

Abstract
The tripeptide backbone of phosphinothricin (PT) tripeptide (PTT), a compound with herbicidal activity from Streptomyces viridochromogenes, is assembled by three stand-alone peptide synthetase modules. The enzyme PhsA (66 kDa) recruits the PT-precursor N-acetyl-demethylphosphinothricin (N-Ac-DMPT), whereas the two alanine residues of PTT are assembled by the enzymes PhsB and PhsC (129 and 119 kDa, respectively). During or after assembly, the N-Ac-DMPT residue in the peptide is converted to PT by methylation and deacetylation. Both phsB and phsC appear to be cotranscribed together with two other genes from a single promoter and they are located at a distance of 20 kb from the gene phsA, encoding PhsA, in the PTT biosynthesis gene cluster of S. viridochromogenes. PhsB and PhsC represent single nonribosomal peptide synthetase elongation modules lacking a thioesterase domain. Gene inactivations, genetic complementations, determinations of substrate specificity of the heterologously produced proteins, and comparison of PhsC sequence with the amino terminus of the alanine-activating nonribosomal peptide synthetase PTTSII from S. viridochromogenes confirmed the role of the two genes in the bialanylation of Ac-DMPT. The lack of an integral thioesterase domain in the PTT assembly system points to product release possibly involving two type II thioesterase genes (the1 and the2) located in the PTT gene cluster alone or in conjunction with an as yet unknown mechanism of product release.
The peptide antibiotic phosphinothricin tripeptide (PTT; bialaphos) consists of the unusual amino acid phosphinothricin (PT) and two alanine residues (Fig. 1). It is produced by Streptomyces viridochromogenes and Streptomyces hygroscopicus (3, 16). PTT penetrates bacterial cells as a prodrug via peptide uptake systems with subsequent release of PT, which, due to its structural similarity to glutamate, inhibits glutamine synthetase, a key enzyme of nitrogen metabolism (10). By contrast, plant cells can take up both PTT and PT directly. The inhibition of glutamine synthetase in their case results in the accumulation of ammonium ions, which are toxic to plant cells (42). Based on this mechanism, both PTT and PT have found commercial applications as broad-spectrum herbicides (known as Herbiace and Basta, respectively).
FIG. 1.

Structure of phosphinothricin tripeptide (PTT) and its precursor N-acetyldemethylphosphinothricin (N-Ac-DMPT).
Previous work on the biosynthesis of PTT indicated that it is derived from a precursor peptide, N-acetyl-demethylphosphinothricin tripeptide (N-Ac-DMPTT), which is assembled in a nonribosomal mechanism (1, 11, 32, 43). In contrast to PTT, N-Ac-DMPTT contains N-acetyldemethylphosphinothricin (N-Ac-DMPT) instead of PT. N-Ac-DMPTT was never isolated, however, the occurrence of DMPT or DMPTT in fermentation broth of PTT-producing S. hygroscopicus and accumulation of N-AcDMPT in a PTT-nonproducing mutant of S. viridichromogenes (NTG1) obtained by mutational cloning point indirectly to its existence (1). Genetic complementation of the latter mutant in trans revealed the gene phsA (1, 43). The deduced gene product of phsA represented a 66-kDa nonribosomal peptide synthetase (NRPS) module consisting of an adenylation (A)-domain and a peptidyl carrier protein (thiolation, T)-domain (32).
Sequence comparison of PhsA with the amino-terminal sequence of the N-Ac-DMPT-activating enzyme (PTT synthetase I) from S. viridochromogenes revealed that PhsA is identical to PTT synthetase I and that it is most probably the initiation module of the PTT assembly system with N-Ac-DMPT representing the formal amino terminus of Ac-DMPTT (11). Analysis of the substrate specificity of PTT synthetase I (PhsA) using several model substrates structurally related to N-Ac-DMPT revealed that PTT synthetase I exclusively activated N-acetylated phosphinoamino acids such as Ac-PT but not deacetylated ones, confirming the presumed role of N-Ac-DMPTT as an intermediate of PTT biosynthesis (11).
Further biochemical analyses of protein extracts of S. viridochromogenes also showed the presence of two alanine-activating enzymes which were able to bind alanine as a thioester and were estimated to have Mrs of 120,000 and 140,000 in their native states. These two peptide synthetases were correlated in their activities with the titers of PTT in different S. viridochromogenes strains and were termed PTT synthetases II and III, respectively, both most probably responsible for recruitment of the two alanine residues of N-Ac-DMPTT (11). However, the two proteins were found to be unstable, and since no sequence data were available, it was not clear how their precise modular structures looked or whether they were distinct protein species or proteolytic fragments of a larger enzyme housing a total of two modules for alanine activation.
Here we show that the PTT biosynthesis gene cluster contains two NRPS genes lying some 20 kb downstream from the phsA gene. These two genes, designated phsB and phsC, were considered candidate genes for PTT synthetases II and III, respectively. In the following we present genetic and biochemical evidence that these genes encode two stand-alone modules which are identical with PTT synthetases II and III, respectively.
MATERIALS AND METHODS
Bacterial strains, plasmids, cosmids, and phages.
The bacterial strains, plasmids, cosmids, and phages used in this study are listed in Table 1.
TABLE 1.
Bacterial strains, phages, cosmids, and plasmids used in this study
| Strain, cosmid, phage, or plasmid | Relevant genotype and phenotype | Source or references |
|---|---|---|
| S. viridochromogenes | ||
| Tü494 | PTT-producing wild type | 3 |
| 707/3 | Gene disruption of the phsC gene, tsr, PTT nonproducing | This study |
| B3-14 | Gene replacement in the phsB gene, apr, PTT nonproducing | This study |
| PHSBC | Gene replacement in the phsB/orfM/phsC genes, apr, PTT nonproducing | This study |
| E. coli | ||
| XL Blue | recA1 endA1 gyrA96 thi-1 hsdr17 supE44 relA1 lac[F′ proAB laclqZΔM15 Tn10(Tetr)] | 7 |
| Phages | ||
| λ-WT8 | λ phage clone carrying approximately 20 kb of the PTT biosynthetic gene cluster from S. viridochromogenes | 33 |
| Plasmids | ||
| pIJ486, pIJ487 | pIJ101 derivative, tsr, promoterless aphII gene, promoter probe vector | 41 |
| pDS5 | pBlueskript SK+ carrying a 10-kb NotI/EcoRI fragment of λ-WT8 | This study |
| pEH13 | pUC21 derivative carrying the apramycin/PermE (aprP) resistance cassette | 12 |
| pDS199 | pK19 derivative; a 4.1-kb Eco47-3/Bst1 107 DNA fragment was replaced by the apramycin/PermE (aprP) resistance cassette (1.6-kb EcoRV/StuI fragment of pEH13) | This study |
| pDS190/191 | E. coli-Streptomyces shuttle vector, promoter probe vector, apr tsr, promoterless neomycin resistance gene, multiple cloning site of pIJ486/487 | This study |
| pGM8 | tsr, temperature-sensitive Streptomyces vector | 21 |
| pEM4 | Streptomyces-E. coli shuttle vector, PermE, trs, bla | 27 |
| pDS207 | pEM4 carrying phsB under the control of PermE located on a 6.2-kb KpnI/Bst107 DNA fragment | This study |
| pDS208 | pEM4 carrying phsC located on a 3.6-kb NruI/EcoRI DNA fragment | This study |
| pDS209 | pEM4 carrying phsB and phsC located on a 8.9-kb KpnI/EcoRI DNA fragment | This study |
| pDS210 | pDS190 carrying the intergenic region of phsB and phsC as a 475-bp ApaI/SmaI DNA fragment | This study |
| pDS211 | pDS191 carrying the promoter region of phsB as a 767-bp PCR DNA fragment | This study |
| pDS104 | pGM8 carrying a 707-bp ApaI fragment of phsC | This study |
| pDS99 | pK19 carrying phsB/orfM as a 6.2-kb KpnI/Bst1 107 fragment cloned into the KpnI/EclI 36 sites | This study |
| pMS99 | pEM4 carrying phsB located on a 4.1-kb KpnI/NcoI fragment | This study |
| pMS100 | pDS99 derivative, a 1.7-kb Eco47-3 fragment was replaced by a 1.6-kb EcoRV/StuI fragment of pEH13 (apr-PermE resistance cassette) | This study |
Chemicals and radiochemicals.
l-[U-14C]alanine (156 mCi/mmol), l-[U-14C]serine (165 mCi/mmol), l-[U-14C]proline (232 mCi/mmol), and [3H-acetyl]coenzyme A (216 mCi/mmol) were from Amersham-Bioscience (Braunschweig, Germany). l-[1-14C]aminobutyrate (26.8 mCi/mmol) was from CEA (Gif-sur-Yvette, France). 3H-labeled N-acetylphosphinothricin ([3H]AcPT) was prepared enzymatically from phosphinothricin and [3H]acetyl coenzyme A using phosphinothricin acetyltransferase purified from wild-type S. viridochromogenes as described (11). Acetylphosphinothricin (acetylglufosinate ammonium) and phosphinothricin (glufosinate ammonium) were kindly provided by Aventis AG (Frankfurt). PTT was purified from aqueous dilutions of Herbiace by repeated precipitation with acetone. All other chemicals were of the highest purity commercially available.
Media and culture conditions.
For the cultivation of Streptomyces viridochromogenes and Streptomyces lividans, YM medium was used (1). Cultures were grown in baffled Erlenmeyer flasks on a rotary shaker for 3 days at 30°C. Escherichia coli XL1 Blue was grown in LB medium at 37°C (29).
Isolation of chromosomal and plasmid DNA.
Isolation of chromosomal and plasmid DNA from Streptomyces strains was carried out according to (14). Plasmids from E. coli were isolated as described (29).
Cloning, restriction mapping, and in vitro manipulation of DNA.
Methods for isolation and manipulation of DNA were as described (14, 29). Restriction endonucleases were purchased from various suppliers and used according to their instructions.
DNA sequencing and analysis.
Subfragments of DNA fragments containing the peptide synthetase genes were subcloned in sequencing vectors pK19, pUC18 and pBlueskript SK+. The sequence of a 9-kb DNA fragment containing phsB and phsC was determined by standard techniques (30). The DNA sequence was examined for open reading frames by applying the codon usage program of Staden and MacLachlan (1982) (38). The programs BLAST (2) and CLUSTAL W (40) were used for homology search. Multiple alignments were generated by the program GENEDOC (22). The deduced amino acid sequence of OrfM was examined for transmembrane helices by the dense alignment surface method (9).
Southern hybridization.
Southern hybridization experiments were carried out using the nonradioactive digoxigenin DNA labeling and detection kit from Roche Diagnostics (Mannheim, Germany). Hybridizations using oligonucleotides (oligonucleotide 1, 2, 5 and 6) were performed at 57°C and a stringent washing step with 1× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)/0.1% sodium dodecyl sulfate (SDS). The oligonucleotides were marked using the digoxigenin oligonucleotide tailing kit from Roche Diagnostics (Mannheim, Germany). Oligonucleotides used for identification of the peptide synthetase genes were derived from conserved core motifs: oligonucleotide 1 (core motif 1, A-domain), oligonucleotide 2 (core motif 2, A-domain), oligonucleotide 4 (core motif 4, A-domain), and oligonucleotide 5 (core motif 5, T-domain) (24) (Table 2). Hybridizations using the phsB gene (including orfM) located on a 6.2-kb KpnI/BstI107 DNA fragment as a probe were performed at 68°C and a stringent washing step with 0.1× SSC/0.1% SDS.
TABLE 2.
Oligonucleotides used in Southern hybridization and PCR experiments in this study
| Oligonucleotide | DNA sequence (5′→3′) |
|---|---|
| Oligo 1 | TTGAAGGCGGGCGGGCCTACGTACCGATCGACCCC |
| Oligo 2 | AGGCCTACATCATCTACACCTCGGGCACGACGGGCAAGCCCAAGGG |
| Oligo 4 | CAGGTCAAGATCCGCGGCTACCGCATCGAGCTCGGCGAGATCGAG |
| Oligo5 | CTCGGCGGGCACTCCCTCAAGGCCT |
| OrflP1 | AGAATTCCCGGATGAGCGGCGGAACG |
| Orf1P2 | TGTCTAGAGTGACGACCCTCGTCCGTT |
Gene replacement/gene disruption mutagenesis and transformation.
Gene disruption mutagenesis in phsC using the temperature-sensitive plasmid pDS104 was performed by a method described previously (32). Mutants PHSB and PHSBC were generated using the nonreplicative plasmids pMS100 and pDS199, respectively (Table 1). Transformations of Streptomyces strains were done according to (14). S. viridochromogenes was transformed with plasmids which were isolated from E. coli ET12567 or S. lividans. The CaCl2 method was applied for transformation of E. coli as described (29). PTT production was examined by a bioassay (1).
Complementation of the mutants PHSB and PHSBC and heterologous expression of phsB/phsC in Streptomyces lividans.
The separate peptide synthetase genes phsB, phsB (including orfM), phsC, and all genes together were subcloned as KpnI/NcoI, KpnI/BstI107, NruI/EcoRI, and KpnI/EcoRI fragments in the expression vector pEM4, resulting in plasmids pMS99, pDS207, pDS208, and pDS209, respectively. The corresponding peptide synthetase genes are transcribed by the constitutive ermE promoter (PermE) (31). After transformation of mutant PHSB with pMS99 and of mutant PHSBC with pDS207, pDS208, and pDS209, the restoration of PTT production was examined as described (1). For heterologous expression of phsB/phsC, S. lividans TK23 was transformed using pDS209.
Promoter probe experiments.
In order to identify a putative promoter in front of phsB, a 0.7-kb DNA fragment upstream of phsB was isolated by PCR. The following reaction mixture was used: 0.7 μg pDS5 as template, 1.0 μM of primers Orf1P1 and Orf1P2 (Table 2), 10 μl 10× reaction buffer (with 2 mM MgCl2), 20 μl Q-solution, 0.2 mM deoxynucleoside triphosphates, and 1.0 μl Pwo DNA polymerase (Roche Diagnostics), The PCR total volume was 100 μl. After denaturation (2 min, 94°C), 25 cycles of amplification (1 min at 94°C, 1.5 min at 58°C, and 1 min at 72°C) were performed in a thermal controller PTC-100 from MJ Research, Inc. (Waltham, MA). PCR products were electrophoretically separated in a 1% agarose gel, isolated by gel elution (Qiaquick, QIAGEN, Hilden, Germany) and directly used for cloning. To identify a putative promoter between orfM and phsC, a 477-bp ApaI/SmaI DNA fragment containing the corresponding intergenic region was used. The putative promoter carrying DNA fragments were cloned upstream of the promoterless neomycin/kanamycin resistance genes of the promoter probe vectors pDS190 or pDS190, respectively. Promoter activity was tested by conferring kanamycin resistance to plasmids carrying S. lividans and S. viridochronogenes cells, respectively.
Enzyme purifications.
PhsA, PhsB, and PhsC activities from disruption mutants or wild-type S. viridochromogenes (as a control) were isolated (11). In the case of each strain, 10 g of mycelium (wet weight) served for preparation of a crude extract and was fractionated on Q-Sepharose (fast flow, Amersham Bioscience) by elution with a salt gradient (0 to 0.25 M NaCl). Individual peaks eluted in the order PTT synthetase I (Ac-PT thioester-forming activity PhsA), PTT synthetase II (alanine thioester-forming activity PhsC), and PTT synthetase III (alanine thioester-forming activity PhsB). Each individual peak was pooled and subjected to ammonium sulfate precipitation (60% saturation). The resulting precipitates were subsequently subjected to gel filtration on Superdex 200 (Amersham, Bioscience). Optionally, PhsB and -C each were purified by one additional round of anion exchange chromatography on MonoQ (HR5/5).
Enzyme assays.
Routine monitoring of PhsA was by the N-Ac-PT dependent ATP-pyrophosphate exchange reaction (11) Monitoring of PhsB or -C was by the thioester formation assay with [14C]alanine, respectively. Quantitation of enzymes was by titration with [3H]N-acetyl-phosphinothricin (PhsA) or [14C]alanine (PhsB or PhsC) and determination of enzyme bound radioactivity by standard protocols. The thioester formation assays consisted of enzyme (PhsA, PhsB or PhsC), 106 cpm [3H]AcPT (or 5 × 105 cpm [14C]alanine, respectively), 5 mM ATP, 20 mM MgCl2 in a total volume of 180 to 250 μl (buffer B (11). Incubation was for 30 min at 30°C. Assays to demonstrate in vitro formation of free or enzyme-bound AcPT-alanine or AcPT-alanyl-alanine contained 9 to 13 pmol PhsA, 3.5 to 5 pmol PhsB, or 2.5 to 3.2 pmol PhsC (or both), 1 μCi [14C]alanine, 0.25 mM AcPT, 5 mM ATP, and 20 mM MgCl2 in total volumes of 250 to 350 μl (buffer B). Incubations were at 30°C for 30 min.
Analytical procedures.
For detection of protein-bound radioactive reaction intermediates, enzyme incubations were subjected to trichloroacetic acid precipitation with 5% trichloroacetic acid. After washing with trichloroacetic acid and ethanol, precipitates were dissolved in 100 μl 0.1 M NaOH. After neutralization with 0.1 M HCl, undissolved protein was removed by centrifugation and supernatants applied to Pasteur pipettes containing freshly regenerated Dowex-50 H+ form (in H2O). Washing with water eluted AcPT and potential AcPT peptides from the columns. Neutral amino acid such as alanine was eluted with 2 M NH3. Further purification of the acidic fraction was on Dowex-1 formate columns (washed with water) and subsequent elution with 1 M ammonium formate, pH 4.5. After freeze drying, acidic and neutral fractions were taken up in minute volumes of water and applied to silica gel 60 thin-layer chromatography sheets and chromatographed using 2-propanol-acetic acid-water (7:2:3, by volume) as the solvent system. Protein determinations and SDS gel electrophoresis were as described (6, 17).
For short-term incorporation studies with whole cells of S. viridochromogenes, 5-ml portions were removed from S. viridochromogenes cultures actively synthesizing PTT (3 days of age) and centrifuged for 5 min at 5,000 × g and room temperature. After washing twice in the same volume of tap water, cells were resuspended in 5 ml of tap water and transferred to a 100-ml Erlenmeyer flask. After addition of 1 μCi of [14C]alanine, the suspension was shaken at 250 rpm in an environmental shaker (New Brunswick, model G 25) for 2 hours at 28°C. After this, mycelium was filtered off and the filtrate was concentrated to a final volume of ca. 250 μl. Aliquots were chromatographed on silica gel sheets along with authentic PTT using 2-propanol-acetic acid-water (7:2:3, by volume) as solvent system. Radioactive PTT was localized on TLC plates by radioscanning as described (11). Labeling of proteins with subsequent separation by SDS-gel electrophoresis and autoradiography was as described (11). Protein microsequencing from protein blots was done on an Applied Biosystems (Foster City, CA) Procise sequencer according to the manufacturer's protocols.
Selectivity conferring code of peptide synthetases.
The selectivity conferring code of peptides synthetases are deposited at the NRPS A-domain database (http://www.tigr.org/jravel/nrps/).
Nucleotide sequence accession number.
The nucleotide sequence data reported have been assigned he accession no. X65195 (as part of the complete PTT biosynthetic gene cluster) in the EMBL data library. In Fig. 2, the biosynthetic gene cluster shown covers a region from bp 1626 to bp 35396 of the deposited sequence data.
FIG. 2.

Localization of the peptide synthetase genes phsA, phsB, and phsC and of the thioesterase genes the1 and the2 in the PTT biosynthetic gene cluster from S. viridochromogenes (A) (34). The replacement of the 4.1-kb Eco47-3/BstI107 fragment by the apramycin/PermE (aprP) cassette resulting in the gene organization of the mutant PHSBC is shown in B. Restriction sites used in subcloning experiments are marked and regions with promoter activity are indicated by arrows. The internal fragments of phsB and phsC used for gene disruption and gene replacement experiments are marked by boxes. DNA fragments used for promoter probe experiments are indicated by black lines. The correct gene replacement in mutant PHSBC is shown by Southern hybridization using phsB/orfM (KpnI/BstI107 DNA fragment) as a probe (C). Lane 1, XhoI-digested chromosomal DNA of wild-type S. viridochromogenes; lane 2, XhoI-digested chromosomal DNA of PHSBC; lane 3, KpnI-digested chromosomal DNA of wild-type S. viridochromogenes; lane 4, KpnI-digested chromosomal DNA of PHSBC; M, DNA size markers.
RESULTS
Identification and characterization of peptide synthetase genes at the left boundary of the PTT biosynthetic gene cluster.
In order to locate and characterize the peptide synthetase genes responsible for recruitment of the alanine residues of PTT in the PTT antibiotic biosynthetic gene cluster, Southern hybridization experiments were performed using oligonucleotides, which were designed from conserved core motifs of peptide synthetases, notably those for the A-domain and T-domains (see Materials and Methods). Two DNA fragments (1.8-kb SacI fragment and 5.5-kb SacI/EcoRI fragment) which hybridized with all of these core sequence-encoding oligonucleotides could be identified (data not shown). These fragments, carrying the genetic information for at least two peptide synthetase modules, were localized in a DNA region approximately 20 kb upstream of the previously described gene phsA (encoding PTT synthetase I).
Sequencing this region along with other regions of the PTT biosynthetic gene cluster (34) revealed four complete open reading frames (ORFs), called orf1, phsB, orfM, and phsC (Fig. 2). orf1 is 210 bp long and encodes a protein of 69 amino acids. The second ORF, phsB, with a size of 3,570 bp, is located immediately downstream of orf1. The gene encodes a protein of 1,189 amino acids (129 kDa). The next ORF (orfM) is 1,662 bp long and the deduced protein consists of 553 amino acids. Whereas the end of phsB and the start of orfM overlap by 8 bp, the following ORF, phsC, is located 72 bp downstream of orfM. It has a size of 3,261 bp and encodes a protein of 1,086 amino acids (119 kDa). The G+C content of the genes (72.6 mol%) is characteristic of Streptomyces DNA (4). In contrast to the phsA gene (1, 36), no rare TTA codon, believed to be involved in regulation of secondary metabolism (18), was found.
Identification of putative promoters.
To examine whether orf1, phsB, orfM, and phsC form a single transcriptional unit, promoter probe experiments were performed. A 767-bp DNA fragment directly upstream of phsB (Fig. 2A) was cloned in front of the promoterless neomycin/kanamycin resistance gene of the promoter probe vector pDS191, resulting in plasmid pDS211. This plasmid was transferred into S. lividans and S. viridochromogenes and conferred a kanamycin resistance level of up to 800 μg/ml in these strains, indicating the localization of a putative promoter on the cloned DNA fragment, and 430 bp in front of orf1 that is translationally coupled with phsB, a sequence (5′ TCTTGACAT 3′) was identified showing similarity to the consensus sequence of E. coli and Streptomyces σ70-dependent promoters (5). In contrast, no promoter region was identified by analysis of the intergenic region between orfM and phsC on a 475-bp ApaI/SmaI DNA fragment cloned in PDS210 (Fig. 2A). Kanamycin resistance only reached a low level of 5 to 10 μg/ml, a value that was also observed in the negative controls (promoter probe vectors without inserts). Regarding these results and the fact that a promoter was already found upstream of the phosphinomethylmalate isomerase gene (pmi), which is 112 bp downstream of phsC (13) (Fig. 2A), the peptide synthetase genes phsB and phsC as well as orf 1 and orfM all appear to be cotranscribed from a single promoter upstream of orf1.
Sequence analysis of the deduced proteins from orf1, phsB, orfM, and phsC.
Comparisons of the deduced amino acid sequences of orf1, orfM, phsB, and phsC with protein sequences in databases revealed that Orf1 (calculated mass, 7.9 kDa) was similar to proteins of similar sizes but unknown function encoded by genes in various NRPS gene clusters such as of mycobactin in Mycobacterium tuberculosis (46% identity) or of enterobactin in E. coli (32% identity to orf1) (25, 28). The deduced gene product of orfM showed weak similarities to putative membrane proteins of unknown function from E. coli and Bacillus subtilis (not shown). To identify putative transmembrane helices, a search was performed using the dense alignments surface (DAS) method (9). By this method, at least 13 transmembrane helices were identified (data not shown) which is consistent with the assumption that OrfM is a membrane protein.
Analysis of the deduced amino acid sequences from phsB and phsC showed that each protein represents a complete elongation module containing well conserved condensation (C-), A-, and T-domains in the order C-A-T. Surprisingly, PhsB was found to possess an extra amino-terminal domain of about 100 amino acids preceding the C-domain. This domain has high similarity to T-domains, indicating a T-C-A-T domain arrangement for PhsC. The alignment of the four T-domains of the PTT assembly system (PhsA, PhsB, and PhsC) showed high conservation of these domains except that the amino-terminal PhsB T-domain deviated from the others in the signature sequence of the 4′-phosphopantheteine attachment site (19) by a change of the double G in L(T,I)GG(D,H)S(L,I) to GA (Fig. 3). Whether this amino acid substitution has significance for the catalytic function of this particular T-domain is not known.
FIG. 3.

Alignment of the T-domains of PhsA, PhsB, and PhsC with the T-domains of NRPS modules from tyrocidine synthetase 2, gramicidin S synthetase 2, and surfactin S synthetase 1 (module 2).
Given that PhsB and PhsC are involved in PTT assembly, they should have preferred substrate specificity for the amino acid alanine and should display sequence similarity to each other. Surprisingly, however, the enzyme sequences differed from each other considerably, not only in their entirety but in particular with respect to their A-domains (38% identity and 52% similarity). The substrate specificity-conferring amino acid residues in the substrate-binding site of these A-domains as predicted from comparison with various A-domains of NRPS (8, 40) indicated specificity of the PhsB A-domain for serine and of the PhsC A-domain for proline (Table 3). This stands in contrast to the observed substrate specificities of PTT synthetases II and III, which both activate alanine as their preferred substrate (Table 4).
TABLE 3.
Selectivity-conferring code of alanine, serine, and proline activating A-domains of peptide synthetase modulesa
| Enzyme | Amino acid at position:
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 235 | 236 | 239 | 278 | 299 | 301 | 322 | 330 | |
| PhsB (Ala) | D | V | W | H | F | S | L | I |
| Hts-2 (Ala) | D | A | G | G | C | A | M | V |
| Hts-3 (Ala) | D | L | L | F | G | I | S | V |
| CssA-11 (Ala) | D | L | W | F | Y | I | A | V |
| PhsC (Ala) | D | V | L | L | V | A | G | V |
| Hts-2 (Ala) | D | A | G | G | C | A | M | V |
| Hts-3 (Ala) | D | L | L | F | G | I | S | V |
| CssA-11 (Ala) | D | L | W | F | Y | I | A | V |
| PhsB (Ala) | D | V | W | H | F | S | L | I |
| EntF, SyrE (Ser) | D | V | W | H | L | S | L | I |
| PhsC (Ala) | D | V | L | L | V | A | G | V |
| Lyss-M3 (Pro) | D | I | T | L | V | A | G | L |
| Lincs-M1 (Pro) | D | V | A | L | V | A | I | G |
The determination of the selectivity-conferring residues in alanine-activating peptide synthetase domains was carried out as described by Stachelhaus et al. (36). In comparison, the code of serine and proline activating domains are shown. The specificity conferring codes were compared with the corresponding codes of PhsB and PhsC. Amino acid positions with 100% similarity are in bold, 66 to 75% similarity is in italic, and 50% similarity is in normal type. PhsB/PhsC, phosphinothricin tripeptide synthetase B/C from Streptomyces viridochromogenes; HtsA-2/HtsA-3, module 2 and module 3 (alanine activating) of the HC-toxin synthetase from Cochliobolus carbonum; CssA-11, domain 11 of the cyclosporin synthetase from Tolypocladium niveum; EntF, enterobactin synthetase from E. coli; SyrE, serine activating xdomain of the syringomycin synthetase from Pseudomonas pv. syringae; Lyss-M3, proline-activating Claviceps purupurea peptide synthetase Lincs-M1, proline-activating lincomycin synthetase LmbC from Streptomyces lincolnensis. Selectivity-conferring codes are deposited at the NRPS A-domain database (http://www.tigr.org/jravel/nrps/).
TABLE 4.
Substrate specificity of heterologously produced PhsB and PhsCa
| Enzyme | Substrate | ATP-pyrophosphate exchange (nmol exchanged) | Thioester formation
|
|
|---|---|---|---|---|
| pmol bound | Rel. act. (%) | |||
| PhsB | l-Ala | 1.55 | 15.8 | 100 |
| l-Abu | 9.3 | 59 | ||
| l-Ser | 7.3 | 46 | ||
| l-Pro | 0 | |||
| H2O | 0.10 | |||
| PhsC | l-Ala | 1.749 | 54.9 | 100 |
| l-Abu | 0.169 | 52.2 | 95 | |
| l-Ser | 0.059 | 18.1 | 33 | |
| l-Val | 0.026 | |||
| l-Pro | 0.010 | 0 | ||
| H2O | 0.009 | |||
The thioester formation assay was preformed as described in Materials and Methods. In the case of PhsC, 3.87 pkat of enzyme was used in each experiment, whereas in the case of PhsB the amount of enzyme was 3.44 pkat. The enzyme was from the Resource Q separation step described in the protocol published previously (11).
Analysis of mutants with disruption or gene replacements of phsB and phsC.
To show the involvement of phsC in PTT biosynthesis an internal fragment of phsC (707-bp ApaI fragment) (Fig. 2A) was cloned into the temperature-sensitive Streptomyces plasmid pGM8, resulting in plasmid pDS104. Use of pDS104 for gene disruption of phsC in wild-type S. viridochromogenes according to a previously published procedure (32) resulted in a PTT-nonproducing mutant (707/3) (Table 1). Southern hybridization analysis confirmed the gene inactivation. Enzymatic analysis of cell extracts of strain 707/3 revealed the presence of N-AcDMPT activating activity (revealed by thioester formation with the model substrate N-[3H]AcPT or the N-AcPT-dependent ATP-pyrophosphate exchange) and alanine-activating activity eluting from anion exchanger columns (Sepharose Q, fast flow) at the same salt strength as PTT synthetase III previously isolated from wild-type S. viridochromogenes (11). The levels of the two activities as revealed by titration with [3H]AcPt or [14C]alanine, respectively, were in the same range as those obtained from wild-type S. viridochromogenes (11). No activity attributable to PTT synthetase II was detected, which suggests that PhsC is identical to PTT synthetase II.
To inactivate the phsB gene, an internal 1.7-kb Eco47-3 fragment (Fig. 2) was replaced by the apramycin/PermE (aprP) resistance cassette (1.6-kb StuI/EcoRV fragment of pEH13) in a gene replacement experiment using plasmid pMS100 (Table 1). To ensure the continuation of orfM and phsC transcription, the ermE promoter (as part of the cassette) had been introduced in the direction of phsB transcription. The successful use of the cassette was shown previously by inactivation of another biosynthetic gene (pmi) gene in the PTT biosynthetic gene cluster (13). The correctness of the gene replacement was checked by Southern hybridization analysis and confirmed by the finding that the resulting phsB null mutant (B3-14) (Table 1) was unable to produce PTT.
Analysis of protein extracts of strain B3-14 revealed that PhsA was present in the same yield as in wild-type S. viridochromogenes and 707/3, but no alanine-activating activity attributable to PTT synthetase III (as judged from the elution behavior from Sepharose Q anion exchange columns) was detectable. Interestingly, the residual alanine-activating activity attributable to PTT synthetase II was only present at a level of 10 to 15% of the level found in the wild type or strain 707/3. The low activity of PTT synthetase II in strain B3-14 may be the result of poor expression of phsC under the control of the ErmE promoter placed in front of orfM/phsC in that construct. On the other hand, it cannot be excluded that expression in part is still triggered by the natural promoter located in front of phsB. However, if it is the ermE promoter, which drives the low expression of phsC in this construct, this should be because it is present in single copy. In high-copy conditions, heterologous expression in S. lividans of both phsB and phsC cloned into plasmid pEM4 is much stronger (see below).
Genetic complementation of B3-14 was achieved by using plasmid pMS99, which carries the native phsB gene under the control of the ermE promoter. Restoration of PTT production in the transformant was detected by the production of a small inhibition zone in the agar plug diffusion assay for PTT which was absent in strain B3-14. From these data, it is concluded that PhsB is identical to PTT synthetase III. Moreover, they indicate that in strain B3-14 both orfM and phsC are functionally expressed and PhsC cannot take on the function of PhsB.
Construction of a triple PTT mutant (phsB, orfM, and phsC) was achieved by replacing a 4.1-kb Eco47-3/BstI107 fragment of the phsB/orfM/phsC region (ΔphsBC, Fig. 2A) with the aprP resistance cassette. Southern hybridizations using genomic DNA from the resulting strain, PHSBC, revealed a double crossover event between the chromosomal copy of phsB/phsC and the mutated fragment in the disruption construct pDS199 (Fig. 2B and C). Accordingly, PHSBC had lost the ability to produce PTT. Transformation of PHSBC with either phsB including orfM (pDS207) or phsC (pDS208) did not restore PTT synthesis, indicating that PhsB could not take on the function of PhsC or vice versa. Transformation of PHSBC with pDS209 containing phsB, orfM, and phsC under the control of the ermE promoter (31), by contrast, resulted in restoration of PTT synthesis indicating the requirement of both PhsB and PhsC for PTT production.
Expression of phsB and phsC in Streptomyces lividans TK23.
Heterologous expression of phsB and phsC in S. lividans strain TK23 was performed by using plasmids pDS209 and pDS207. pDS209 contains an insert encompassing both genes phsB and phsC (together with orfM) cloned behind the ermE promoter of plasmid pEM4. Testing protein extracts of S. lividans TK23(pDS209) for enzyme activities catalyzing enzyme-alanine thioester formation revealed alanine-activation which was not seen in the control extracts derived from strain S. lividans TK23 containing plasmid pEM4 (data not shown).
Purification according to the published protocol for the phosphinothricin peptide synthetases from S. viridochromogenes by ion exchange chromatography on Sepharose Q, gel filtration on Superdex 200, and anion exchange chromatography (Mono Q HR5/5) afforded a clear separation of the alanine thioester-forming activities from S. lividans TK23 transformed with pDS209 in two separate peaks identical to those isolated by the purification of the two original enzymes from wild-type S. viridochromogenes (11) (not shown). Covalent labeling of enzyme in the two peak fractions with radioactive alanine, gel electrophoretic separation by SDS-polyacrylamide gel electrophoresis (PAGE), and autoradiography permitted visualization of the two proteins (Fig. 4).
FIG. 4.
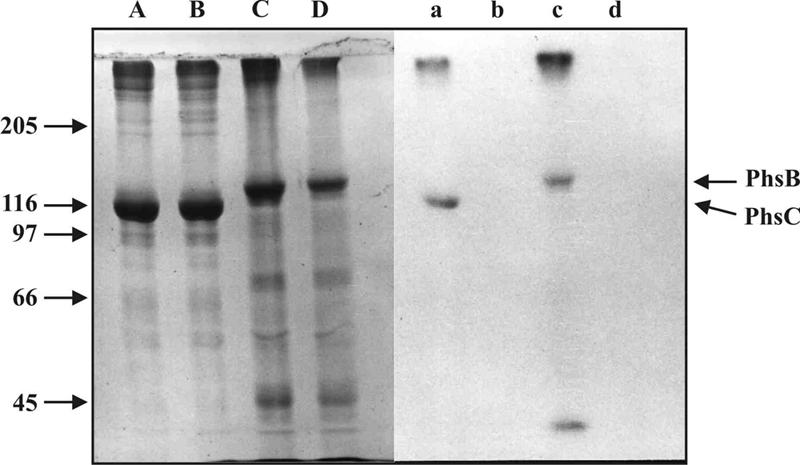
Covalent labeling of PhsB and PhsC. Partially purified PhsB or PhsC was incubated at 30°C for 30 min with [14C]alanine as described in Materials and Methods. In order to concentrate protein samples, incubations were treated with trichloroacetic acid (7%, wt/vol) and precipitates were dissolved in sample buffer (125 mM Tris-HCl, pH 6.8/10% 2-mercaptoethanol/10% SDS/10% glycerol) and subjected to SDS-PAGE (7.5%). Lanes: A: PhsB; B: PhsB without ATP; C: PhsC; D: PhsC without ATP. Right panel: autoradiograph of the same gel. A major fraction not dissolved in sample buffer stays at the top of the gels. A minor band of low Mr in the PhsB lane is a degradation product which, like PhsB itself, was not seen in plasmid-free S. lividans TK23.
Careful analysis of the protein elution patterns from the Mono Q column showed that, as in S. viridochromogenes extracts, the larger species eluted at higher salt strength from the column and therefore was PTT synthetase III (PhsB). The protein eluting at lower salt strength with a lower mass (in the case of S. viridochromogenes previously named PTT synthetase II) was presumed to be the gene product of phsC. Sequence analysis of PTT synthetase II from S. viridochromogenes revealed the amino-terminal sequence A-A-P-G-P-D-P-A-I. This sequence was found to represent amino acid positions 27 to 35 of the protein encoded by phsC. A protein sequence of PTT synthetase III from S. viridochromogenes could not be obtained due to severe losses of intact enzyme during purification. Nevertheless, since the data clearly indicate that PTT synthetase II is identical with PhsC by exclusion PhsB must be PTT synthetase III (for clarity, the two proteins along with the gene product of phsA were subsequently named PhsA, PhsB, and PhsC).
In S. lividans, levels of expressed PhsB and PhsC from pDS209 (with the ermE promoter) were two to threefold higher than in S. viridochromogenes (based on yield from wet weight of mycelium). Similarly, high expression of PhsB was also observed from pDS207. These high expression levels were apparently due to the high copy number of the plasmids. In contrast, a lower level of PhsB and PhsC production was found in mutant 707/3 and B3-14, where both the corresponding genes and their promoters (native promoter in 707/3, PermE in B3-14) are only present in single copy within the genome. Interestingly, PhsB purified from S. lividans, like the enzyme purified from S. viridochromogenes, also showed slow degradation, as demonstrated by the gradual disappearance of the 140-kDa gel band from the preparations. PhsC, in contrast, was stable.
Measurements of the loading reaction of freshly prepared PhsB with alanine indicated that its specific activity is less than that of PhsC (ca. 30 to 40%) whereas the activities in the alanine-dependent ATP-pyrophosphate exchange were comparable. The migration behavior of both recombinantly produced proteins in gel filtrations on a calibrated Superdex 200 column indicated that they were monomers like the wild-type enzymes from S. viridochromogenes (11).
Testing thioester formation ability with several amino acid substrates revealed, in contrast to the predicted substrate specificity of PhsB and PhsC for serine and proline, respectively (Table 3), alanine was the preferred substrate for both enzymes (Table 4). PhsB activated serine as thioester to an extent of about 50% of that of alanine. PhsC activated serine also, but only at 30% of the amount of alanine thioester. Remarkably, proline was not activated by either enzyme to any extent. Both heterologously expressed PhsB and PhsC were indistinguishable in their substrate specificities and relative specific activities from their wild-type counterparts isolated from S. viridochromogenes (11).
The A-domains of NRPS often have broad substrate specificities enabling them to activate structurally similar amino acid substrates (19). From this, the observed discrepancy between the predicted and the experimentally determined amino acid specificities of PhsB can still be considered acceptable because serine has structural similarity to alanine in that both are unbranched and have no charge in their side groups. However, in the case of PhsC the discrepancy between the predicted and observed substrate specificities cannot be explained in terms of structural similarity between the predicted substrate, proline, and the actual substrate, alanine. This behavior of PhsC (and also that of PhsB) represents an important exception from the established substrate specificity prediction rules of NRPS A-domains (8, 41) and may indicate differences between the structural determinants of the amino acid binding pockets of PhsC and PhsB and the majority of A-domains in the NRPS systems.
To address the question of alanylation of the model substrate AcPT, recombinantly produced PhsB and PhsC individually and in combination were incubated with wild-type PhsA from S. viridochromogenes in the presence of AcPT, [14C]alanine, and ATP. Although PhsA activates AcPT, a nonnatural substrate of PhsA, as adenylate and binds it as thioester as measured by the covalent binding of [3H]Ac-PT to PhsA in the presence of ATP (11), no acidic alanine-containing peptide intermediates bound to proteins were detected in ion exchange chromatography analyses of hydrolysates of protein-substrate complexes. In all cases [14C]alanine was the only covalently bound radioactive material recovered.
To exclude the possibility that failure of PhsB or PhsC to catalyze N-AcPT peptide formation could be attributed to improper folding or insufficient modification with 4′-phosphopantetheine cofactor in the proteins from S. lividans, the same experiments were repeated with these enzymes from wild-type S. viridochromogenes (PhsA plus PhsB plus PhsC), strain 707/3 (PhsA plus PhsB), and strain B3-14 (PhsA plus PhsC). In all cases, formation of any acidic reaction products between AcPT and alanine, either as covalently bound intermediates or free products, could not be detected. Similarly, no formation of acidic peptides were observed in nonfractionated extracts or homogenates of S. viridochromogenes or mutant 707/3 or B3-14. Short-term labeling experiments with whole cells using [14C]alanine as the radiolabel to detect labeled intermediates in these strains revealed the formation of detectable amounts of radioactive PTT in wild-type S. viridochromogenes but no detectable acidic peptides formed in the mutants.
DISCUSSION
The earlier identification of PhsA (previously named PTT synthetase I) encoded by the gene phsA of the PTT biosynthesis gene cluster and characterization of the enzyme revealed that this enzyme is a nonribosomal peptide synthetase module consisting of an A- and a T-domain. It activates exclusively N-acetylated phosphinoamino acids and structural analogs such as N-acetylglutamate or -aspartate as adenylate and thioester but not the deacetylated ones (11). This was consistent with the observed accumulation of N-Ac-DMPT observed in S viridochromogenes phsA mutants and indicated that N-Ac-DMPT is the original substrate of PhsA (1, 43). Apparently, steps of N-Ac-DMPT methylation and deacetylation take place after the thioester activation of N-Ac-DMPT or after assembly and release of the completed tripeptide (Fig. 5) (1, 43). Most probably, therefore, PhsA represents the initiation module of the N-Ac-DMPT assembly system (11, 32).
FIG. 5.

Hypothetical model of PTT formation based on the modular and domain organization of the PTT assembly system from the PTT gene cluster. Various possibilities for the stage of assembly at which methylation is possible are indicated. C, NRPS condensation domain; A, NRPS adenylation domain; T, NRPS thiolation domain; Te, thioesterase). This is one model of just two: another possibility would place PhsC between PhsA and PhsB. Roles of both thioesterases of type 2 are still speculative. For further explanation see text.
We show here that the two elongation modules necessary for bi-alanylylation of N-Ac-DMPT are encoded by the genes phsB and phsC. They lie about 20 kb from phsA in the PTT biosynthesis gene cluster (34) and in addition are separated by a gene, orfM, encoding a membrane protein of unknown function (Fig. 2). The two genes phsB and phsC, together with orfM and orf1 (Fig. 2), form a transcriptional unit and therefore are apparently coregulated. In addition to the effect of mutagenesis of phsB and phsC by gene replacement or disruption on PTT biosynthesis in S. viridochromogenes, heterologous expression of the two genes in S. lividans and analysis of their gene products clarified their role as the alanine-activating modules of PTT assembly. It turned out that the two previously isolated alanine-activating NRPS enzymes (11) have the same sizes as the deduced gene products of phsB and phsC and one of them (PTT synthetase II) has an amino-terminal sequence identical to amino acid positions 27 to 35 of PhsC.
The data shown here reveal further that these two enzymes are not derived from a larger bimodular NRPS by proteolytic cleavage but instead are stand-alone modules involved in N-Ac-DMPTT assembly. The involvement of the two modules in PTT synthesis was demonstrated by gene disruption and complementation in S. viridochromogenes. Although from the sequences of their A-domains the enzymes can be predicted to prefer amino acid substrates other than alanine, expression of phsB and phsC in S. lividans and testing of the gene products in vitro revealed that the substrate specificity of both proteins is alanine, in agreement with the results obtained with the corresponding enzymes from S. viridochromogenes. Nevertheless, both enzymes showed activity for the structurally related amino acids serine and aminobutyric acid but not, as predicted from the alignment of residues of the active site pocket residues, for proline (8, 36).
As yet it is not possible to assign which of the two alanylylation steps are catalyzed by PhsB and PhsC. The fact that neither of the two enzymes can replace each other suggests a defined positioning for each protein in the PTT assembly line. From their architectures, phsB and phsC differ from each other mainly by the presence of the amino-terminal T-domain in phsB, and this may be a clue to PhsB's positioning in the PTT synthetase assembly line either after phsA or after phsC. For example, proper module-module interactions are mediated by the E-domain of gramicidin S synthetase 1 (GrsA), which is not only involved in the epimerization of substrate but is important for interaction of GrsA with the next module in the assembly line of gramicidin S synthesis (36). In the case of tyrocidine synthetases, short protein-protein communication-mediating domains were shown to mediate interaction between the tyrocidine synthetases TycA, TycB, and TycC (12). However, in the PTT synthetases described here, such short sequences responsible for protein-protein interactions could not be identified.
Possible reasons for failure to demonstrate cell-free peptide bond formation may lie in the fact that PhsB is extremely susceptible to degradation, leading to loss of putative amino-terminal recognition motifs, rendering the protein unable to interact with its protein partners in vitro. Moreover, the loss of amino acid residues in the amino terminus of PhsC indicated the determined shortened amino-terminal sequence (see above) may be a constraint for the accomplishment of productive protein-protein interactions of this protein. The membrane protein OrfM, the gene for which lies between phsB and phsC and is cotranscribed with the two NRPS genes, may also be crucial for the formation of a productive complex. A role of OrfM in protein association, such as directing both PhsB and PhsC to the cellular membrane in the form of a membrane-associated multienzyme complex, could be envisaged.
Interestingly, a role of OrfM in PTT synthesis was demonstrated by disruption of orfM, which led to a block in PTT production in S. viridochromogenes (34). As a matter of fact, the biochemical data shown here clearly point to the fact that the functional interactions of the PPT synthases are governed by the structural intactness of the cellular organization because neither purified enzymes nor crude extracts of dialyzed cell homogenates displayed synthetic activity in terms of formation of free or enzyme-bound reaction products. Further experiments are therefore necessary to clarify the biochemical role of OrfM in PTT biosynthesis.
In conclusion, the data here show that the PTT assembly system featuring three stand-alone modules is unique among NRPS systems and represents the most drastic contrast to the well-known tripeptide synthetase aminoadipyl-cysteinyl-d-valine synthetase (20) in which all three modules are covalently joined with the domain order A-T-C-A-T-C-A-T-E-Te. To our knowledge, all NRPS systems synthesizing peptides with more than two amino acids contain at least one bimodular NRPS or one with bimodular characteristics (26). The lack of an integral thioesterase domain is an additional peculiarity of this system, leading to speculations on the role of two genes, the1 and the2, of the PTT gene cluster, each encoding stand-alone thioesterase domains of type II. Such thioesterase domains of type II were also observed in other NRPS systems and were shown to be involved in the regeneration of mischarged NRPS enzymes (35). The lack of an integral Te-domain in the PTT system may point to a mechanism of product release different from that observed in the majority of NRPSs (19). More detailed analyses at both the genetic and enzymatic levels are therefore required to resolve the mechanism of peptide formation and product release in this unique NRPS system.
Interestingly, PT is also found as a part of other prodrug peptide forms such as the peptide antibiotic trialaphos (phosphinothricylalanylalanylalanine) from Streptomyces hygroscopicus KSB1285 (15) and phosalacine (phosphinothricylalanylleucine) from Kitasatospora phosalacinea (23). A comparison of the module arrangements in the PTT biosynthesis gene cluster of S. viridochromogenes to the corresponding regions in the as yet uncharacterized biosynthetic gene clusters of these antibiotics could also be helpful to understand the evolution of the various enzyme systems which are destined to channel the unique metabolite PT into its various prodrug forms.
Acknowledgments
We thank S. J. Lucania for samples of thiostrepton. The oligonucleotides derived from peptide synthetase core motifs were kindly provided by S. Pelzer.
Part of the work was financed by a grant of the Fond der Chemischen Industrie (163607). This research has been supported by the Deutsche Forschungsgemeinschaft (Wo 485/2-1, Ke 452/11-3, and SPP1152).
REFERENCES
- 1.Alijah, R., J. Dorendorf, S. Talay, A. Pühler, and W. Wohlleben. 1991. Genetic analysis of the phosphinothricin-tripeptide biosynthetic pathway of Streptomyces viridochromogenes Tü494. Appl. Microbiol. Biotechnol. 34:749-755. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 3.Bayer, E., K. H. Gugel, K. Hagele, H. Hagemaier, S. Jassipow, W. A. König, and H. Zähner. 1972. Stoffwechselprodukte von Mikroorganismen. Phosphinothricin und Phosphinothricyl-Alanyl-Alanin. Helv. Chim. Acta 55:224-239. [DOI] [PubMed] [Google Scholar]
- 4.Bibb, M. J., P. R. Findlay, and M. W. Johnson. 1984. The relationship between base composition and codon usage in bacterial genes and its use for the simple and reliable identification of protein-coding sequences. Gene 30:157-166. [DOI] [PubMed] [Google Scholar]
- 5.Bourn, W. R., and B. Babb. 1995. Computer assisted identification and classification of streptomycete promoters. Nucleic Acids Res. 23:3696-3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microorganism quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 7.Bullock, W. O., J. M. Fernandez, and J. M. Short. 1987. Xl1-Blue, a high efficiency plasmid transforming recA Escherichia coli strain with beta galactosidase selection. Focus 5:376-378. [Google Scholar]
- 8.Challis, G. L., J. Ravel, and C. A. Townsend. 2000. Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem. Biol. 7:211-224. [DOI] [PubMed] [Google Scholar]
- 9.Cserzo, M., E. Wallin, I. Simon, G. von Heijne, and A. Elofsson. 1997. Prediction of transmembrane alpha-helices in procariotic membrane proteins: the dense alignment surface method. Protein Eng. 10:673-676. [DOI] [PubMed] [Google Scholar]
- 10.Diddens, H., H. Zähner, E. Kraas, W. Goehring, and G. Jung. 1976. On the transport of tripeptide antibiotics in bacteria. Eur. J. Biochem. 66:11-23. [DOI] [PubMed] [Google Scholar]
- 11.Grammel, N., D. Schwartz, W. Wohlleben, and U. Keller. 1998. Phosphinothricin-tripeptide synthetases from Streptomyces viridochromogenes. Biochemistry 37:1596-1603. [DOI] [PubMed] [Google Scholar]
- 12.Hahn, M., and T. Stachelhaus. 2004. Selective interaction between nonribosomal peptide synthetases is facilitated by short communication-mediating domains. Proc. Natl. Acad. Sci. USA 101:15585-15590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heinzelmann. E., G. Kienzlen, S. Kaspar, J. Recktenwald, W. Wohlleben, and D. Schwartz. 2001. The phosphinomethylmalate isomerase gene pmi, encoding an aconitase-like enzyme, is involved in the synthesis of phosphinothricin tripeptide in Streptomyces viridochromogenes. Appl. Environ. Microbiol. 67:3603-3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hopwood, D. A., M. J. Bibb, K. F. Chater, T. Kieser, C. J. Bruton, H. M. Kieser, D. J. Lydiate, C. P. Smith, J. M. Ward, and H. Schrempf. 1985. Genetic manipulation of Streptomyces: a laboratory manual. John Innes Foundation, Norwich, United Kingdom.
- 15.Kato, H., K. Nagayama, H. Abe, R. Kobayashi, and E. Ishihara. 1991. Isolation, structure and biological activity of trialaphos. Agric. Biol. Chem. 55:1133-1134. [Google Scholar]
- 16.Kondo, Y., T. Shomura, Y. Ogawa, T. Tsuruoka, H. Watanabe, K. Totsukawa, T. Suzuki, C. Moriyama, J. Yoshida, S. Inouye, and T. Niida. 1973. Studies on a new antibiotic SF-1293, I. Isolation and physico-chemical and biological characterization of SF-1293 substances. Sci. Rep. Meiji Seika 13:34-41. [Google Scholar]
- 17.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 18.Leskiw, B. K., E. J. Lawlor, J. M. Fernandez-Abalos, and K. F. Chater. 1991. TTA codons in some genes prevent their expression in a class of developmental, antibiotic-negative, Streptomyces mutants. Proc. Natl. Acad. Sci. USA 88:2461-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marahiel, M. A. 1997. Protein templates for the biosynthesis of peptide antibiotics. Chem. Biol. 4:561-567. [DOI] [PubMed] [Google Scholar]
- 20.Martin, J. F. 1998. New aspects of genes and enzymes for beta-lactam antibiotic biosynthesis. Appl. Microbiol. Biotechnol. 50:1-15. [DOI] [PubMed] [Google Scholar]
- 21.Muth, G., B. Nussbaumer, W. Wohlleben, and A. Pühler. 1989. A vector system with temperature-sensitive replication for gene disruption and mutational cloning in streptomycetes. Mol. Gen. Genet. 219:341-348. [Google Scholar]
- 22.Nicholas, K. B., H. B. Nicholas, Jr., and D. W. Deerfield II. 1997. GeneDoc: analysis and visualization of genetic variation. EMBNEW News 4:14. [Google Scholar]
- 23.Omura, S., K. Hinotozawa, N. Imamura, and M. Murata. 1984. The structure of phoasalacine, a new antibiotic containing phosphinothricin. J. Antibiot. (Tokyo) 37:939-940. [DOI] [PubMed] [Google Scholar]
- 24.Pelzer, S., D. Heckmann, M. Huppert, W. Reichert, and W. Wohlleben. 1997. Cloning and analysis of a peptide synthetase gene of the balhimycin producer Amycolatopsis mediterranei DSM 5908 and development of a gene disruption/replacement system. J. Biotechnol. 56:115-128. [DOI] [PubMed] [Google Scholar]
- 25.Pettis, G. S., T. J. Brickman, and M. A. McIntosh. 1988. Transcriptional mapping and nucleotide sequence of the Escherichia coli fep-fes enterobactin region. J. Biol. Chem. 263:18857-18863. [PubMed] [Google Scholar]
- 26.Quadri, L. E. 2000. Assembly of aryl-capped siderophores by modular peptide synthetases and polyketide synthases. Mol. Microbiol. 37:1-12. [DOI] [PubMed] [Google Scholar]
- 27.Quiros, L. M., I. Aguirrezabalaga, C. Olano, C. Mendez, and J. A. Salas. 1998. Two glycosyltransferases and a glycosidase are involved in oleandomycin modification during its biosynthesis by Streptomyces antibioticus. Mol. Microbiol. 28:1177-1185. [DOI] [PubMed] [Google Scholar]
- 28.Rusnak, F., M. Sakaitani, D. Drueckhammer, J. Reichert, and C. T. Walsh. 1991. Biosynthesis of the Escherichia coli siderophore enterobactin: sequence of the entF gene, expression and purification of EntF, and analysis of covalent phosphopantetheine. Biochemistry 30:2916-2927. [DOI] [PubMed] [Google Scholar]
- 29.Sambrook, J., T. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 30.Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain termination inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitt-John, T., and J. W. Engels. 1992. Promoter constructions for efficient secretion expression in Streptomyces lividans. Appl. Microbiol. Biotechnol. 36:493-498. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz, D., R. Alijah, B. Nussbaumer, S. Pelzer, and W. Wohlleben. 1996. The peptide synthetase gene phsA from Streptomyces viridochromogenes is not juxtaposed with other genes involved in nonribosomal biosynthesis of peptides. Appl. Environ. Microbiol. 62:570-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwartz, D., J. Recktenwald, S. Pelzer, and W. Wohlleben. 1998. Isolation and characterization of the PEP-phosphomutase and the phosphonopyruvate decarboxylase genes from the phosphinothricin tripeptide producer Streptomyces viridochromogenes Tü 494. FEMS Microbiol. Lett. 163:149-157. [DOI] [PubMed] [Google Scholar]
- 34.Schwartz, D., S. Berger, E. Heinzelmann, K. Muschko, K. Wetzel, and W. Wohlleben. 2004. Biosynthetic gene cluster of the herbicide phosphinothricin tripeptide from Streptomyces viridochromogenes Tü494. Appl. Environ. Microbiol. 70:7093-7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarzer, D., H. D. Mootz, U. Linne, and M. A. Marahiel. 2002. Regeneration of misprimed nonribosomal peptide synthetases by type II thioesterases Proc. Natl. Acad. Sci. USA 99:14083-14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stachelhaus, T., H. Mootz, and M. A. Marahiel. 1999. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 6:493-505. [DOI] [PubMed] [Google Scholar]
- 37.Stachelhaus, T., and C. T. Walsh. 2000. Mutational analysis of the epimerization domain in the initiation module PheATE of gramicidin S synthetase. Biochemistry 39:5775-5787. [DOI] [PubMed] [Google Scholar]
- 38.Staden, R., and A. D. McLachlan. 1982. Codon preference and its use in identifying protein coding regions in large DNA sequences. Nucleic Acids Res. 10:141-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thompson, C. J., and H. Seto. 1995. Bialaphos, p. 197-222. In L. C. Vining and C. Stuttard (ed.), Genetics and biochemistry of antibiotic production. Butterworth Heinemann Biotechnology, Oxford, United Kingdom.
- 40.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. ClustalW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ward, J. M., G. R. Janssen, T. Kieser, M. J. Bibb, and M. J. Buttner. 1986. Construction and characterization of a series of multicopy promoter-probe plasmid vectors for Streptomyces using the aminoglycoside phosphotransferase gene from Tn5 as indicator. Mol. Gen. Genet. 203:468-478. [DOI] [PubMed] [Google Scholar]
- 42.Wild, A., and R. Manderscheid. 1984. The effect of phosphinothricin an the assimilation of ammonia in plants. Z. Naturforsch. 39c:500-504. [Google Scholar]
- 43.Wohlleben, W., R. Alijah, J. Dorendorf, D. Hillemann, B. Nuβbaumer, and S. Pelzer. 1992. Identification and characterization of phosphinothricin-tripeptide biosynthetic genes in Streptomyces viridochromogenes. Gene 115:127-132. [DOI] [PubMed] [Google Scholar]