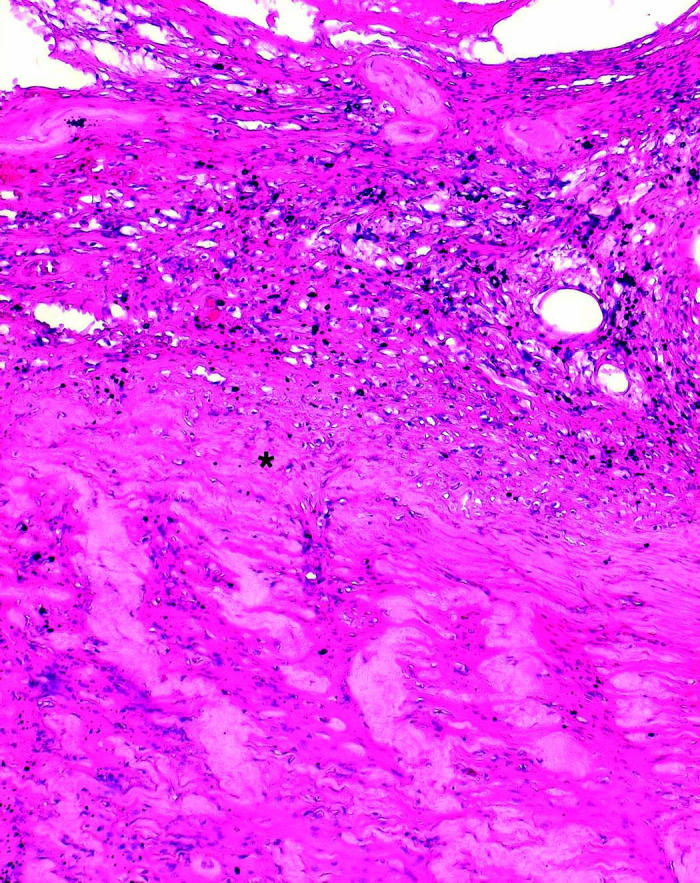
Microphotographs showing optic nerve hemangiomas located inside the optic nerve (A) and above the lamina cribrosa (asterisk, B) in two patients with von Hippel–Lindau disease (hematoxylineosin, original magnification ×200).
Three of the four cases were informative and showed LOH in the cells of the hemangiomas. One case showed slightly lower intensity in one allele as compared to the other allele in autoradiography (Figure 3). Retention of heterozygosity was shown in the normal cells. The one noninformative case (the orbital tissue specimen) previously had had a decalcified procedure and did not yield polymerase chain reaction products.
Figure 3.

Autoradiographs showing allelic imbalance indicative of loss of heterozygosity (T) after amplification with VHL gene flanking primers D3S1038, D3S1110, and D3S656. Matched adjacent normal cells (N) show preservation of both alleles of the VHL gene.
HIF-1 and ubiquitin were stained intensely in all four hemangiomas located juxtapapillary and inside the optic nerve regions. The staining pattern was even throughout each entire VHL lesion (Figure 4).
Figure 4.
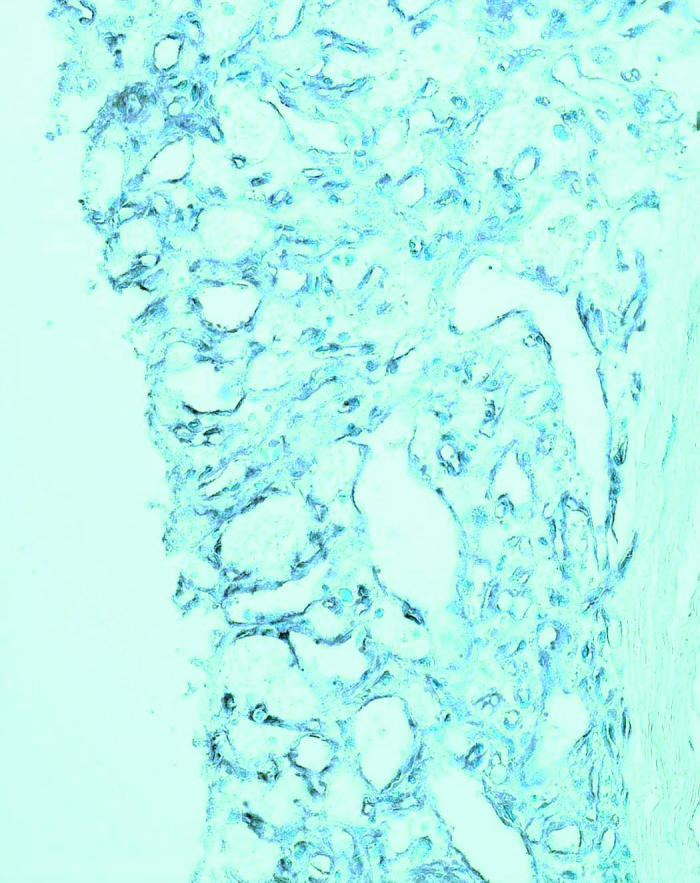
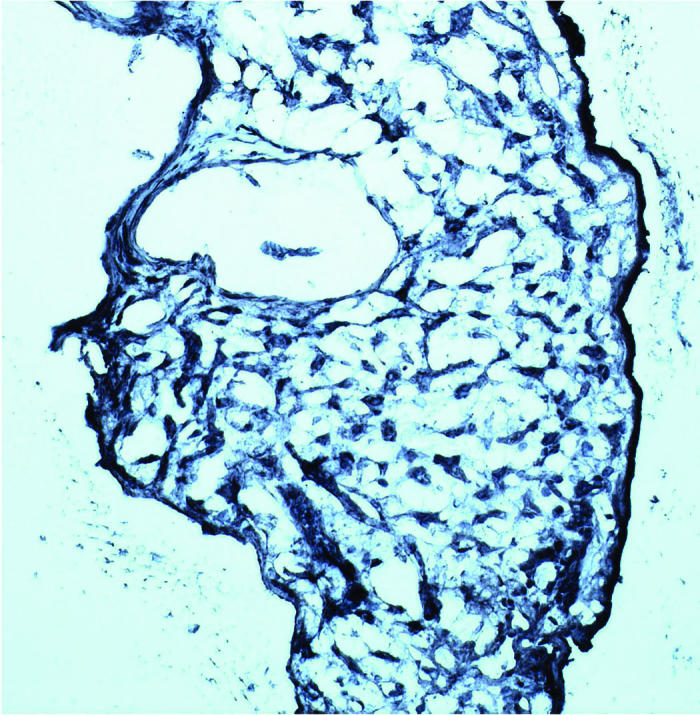
Microphotographs showing positive staining for hypoxia-inducible factor-α (A) and ubiquitin (B) in two von Hippel–Lindau hemangioblastomas (avidin-biotin-complex immunoperoxidase, original magnification ×200).
DISCUSSION
Although optic nerve lesions associated with VHL are a rare cause of blindness, they may be associated with higher morbidity and reduced life expectancy. In a recent study of 60 eyes with juxtapapillary capillary hemangioma and VHL, McCabe and associates10 observed that these patients more often presented at a younger age, had tumors with an endophytic growth pattern, and had bilateral, multiple lesions. Furthermore, in long-term follow-up, visual acuity was generally worsened in spite of laser photocoagulation therapy. Our four patients presented similarly complicated clinical courses and poor outcome.
The morphology of the optic nerve hemangiomas in our patients more closely resembles central nervous system hemangiomas than retinal angiomas. Small vascular components with hemorrhages are prominent, and the vascular channels seem more closely bundled together, thus making it difficult to identify the typical vacuolated “stromal” cells. These features may explain the aggressive nature of VHL lesions in the optic nerve.
Von Hippel–Lindau disease is a heritable multisystem cancer syndrome that is associated with a germline mutation of the VHL tumor suppressor gene. Similar to the “stromal” cells of the VHL tumors in various systemic organs, the present study has documented LOH of the VHL gene in the optic nerve hemangiomas. Again, the “stromal” cells of the hemangiomas are the true neoplastic component of the VHL lesions.
According to the two-hit hypothesis, dominantly inherited predisposition to cancer entails a germline mutation, whereas tumorigenesis requires a second somatic genetic alteration.11 Typically, patients with VHL disease have inherited an inactive VHL allele from an affected parent. In other words, they are VHL +/− heterozygous. Some VHL patients without a positive family history have, upon further investigation, been found to have a parent who is mosaic for a VHL mutation.12 This event has been documented in very early, premalignant lesions in the kidneys of the patients with VHL disease.13,14 It is presumed that mutations that affect one or more other genes are required for conversion of these premalignant renal lesions to frank renal cell carcinoma. Tumor development in VHL disease is linked to inactivation or loss of the remaining wild-type VHL allele in a susceptible cell or LOH (VHL −/−), which leads to loss of the VHL gene product, VHL protein. Indeed, the VHL gene is frequently inactivated, whether as a result of mutation or hypermethylation, in nonhereditary clear cell renal carcinomas and hemangioblastomas.15 In these settings, VHL gene alterations are not inherited but rather occur somatically. The mechanism of tumor development at different sites and organs after VHL gene inactivation is still unknown.
TheVHLmRNAencodesaVHLproteinthatcontains 213 amino acids. Tumor formation by VHL protein–defective renal cell carcinoma can be suppressed after restoration of wild-type VHL protein function.16–18 Therefore, VHL protein is a tumor suppressor protein based on both genetic and functional criteria. The VHL protein forms stable complexes in mammalian cells with other proteins, including elongin B, elongin C, Cul2, and Rbx.17,19–21 Elongin C and Cul2 regulate protein turnover. Many proteins that undergo regulated destruction are first covalently modified by the attachment of a polyubiquitin tail, which serves as a signal for degradation by a multiprotein complex called a proteasome. The VHL protein ubiquitin ligase complex polyubiquitinylates and targets proteasomal degradation and, when oxygen is present, HIF-α subunits. In the absence of oxygen, HIF-α subunits accumulate and activate hypoxia-inducible genes.15 During normoxia, translated HIF-α subunits are hydroxylated, which results in their subsequent ubiquitination and degradation. During hypoxia, the hydroxylation is inhibited. The HIF-α subunits are therefore not ubiquitinated and thus accumulate, regulating transcription of the HIF-responsive genes.22 High expression of HIF and ubiquitin in our patients’ optic nerve hemangiomas and juxtapapillary angiomas is most likely due to the failure of ubiquitination in the VHL lesions.
HIF-1 is a heterodimer consisting of a constitutively expressed HIF-1β subunit and an oxygen- and growth factor–regulated HIF-1α subunit.23 Increased HIF-1 activity provides a molecular basis for VEGF-induced angiogenesis and other cancer cell adaptations to hypoxia critical for the establishment of a primary tumor and the eventual progression to the lethal phenotype. Retinal angiomas, central nervous system hemangioblastomas, and renal cell carcinomas in VHL disease are highly vascular and frequently overproduce angiogenic factors such as VEGF.4,24,25 The production of HIF mRNA is uncoupled from changes in ambient oxygen in VHL protein–defective tumor cell lines. This defect can be corrected by restoration of wild-type VHL protein function.26,27 Furthermore, the products of some HIF-1–inducible genes might be suitable targets for therapeutic approaches to VHL tumors, including retinal angioma and optic nerve hemangioma. Recently, Aiello and colleagues28 reported successful treatment in a patient with VHL and optic nerve hemangioma after systemic therapy with VEGF receptor inhibitor SU5416. On the other hand, Girmens and associates29 have shown that SU5416 is more effective for VHL-associated macular edema than for hemangiomas. Further clinical studies with careful monitoring and long-term follow-up of such treatments are required.
DISCUSSION
Dr Hans E. Gossniklaus
If one graphs the age at diagnosis of retinal von Hippel Lindau (VHL) hemangioma versus percent of undiagnosed cases, two curves emerge, similar to the two curves that Knudson found when he derived his two-hit hypothesis for retinoblastoma. Like the retinoblastoma gene, the VHL gene is a tumor suppressor gene. The VHL gene has been mapped to 3p25.5 and cloned. In heritable VHL disease, patients inherit an inactive VHL allele and the other allele is inactivated or there is loss of heterozygosity (LOH). The VHL protein ubiquitin ligase complex polyubiquitinylates hypoxia inducible factor (HIF-α) subunits during normoxia. During hypoxia, the hydroxylation is inhibited, HIF-α subunits are not ubiquitinated, and VEGF is produced. Abnormalities in the VHL protein result in absence of ubiquitination, increased HIF-α subunits during normoxia and hyperoxia, and VEGF production.
In this study, Chan and coworkers used laser-capture microdissection to extract cells from optic nerve VHL hemangiomas and an enucleated eye with a peripapillary VHL hemangioma. They demonstrated LOH for the VHL gene in 3 of 4 cases and found HIF-1 and ubiquitin overexpression in all four cases, thus indicating loss of function of the VHL protein. This LOH was present in the tumor stromal cells, but not in endothelial cells. This reaffirms Chan’s previous work demonstrating LOH in the stromal cells of retinal VHL hemangiomas. This work is an example of how utilization of modern technologies advances the field of ophthalmic pathology.
Pathologists had previously noted two components in VHL hemangiomas: endothelial-lined vascular channels and vacuolated stromal cells. The lesion in retinal VHL hemangioma is identical to optic nerve hemangioblastoma and cerebellar hemangioblastoma. The work of Chan and coworkers definitively shows that the retinal and optic nerve lesions in VHL are primary stromal cell tumors with secondary vascularization due to increased VEGF production in the tumor. Hence, “hemangioma” and “angioma” are misnomers and should be supplanted. A better term is retinal hemangioblastoma, since it is identical to cerebellar hemangioblastoma.
The clever work by Chan and coworkers offers the opportunity for further discussion. What is the cell of origin in optic nerve hemangioblastoma? Is it intrinsic in the optic nerve, in the meninges in cases of the hemangioblastoma variant of meningioma, or both? Did any of the patients in the current study develop VHL syndrome? Also, do the authors recommend intravitreal injection of VEGF inhibitors for optic nerve or peripapillary hemangioblastoma?
Dr Robert N. Frank
In von Hippel–Lindau syndrome and in all the other VEGF-related diseases such as diabetic retinopathy and age-related macular degeneration, it should be noted that the major source of the VEGF is not the vascular cells; it is other cells, for example neuronal and glial stromal cells of retinal tissue, which then act secondarily on the vascular endothelium. Dr Chan’s presentation also emphasizes that it’s not just VEGF, but it’s what up-regulates VEGF, hypoxia-inducible factor, which is a transcription factor that hooks onto the DNA and stimulates it to make more mRNA and hence VEGF protein. Hence, the injection of a VEGF inhibitor alone may not be sufficient for therapy. Perhaps an approachable goal may be to develop technologies to down-regulate these transcription factors.
Dr Robert B. Welch
von Hippel–Lindau disease and the AOS have a long history of association. Almost three-quarters of a century ago, Dr Harvey Cushing, who was at Peter Brigham Hospital, presented a fascinating case at this meeting. This was shortly after Dr Lindau had defined the syndrome in 1926. At that meeting, Dr Wilmer discussed this paper and presented a beautiful fundus painting by Annette Burgess, our wonderful artist for so many years at Wilmer. Thirty-four years ago, my thesis for the AOS was on von Hippel–Lindau disease, in which I discussed the early lesions and treatment. Dr Chan is now advancing this disease another step forward on the molecular level. I hope this will provide us with new means of therapy, because this is a potentially blinding disease, especially for those with hemangioblastomas of the optic nerve.
Dr John T. Flynn
It is the hypoxia insult that produces VEGF that produces the tumor. What if you could treat these patients with hyperbaric oxygen so that their blood-oxygen was always saturated at a PAO2 of 120? Would that damage them vascularly, or could they survive and could that suppress the tumor?
Dr Chi-Chao Chan
I appreciate the thoughtful discussion by Dr Grossniklaus. There are debates on the cell of origin of optic nerve hemangioblastoma and retinal hemangioblastoma. Recent publications by Vortmyler and colleagues have demonstrated that the “stromal” or VHL cells may be developmentally arrested angioblasts1,2 that can co-express erythropoietin (Epo) and Epo receptor (EpoR). These hemangioblastoma cells have the potential to develop into primitive blood vessels and erythrocytes (extramedullary hematopoiesis). The hypothesis is that deletion of the VHL gene appears to be primarily responsible for the arrest of differentiation and possible up-regulation of Epo in EpoR expressing angiomesenchyme during embryonic development. In the preliminary study, we also found co-expression of Epo and EpoR messengers and protein in VHL retinal and optic nerve hemangioblastoma. Furthermore, we even detected many “stromal” cells bearing hematogenous and vascular stem cell markers such as CD31, CD34, and CD133 in the eyes with VHL disease. So, I agree with the proposal of Dr Grossniklaus to rename the retinal angioma or hemangioma associated with von Hippel-Lindau disease to retinal hemangioblastoma.
All four patients in the current study had VHL syndrome. Two presented with VHL lesions in the eye, CNS, and pancreas; one with VHL lesions in the eye, CNS, head, kidney, pancreas, and adrenal gland; and one in the eye, head, and kidney.
Macugen (intravitreal EYE001, 3 mg, q 6 wks, x6) had no effect on retinal hemangioblastoma in five VHL patients. However, the therapy may decrease retinal hard exudates and macular edema, and thus may increase visual acuity. We suggest that Macugen may be useful as an adjunct therapy in patients with retinal hemangioblastoma to decrease macular thickening and exudates.3
I agree with Dr Frank that we should think about targeting other cell products in addition to VEGF since there are other transcription factors that are targeted by VHL and/or HIF. We have a proposal awaiting NIH approval where we plan to target Epo and EpoR. The problem with systemic treatment is that it may affect erythroid progenitor cells in the bone marrow. This will have problems of erythrocyte regeneration. We should consider locally targeted drug treatment or drug delivery.
Dr Welch is the person who defined von Hippel–Lindau disease for the ophthalmologists; I am very thankful for his remarks and kind words. Concerning Dr John Flynn’s comment, hyperbaric oxygen might be tried and proposed for a new therapeutic protocol.
REFERENCES
- 1.Vortmeyer AO, Frank S, Jeong SY, et al. Developmental arrest of angioblastic lineage initiates tumorigenesis in von Hippel-Lindau disease. Cancer Res. 2003;63:7051–7055. [PubMed] [Google Scholar]
- 2.Vortmeyer AO, Yuan Q, Lee YS, et al. Developmental effects of von Hippel–Lindau gene deficiency. Ann Neurol. 2004;55:721–728. doi: 10.1002/ana.20090. [DOI] [PubMed] [Google Scholar]
- 3.Cusik M., Dahr S, Srivastava S, et al. Intravitreal anti-VEGF therapy for advance ocular disease of von Hippel-Lindau (VHL) disease. ARVO, 2004.
REFERENCES
- 1.Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 2.Melmon KL, Rosen SW. Lindau’s disease: review of the literature and study of a large kindred. Am J Med. 1964;36:595–617. doi: 10.1016/0002-9343(64)90107-x. [DOI] [PubMed] [Google Scholar]
- 3.Maher ER, Yates JR, Harries R, et al. Clinical features and natural history of von Hippel-Lindau disease [see comments] Q J Med. 1990;77:1151–1163. doi: 10.1093/qjmed/77.2.1151. [DOI] [PubMed] [Google Scholar]
- 4.Chan CC, Vortmeyer AO, Chew EY, et al. VHL gene deletion and enhanced VEGF gene expression detected in the stromal cells of retinal angioma. Arch Ophthalmol. 1999;117:625–630. doi: 10.1001/archopht.117.5.625. [DOI] [PubMed] [Google Scholar]
- 5.Wizigmann-Voos S, Plate KH. Pathology, genetics and cell biology of hemangioblastomas. Histol Histopathol. 1996;11:1049–1061. [PubMed] [Google Scholar]
- 6.Harris AL. Von Hippel-Lindau syndrome: target for anti-vascular endothelial growth factor (VEGF) receptor therapy. Oncologist. 2000;5((suppl 1)):32–36. doi: 10.1634/theoncologist.5-suppl_1-32. [DOI] [PubMed] [Google Scholar]
- 7.Sano T, Horiguchi H. Von Hippel-Lindau disease. Microsc Res Tech. 2003;60:159–164. doi: 10.1002/jemt.10253. [DOI] [PubMed] [Google Scholar]
- 8.Kuznetsova AV, Meller J, Schnell PO, et al. Von Hippel-Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc Natl Acad Sci U S A. 2003;100:2706–2711. doi: 10.1073/pnas.0436037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohh M, Park CW, Ivan M, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 10.McCabe CM, Flynn HW, Jr, Shields CL, et al. Juxtapapillary capillary hemangiomas. Clinical features and visual acuity outcomes. Ophthalmology. 2000;107:2240–2248. doi: 10.1016/s0161-6420(00)00422-x. [DOI] [PubMed] [Google Scholar]
- 11.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murgia A, Martella M, Vinanzi C, et al. Somatic mosaicism in von Hippel-Lindau Disease. Hum Mutat. 2000;15:114. doi: 10.1002/(SICI)1098-1004(200001)15:1<114::AID-HUMU20>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 13.Zhuang Z, Bertheau P, Emmert-Buck MR, et al. A microdissection technique for archival DNA analysis of specific cell populations in lesions <1 mm in size. Am J Pathol. 1995;146:620–625. [PMC free article] [PubMed] [Google Scholar]
- 14.Mandriota SJ, Turner KJ, Davies DR, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell. 2002;1:459–468. doi: 10.1016/s1535-6108(02)00071-5. [DOI] [PubMed] [Google Scholar]
- 15.Kaelin WG., Jr The von Hippel-Lindau gene, kidney cancer, and oxygen sensing. J Am Soc Nephrol. 2003;14:2703–2711. doi: 10.1097/01.asn.0000092803.69761.41. [DOI] [PubMed] [Google Scholar]
- 16.Iliopoulos O, Kaelin WG., Jr The molecular basis of von Hippel-Lindau disease. Mol Med. 1997;3:289–293. [PMC free article] [PubMed] [Google Scholar]
- 17.Lonergan KM, Iliopoulos O, Ohh M, et al. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol. 1998;18:732–741. doi: 10.1128/mcb.18.2.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schoenfeld A, Davidowitz EJ, Burk RD. A second major native von Hippel-Lindau gene product, initiated from an internal translation start site, functions as a tumor suppressor. Proc Natl Acad Sci U S A. 1998;95:8817–8822. doi: 10.1073/pnas.95.15.8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duan DR, Humphrey JS, Chen DY, et al. Characterization of the VHL tumor suppressor gene product: localization, complex formation, and the effect of natural inactivating mutations. Proc Natl Acad Sci U S A. 1995;92:6459–6463. doi: 10.1073/pnas.92.14.6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamura T, Koepp DM, Conrad MN, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999;284:657–661. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- 21.Pause A, Lee S, Worrell RA, et al. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci U S A. 1997;94:2156–2161. doi: 10.1073/pnas.94.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 23.Semenza GL. HIF-1: using two hands to flip the angiogenic switch. Cancer Metastasis Rev. 2000;19:59–65. doi: 10.1023/a:1026544214667. [DOI] [PubMed] [Google Scholar]
- 24.Wizigmann-Voos S, Breier G, Risau W, et al. Up-regulation of vascular endothelial growth factor and its receptors in von Hippel-Lindau disease-associated and sporadic hemangioblastomas. Cancer Res. 1995;55:1358–1364. [PubMed] [Google Scholar]
- 25.Takahashi A, Sasaki H, Kim SJ, et al. Markedly increased amount of messenger RNAs for vascular endothelial associated factor and placenta growth factor in renal cell carcinoma associated angiogenesis. Cancer Res. 1994;54:4233–4237. [PubMed] [Google Scholar]
- 26.Gnarra JR, Zhou S, Merrill MJ, et al. Post-transcriptional regulation of vascular endothelial growth factor mRNA by the product of the VHL tumor suppressor gene. Proc Natl Acad Sci U S A. 1996;93:10589–10594. doi: 10.1073/pnas.93.20.10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iliopoulos O, Levy AP, Jiang C, et al. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A. 1996;93:10595–10599. doi: 10.1073/pnas.93.20.10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aiello LP, George DJ, Cahill MT, et al. Rapid and durable recovery of visual function in a patient with von Hippel-Lindau syndrome after systemic therapy with vascular endothelial growth factor receptor inhibitor su5416. Ophthalmology. 2002;109:1745–1751. doi: 10.1016/s0161-6420(02)01159-4. [DOI] [PubMed] [Google Scholar]
- 29.Girmens JF, Erginay A, Massin P, et al. Treatment of von Hippel-Lindau retinal hemangioblastoma by the vascular endothelial growth factor receptor inhibitor SU5416 is more effective for associated macular edema than for hemangioblastomas. Am J Ophthalmol. 2003;136:194–196. doi: 10.1016/s0002-9394(03)00101-6. [DOI] [PubMed] [Google Scholar]