

Abstract
Indolyl aryl sulfone (IAS) nonnucleoside inhibitors have been shown to potently inhibit the growth of wild-type and drug-resistant human immunodeficiency virus type 1 (HIV-1), but their exact mechanism of action has not been elucidated yet. Here, we describe the mechanism of inhibition of HIV-1 reverse transcriptase (RT) by selected IAS derivatives. Our results showed that, depending on the substitutions introduced in the IAS common pharmacophore, these compounds can be made selective for different enzyme-substrate complexes. Moreover, we showed that the molecular basis for this selectivity was a different association rate of the drug to a particular enzymatic form along the reaction pathway. By comparing the activities of the different compounds against wild-type RT and the nonnucleoside reverse transcriptase inhibitor-resistant mutant Lys103Asn, it was possible to hypothesize, on the basis of their mechanism of action, a rationale for the design of drugs which could overcome the steric barrier imposed by the Lys103Asn mutation.
Anti-AIDS therapy is actually based on three classes of anti-human immunodeficiency virus (HIV) drugs, the nucleoside reverse transcriptase inhibitors (NRTIs), the nonnucleoside reverse transcriptase inhibitors (NNRTIs), and the protease inhibitors. More recently, enfuvirtide, a 36-amino-acid residue peptide acting as a viral entry inhibitor, has been licensed for the treatment of HIV infection (7, 8). NRTIs, NNRTIs, and protease inhibitors are mixed in highly active antiretroviral therapy, which dramatically slows down viral replication, but they are unable to eradicate the viral infection (29). Moreover, the rapid development of drug resistance and toxicity problems make urgent the discovery of novel anti-HIV agents effective against resistant mutants and without unpleasant side effects (14).
NNRTI interaction with HIV-1 reverse transcriptase (RT) is a highly dynamic process (6). Crystal structures of RT-NNRTI complexes (19) showed that the drugs interacted with a hydrophobic pocket (nonnucleoside binding site [NNBS]) on the enzyme in a “butterfly-like” mode. One of the “wings” of this butterfly is made of a π-electron-rich moiety (phenyl or allyl substituents) that interacts through π-π interactions with a hydrophobic pocket formed mainly by the side chains of aromatic amino acids (Tyr181, Tyr188, Phe227, Trp229, and Tyr318). On the other hand, the other wing is normally represented by a heteroaromatic ring bearing at one side a functional group capable of donating and/or accepting hydrogen bonds with the main chain of Lys101 and Lys103. Finally, on the butterfly body, a hydrophobic portion fills a small pocket formed mainly by the side chains of Lys103, Val106, and Val179. Upon complexation, the NNBS hydrophobic pocket changes its own conformation, leading to the inactivation of the enzyme itself. Because of the different chemical and structural features of the inhibitors and the side chain flexibility, the bound NNBS adopts different conformations (28). Moreover, mutations of some amino acids cause variation of the NNBS properties, thus decreasing affinities of most of the inhibitors (12, 24, 25). In particular, the NNRTI resistance mutations Tyr188Leu and Tyr181Ile/Cys reduce π-π interactions, the Gly190Ala mutation leads to a smaller active site space because of a steric conflict between the methyl side chain and the inhibitor, and the formation of an additional hydrogen bond when amino acid 103 is mutated from Lys to Asn reduces inhibitor entrance into the NNBS.
However, HIV-1 RT itself also undergoes a conformational reorganization upon interaction with its substrates template-primer (TP) and deoxynucleoside triphosphate (dNTP), so that three structurally distinct mechanistic forms can be recognized in the reaction pathway catalyzed by HIV-1 RT (1, 11): the free enzyme, the binary complex of RT with the template-primer (RT/TP), and the catalytically competent ternary complex of RT with both nucleic acid and dNTP (RT/TP/dNTP). This means that, in principle, the NNBS might not be identical in these three mechanistic forms. Several kinetic studies have shown that this is indeed the case, so that some NNRTIs selectively target one or a few of the different enzymatic forms along the reaction pathway (5, 13, 15). This observation likely reflects the different spatial rearrangements not only of the NNBS itself but also of the adjacent nucleotide binding site (3, 20, 26, 27). Indeed, it has been shown that a “communication” exists between the NNBS and the nucleotide binding site, so that some NRTI resistance mutations can influence NNRTI binding and vice versa (2, 4, 20). Thus, understanding the molecular determinants governing the selective interaction of a drug with the three different NNBS structures present along the RT reaction pathway will be important for the design of novel, highly selective, and potent NNRTIs.
During extensive structure-activity relationship studies on diarylsulfones, we identified pyrryl sulfones and the novel indolyl aryl sulfones (IASs) as highly potent NNRTIs (18, 22, 23). In particular, indole derivatives bearing 2-methylphenylsulfonyl or 3-methylphenylsulfonyl moieties were found to inhibit HIV-1 at nanomolar concentrations. Furthermore, the introduction of a 3,5-dimethylphenylsulfonyl moiety led to a compound displaying high activity and selectivity not only against the wild-type strain but also against the Tyr181Cys and Lys103Asn-Tyr181Cys viral variants and the efavirenz-resistant mutant Lys103Arg-Val179Asp-Pro225His.
In light of their extremely potent activities, especially towards NNRTI-resistant mutants, we sought to investigate in detail the mechanism of action of some selected IAS derivatives. In this work, we show that IASs do not display a unique mode of action but rather that they can selectively bind different mechanistic forms of the enzyme, depending on the substituents introduced on the drug pharmacophore. These results suggest that IASs are very flexible molecules, interacting dynamically with the viral RT.
MATERIALS AND METHODS
Chemicals.
[3H]dTTP (40 Ci/mmol) was from Amersham, and unlabeled dNTPs were from Boehringer. Whatman was the supplier of the GF/C filters. All other reagents were of analytical grade and purchased from Merck or Fluka.
Chemistry.
5-Chloro-3-(phenyl)sulfonyl-1H-indole-2-carboxamide (RS1202) (23, 30), 5-chloro-3-[(3,5-dimethylphenyl)sulfonyl]-1H-indole-2-carboxamide (RS1588) (21), N-{3-[(3,5-dimethylphenyl)sulfonyl]-5-chloro-1H-indole-2-carbonyl}-d,l-alanylamide (RS1980) (21), and 4,5-difluoro-3-[(3,5-dimethylphenyl)sulfonyl]-1H-indole-2-carboxamide (RS1866) (R.S., unpublished data) were obtained by heating the corresponding esters with concentrated ammonium hydroxide in a sealed tube. The starting esters needed for the preparation of RS1202, RS1588, and RS1866 were obtained by oxidation of the corresponding 3-arylthio-1H-indole-2-carboxylates using 3-chloroperoxybenzoic acid (MCPBA). The ethyl ester of N-{3-[(3,5-dimethylphenyl)sulfonyl]-5-chloro-1H-indole-2-carbonyl}-d,l-alanine for the synthesis of RS1980 was obtained by lithium hydroxide of ethyl 5-chloro-3-[(3,5-dimethylphenyl)sulfonyl]-1H-indole-2-carboxylate and subsequent condensation of d,l-alanine ethyl ester in the presence of O-benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate and triethylamine. The required 3-arylthio-1H-indole-2-carboxylates were prepared by reaction of proper arylthiodisulfides with 1H-indole-2-carboxylic acids in the presence of sodium hydride according to the Atkinson method and subsequent esterification of the 3-arylthio-1H-indole-2-carboxylic acids with (trimethylsilyl)diazomethane. Alternatively, these esters were obtained by reaction of methyl or ethyl 1H-indole-2-carboxylates with N-(arylthio)succinimides in the presence of boron trifluoride diethyl etherate.
Nucleic acid substrates.
The homopolymer poly(rA) (Pharmacia) was mixed at weight ratios in nucleotides of 10:1 to the oligomer oligo(dT)12-18 (Pharmacia) in 20 mM Tris-HCl (pH 8.0) containing 20 mM KCl and 1 mM EDTA, heated at 65°C for 5 min, and then slowly cooled at room temperature.
Expression and purification of recombinant HIV-1 RT forms.
Recombinant heterodimeric wild-type RT and the Lys103Asn and Tyr181Ile variants were expressed and purified to >95% purity (as judged by sodium dodecyl sulfate-polyacrylamide gel electrophoresis) as described previously (12).
HIV-1 RT RNA-dependent DNA polymerase activity assay.
RNA-dependent DNA polymerase activity was assayed as follows. A final volume of 25 μl contained buffer A (50 mM Tris-HCl, pH 7.5, 1 mM dithiothreitol, 0.2 mg/ml bovine serum albumin, 4% glycerol), 10 mM MgCl2, 0.5 μg of poly(rA)/oligo(dT)10:1 (0.3 μM 3′-OH ends), 10 μM [3H]dTTP (1 Ci/mmol), and 5 to 10 nM RT. Reactions were incubated for 10 min at 37°C. Aliquots (20 μl) were then spotted on glass fiber filters (GF/C filters), which were immediately immersed in 5% ice-cold trichloroacetic acid. Filters were washed twice in 5% ice-cold trichloroacetic acid and once in ethanol for 5 min and dried, and acid-precipitable radioactivity was quantitated by scintillation counting.
Inhibition assays.
Reactions for inhibition assays were performed under the conditions described for the HIV-1 RT RNA-dependent DNA polymerase activity assay. Incorporation of radioactive dTTP into poly(rA)/oligo(dT) at different concentrations of DNA or dNTP was monitored in the presence of increasing amounts of inhibitor as indicated in the figure legends.
Kinetics of inhibitor binding.
Kinetics of inhibitor binding experiments were as described previously (12). Briefly, HIV-1 RT (20 to 40 nM) was incubated for 2 min at 37°C in a final volume of 4 μl in the presence of buffer A, with 10 mM MgCl2 alone or with 100 nM 3′-OH ends (for the formation of the RT/TP complex), or in the same mixture complemented with 10 μM unlabeled dTTP (for the formation of the RT/TP/dNTP complex). The inhibitor to be tested was then added to a final volume of 5 μl at a concentration at which [EI]/[E0] = [1 − 1/(1 + [I]/Ki)] > 0.9. Then, 145 μl of a mix containing buffer A, 10 mM MgCl2, and 10 μM [3H]dTTP (5 Ci/mmol) was added at different time points. After an additional 10 min of incubation at 37°C, 50-μl aliquots were spotted on GF/C filters, and acid-precipitable radioactivity was measured as described for the HIV-1 RT RNA-dependent DNA polymerase activity assay. The quantity vt/v0, representing the normalized difference between the amount of dTTP incorporated at the zero time point and at different time points, was then plotted against time. The kapp values were determined by fitting the experimental data to the single-exponent equation vt/v0 =  , where t is time. The kon and koff values were calculated according to the relationships (27) kapp = kon(Ki + [I]) and Kd = koff/kon.
, where t is time. The kon and koff values were calculated according to the relationships (27) kapp = kon(Ki + [I]) and Kd = koff/kon.
Kinetic model.
The mechanism of action of IASs was found to be either fully noncompetitive or partially mixed. A schematic drawing of the different equilibria is depicted in Fig. 1. According to the ordered mechanism of the polymerization reaction, whereby template-primer (TP) binds first followed by the addition of dNTP, HIV-1 RT can be present in three different catalytic forms as reported in Fig. 1A: as a free enzyme, in a binary complex with the TP, and in a ternary complex with TP and dNTP. The resulting rate equation for such a system is very complex and impractical to use. For these reasons, the general steady-state kinetic analysis was simplified by varying one of the substrates (either TP or dNTP) while the other was kept constant. When the TP substrate was held constant at saturating concentration and the inhibition at various concentrations of dNTPs was analyzed, at the steady state all of the input RT was in the form of the RT/TP binary complex and only two forms of the enzyme (the binary complex and the ternary complex with dNTP) could react with the inhibitor, as shown in the left part of Fig. 1B. Similarly, when the dNTP concentration was kept constant at saturating levels and the inhibition at various TP concentrations was analyzed, RT was present either as a free enzyme or in the ternary complex with TP and dNTP, as shown in the right part of Fig. 1B.
FIG. 1.

Schematic diagram of the HIV-1 reverse transcriptase RNA-dependent DNA polymerization reaction pathway. (A) Full reaction pathway. (B) The general steady-state kinetic analysis was simplified by varying one of the substrates (either TP or dNTP) while the other was kept constant. When the TP substrate was held constant at saturating concentration and the inhibition at various concentrations of dNTPs was analyzed, at the steady state all of the input RT was in the form of the RT/TP binary complex and only two forms of the enzyme (the binary complex and the ternary complex with dNTP) could react with the inhibitor (left part of the panel). Similarly, when the dNTP concentration was kept constant at saturating levels and the inhibition at various TP concentrations was analyzed, RT was present either as a free enzyme or in the ternary complex with TP and dNTP (right part of the panel). For details, see Materials and Methods.
The steady-state rate equation used for the partially mixed inhibition was the one describing a reaction involving only two mechanistic forms of the enzyme (according to the simplified pathways in Fig. 1B) (9)
 |
(1) |
where kcat is the apparent reaction rate in the absence of the inhibitor, k′cat is the reaction rate at infinite inhibitor concentration, E0 is the total enzyme concentration, I is the inhibitor concentration, S is the substrate concentration, and Ki and K′i are the equilibrium dissociation constants for the different enzyme-substrate complexes. According to Fig. 1B, when TP was held constant and only the dTTP concentration was varied, Ki = Kbini (for the binding of the inhibitor to the binary complex RT/TP), whereas K′i = Kteri (for the binding of the inhibitor to the ternary RT/TP/dNTP complex). Conversely, when dTTP was held constant, Ki referred to the binding of the inhibitor to the free enzyme, whereas K′i = Kteri (for binding to the ternary RT/TP/dNTP complex).
The partially mixed mode of inhibition predicts that the enzyme-inhibitor complex is still able to bind the substrate but with a lower affinity constant K′s. Conversely, the enzyme-substrate complex binds the inhibitor but with a lower inhibition constant K′i. Moreover, the enzyme-inhibitor-substrate complex can still break down to give products but with a reduced catalytic rate k′cat (9). Thus, equation 1 was used to derive the apparent reaction rate, kcat(app), and affinity, Ks(app), for the TP and dNTP substrates of the reaction at different inhibitor concentrations. Then, Ks, Ki, K′i, and K′s were derived by computer fitting of the variation of the Ks(app) values as a function of the inhibitor concentrations, according to the equations (9)
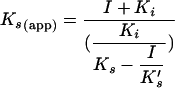 |
(2) |
and
 |
(3) |
The K′i and k′cat values were derived by computer fitting of the variation of the kcat(app) values as a function of the inhibitor concentrations, according to the equation (9)
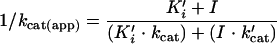 |
(4) |
For the fully noncompetitive case, k′cat = 0, Ks = K′s, and Ki = K′i, and thus equation 1 simplifies to
 |
(5) |
Similarly, equation 4 simplifies to
 |
(6) |
Initial velocities of the reaction were determined after 10 min of incubation at 37°C, which represents the midpoint of the linear range of the reaction, as determined in separate experiments (data not shown). Each experiment was done in triplicate, and mean values were used for the analysis. Curve fitting was done with the computer program GraphPad Prism.
RESULTS
IAS derivatives show high potencies of inhibition of HIV-1 wild-type RT and NNRTI-resistant mutants.
The IAS derivatives RS1202, RS1588, RS1866, and RS1980 (Fig. 2) were tested against HIV-1 wild-type RT and different NNRTI-resistant RT mutants. As summarized in Table 1, all four derivatives showed potent inhibition towards wild-type RT. In general, the inhibition activities were higher towards the mutants Leu100Ile, Val106Ala, and Val179Asp than towards the two mutants Tyr181Ile and Tyr188Leu. Interestingly, the compounds RS1202 and RS1588 showed little or no loss of potency towards the Lys103Asn mutant, being 400- to 600-fold more potent than nevirapine and efavirenz against this mutant. Given their interesting activity profile, the mechanism of inhibition of these IAS derivatives was studied in more detail. Increasing concentrations of either the dTTP or the poly(rA)/oligo(dT) substrates were titrated into the reaction in the presence of increasing fixed amounts of inhibitors. From the variation of the apparent affinity of the enzyme for its substrates, Ks(app), and the apparent reaction rate, kcat(app), as a function of the inhibitor concentration, all of the kinetic parameters for drug binding were determined, as explained in Materials and Methods and in Fig. 1.
FIG. 2.

Structures of the compounds used in this study.
TABLE 1.
Inhibition potencies of IAS derivatives against HIV-1 wild-type RT and NNRTI-resistant mutants
| Compound | ID50 (nM) fora
|
||||||
|---|---|---|---|---|---|---|---|
| wt | Leu100Ile | Lys103Asn | Val106Ala | Val179Asp | Tyr181Ile | Tyr188Leu | |
| RS1202 | 1 (±0.1) | 100 (±20) | 5 (±1) | 75 (±8) | 35 (±3) | 140 (±10) | 1,000 (±100) |
| RS1588 | 3 (±0.1) | 60 (±6) | 3 (±0.5) | 90 (±8) | 50 (±5) | 140 (±10) | 200 (±20) |
| RS1866 | 2.2 (±0.1) | n.d. | 750 (±60) | n.d. | n.d. | 1,500 (±100) | n.d. |
| RS1980 | 3 (±0.2) | n.d. | 500 (±50) | n.d. | n.d. | 130 (±10) | n.d. |
| Nevirapine | 400 (±50) | 900 (±90) | 2,000 (±80) | 2,000 (±300) | 300 (±25) | 5,000 (±400) | 5,000 (±400) |
| Efavirenz | 40 (±0.5) | n.d. | 1,000 (±40) | n.d. | n.d. | 400 (±50) | n.d. |
ID50, concentration of the inhibitor that reduced the in vitro activity of HIV-1 RT by 50% under the conditions described in Materials and Methods. Numbers are the mean values of three independent experiments (standard deviations are in brackets). wt, wild type; n.d., not determined.
RS1866 is a fully noncompetitive inhibitor of HIV-1 RT.
As shown in Fig. 3A, the IAS derivative RS1866 showed a fully noncompetitive mechanism of action, since it did not influence the affinity of the enzyme for the substrates, affecting only the rate of the reaction. Accordingly, the reciprocals of the kcat(app) values followed linear relationships as a function of the inhibitor concentrations, as described by equation 6 in Materials and Methods.
FIG. 3.

Mechanisms of inhibition of HIV-1 RT by the IAS derivatives. (A) Increasing concentrations of RS1866 were titrated in the presence of 5 nM RT and either 2 μM, 4 μM, 10 μM, or 20 μM dTTP or 0.04 μM, 0.08 μM, 0.2 μM, and 0.4 μM (as 3′-OH ends) poly(rA)/oligo(dT) under the conditions described in Materials and Methods. Initial velocities of the reaction were plotted as a function of the substrate concentration. The variations of the kcat(app) values for the reaction derived from these experiments were plotted as a function of the RS1866 concentrations. Data were fitted to equation 6 as described in Materials and Methods. (B) Increasing concentrations of RS1588 were titrated in the presence of 5 nM RT and 2 μM, 4 μM, 10 μM, and 20 μM dTTP or 0.04 μM, 0.08 μM, 0.2 μM, and 0.4 μM (as 3′-OH ends) of poly(rA)/oligo(dT) under the conditions described in Materials and Methods. Initial velocities of the reaction were plotted as a function of the substrate concentration. The variation of the kcat(app) values for the reaction derived from these experiments was plotted as a function of the RS1588 concentrations. Data were fitted to equation 4 as described in Materials and Methods. (C) Increasing concentrations of RS1202 were titrated in the presence of 5 nM RT and 2 μM, 4 μM, 10 μM, and 20 μM dTTP or 0.04 μM, 0.08 μM, 0.2 μM, and 0.4 μM (as 3′-OH ends) poly(rA)/oligo(dT) under the conditions described in Materials and Methods. Initial velocities of the reaction were plotted as a function of the substrate concentration. The values of the apparent affinity for the nucleic acid substrate (Ks3′-OH) derived from these experiments were plotted as a function of the inhibitor concentration. Data were fitted to both equation 2 and equation 3, as described in Materials and Methods. (D) The variation of the kcat(app) values for the reaction derived as described for panel C was plotted as a function of the RS1202 concentrations. Data were fitted to equation 4 as described in Materials and Methods. (E) Increasing concentrations of RS1980 were titrated in the presence of 5 nM RT and 2 μM, 4 μM, 10 μM, and 20 μM dTTP or 0.04 μM, 0.08 μM, 0.2 μM, and 0.4 μM (as 3′-OH ends) poly(rA)/oligo(dT) under the conditions described in Materials and Methods. Initial velocities of the reaction were plotted as a function of the substrate concentration. The values of the apparent affinity for the nucleotide (KsTTP, circles) derived from these experiments were plotted as a function of the inhibitor concentration. Data were fitted to both equation 2 and equation 3, as described in Materials and Methods. (F) The variation of the kcat(app) values for the reaction derived as described for panel E was plotted as a function of the RS1980 concentrations. Data were fitted to equation 4 as described in Materials and Methods.
RS1588 is a partially noncompetitive inhibitor of HIV-1 RT which shows preferential binding to the free enzyme.
The same kind of analysis was applied to the compound RS1588. As shown in Fig. 3B, this inhibitor did not influence the affinity of wild-type RT for the reaction substrates, affecting only the reaction rate. However, the reciprocals of the kcat(app) values did not follow a linear relationship with the inhibitor concentrations, as expected for a fully noncompetitive inhibitor, but instead followed the nonlinear relationship described by equation 4 of Materials and Methods, diagnostic of a partially noncompetitive type of inhibition (Fig. 3B). Thus, the reaction rate extrapolated at infinite inhibitor concentration (k′cat) was found to be 2.5 to 3% of the uninhibited reaction, indicating a maximum of 97% to 98% inhibition. Interestingly, from the variation of the kcat(app) values measured in the presence of various concentrations of the poly(rA)/oligo(dT) substrate, an inhibition constant (Ki) value of 0.4 nM was calculated (Fig. 3B, circles), whereas the same analysis performed with the kcat(app) values measured in the presence of various concentrations of the dTTP substrate yielded a Ki of 2.7 nM (Fig. 3B, triangles). According to the scheme outlined in Fig. 1B, these values represent the apparent binding constants of the inhibitor to the free enzyme and the ternary RT/TP/dNTP complex, respectively. Thus, even though RS1588 binding did not affect the affinity of RT for the substrates, the binding of the substrates to the enzyme reduced the apparent Ki of the inhibitor.
RS1202 is a mixed inhibitor which binds preferentially to the free enzyme and is partially competitive with the nucleic acid substrate.
When the IAS derivative RS1202 was studied, a strong reduction of the apparent affinity for the nucleic acid substrate but not for dTTP was noted. The increase in the Ks(app)3′-OH (apparent affinity for the nucleic acid substrate) values as a function of increasing inhibitor concentration (Fig. 3C) suggested a partially competitive inhibition. In addition, the reciprocal of the variation of the kcat(app) values followed a nonlinear relationship, as in the case of RS1588, indicating a partially noncompetitive mechanism (Fig. 3D) with a maximal reduction of the reaction rate of 95%. Thus, RS1202 behaved as a mixed inhibitor, and according to the kinetic model outlined in Fig. 1B, all of the kinetic parameters were determined as described in Materials and Methods. As a result, it was found that RS1202 interacted preferentially with the free enzyme (similarly to RS1588), with a Ki of 0.3 nM. Binding of the nucleic acid substrate to the enzyme reduced the affinity of the inhibitor by fourfold, giving a Kibin/ter of 1.2 nM.
RS1980 is a mixed inhibitor which is partially competitive with the nucleotide substrate.
Similar experiments were conducted with the compound RS1980 (Fig. 3E and F). This inhibitor showed a partially competitive mechanism with the nucleotide substrate, as shown by the increase in the Ks(app)TTP (apparent affinity for the nucleotide substrate) values as a function of increasing inhibitor concentrations (Fig. 3E). Again, the reciprocal of the variation of the kcat(app) values followed a nonlinear relationship, as in the case of RS1588, indicating a partially noncompetitive mechanism (Fig. 3F) with a maximal inhibition of 92%. Determination of the kinetic constant according to the reaction scheme outlined in Fig. 1 revealed that RS1980 binding to the ternary RT/TP/dNTP complex was reduced 10-fold with respect to either the free enzyme or the binary RT/TP complex (compare Ki and Kibin with Kiter values).
Preferential binding of IAS derivatives to specific reaction intermediates is mainly driven by differences in the association rates.
The differences in the equilibrium dissociation constants Ki, Kibin, and Kiter of the inhibitors for the different reaction intermediates could reflect either slower association (kon) or faster dissociation (koff) rates or both. In order to more quantitatively address which step of inhibitor binding was affected by the nucleic acid or the nucleotide substrates, the association and dissociation rates of the different IAS derivatives were determined for the free enzyme and the binary RT/TP and ternary RT/TP/dNTP complexes. Experiments were performed as described in Materials and Methods, and the calculated rate constants are summarized in Table 2. As can be seen, the preference of RS1202 for the free enzyme, as well as the improved binding of RS1980 to both the free enzyme and the binary complex, was due to a faster association rate, whereas the dissociation step was not significantly affected. Notably, RS1588 showed a threefold reduction of its kon and a concomitant threefold increase in its koff values for the binary complex with respect to the free enzyme. Taken together, these results indicate that binding of either the nucleic acid or the nucleotide substrate to HIV-1 RT imposed some steric and/or thermodynamic barrier to subsequent inhibitor binding. As a comparison, the association and dissociation rates were also determined for the two clinically approved NNRTIs nevirapine and efavirenz. It can be seen that all of the IAS derivatives were superior to both reference drugs in terms of binding, showing much faster association rates, whereas the dissociation rates were of the same order of those of efavirenz but significantly better (i.e., slower) than those of nevirapine.
TABLE 2.
Association and dissociation rates for the IAS derivatives to the different enzyme-substrate complexes for wild-type RT and the Lys103Asn mutanta
| RT type and inhibitor | Rate for:
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| RT
|
RT/TP
|
RT/TP/dNTP
|
|||||||
| Ki (nM) | kon (s−1M−1) | koff (s−1) | Kibin (nM) | konbin (s−1M−1) | koffbin (s−1) | Kiter (nM) | konter (s−1M−1) | koffter (s−1) | |
| Wild-type RT | |||||||||
| RS1202 | 0.3 | (11 ± 1) × 105 | (4 ± 0.5) × 10−4 | 1.6 | (3 ± 0.5) × 105 | (5 ± 0.5) × 10−4 | 1.2 | (2 ± 0.3) × 105 | (3 ± 0.3) × 10−4 |
| RS1588 | 0.3 | (6 ± 0.5) × 105 | (2 ± 0.3) × 10−4 | 2.7 | (2 ± 0.5) × 105 | (6 ± 0.5) × 10−4 | n.d. | n.d. | n.d. |
| RS1866 | 2.2 | (4 ± 0.6) × 105 | (9 ± 1) × 10−4 | 2.3 | (3 ± 0.2) × 105 | (7 ± 1) × 10−4 | n.d. | n.d. | n.d. |
| RS1980 | 0.3 | (6 ± 0.5) × 105 | (2 ± 0.3) × 10−4 | 0.4 | (7 ± 0.5) × 105 | (3 ± 0.3) × 10−4 | 3 | (1 ± 0.3) × 105 | (3 ± 0.3) × 10−4 |
| EFV | 30 | (0.1 ± 0.02) × 105 | (4 ± 1) × 10−4 | .30 | (0.1 ± 0.02) × 105 | (4 ± 1) × 10−4 | 4 | (0.4 ± 0.02) × 105 | (1.6 ± 0.3) × 10−4 |
| NVP | 400 | (0.04 ± 0.01) × 105 | (16 ± 1) × 10−4 | 500 | (0.03 ± 0.01) × 105 | (15 ± 1) × 10−4 | 375 | (0.04 ± 0.01) × 105 | (15 ± 1) × 10−4 |
| Lys103Asn mutant RT | |||||||||
| RS1202 | 1.3 | (1.9 ± 0.2) × 105 | (2.5 ± 0.5) × 10−4 | 5 | (5 ± 1) × 104 | (2.5 ± 0.5) × 10−4 | 5 | (5 ± 1) × 104 | (2.5 ± 1) × 10−4 |
| RS1588 | 0.3 | (7.1 ± 0.5) × 104 | (2 ± 0.2) × 10−5 | 3 | (4.5 ± 0.5) × 104 | (1.5 ± 0.2) × 10−4 | n.d. | n.d. | n.d. |
| EFV | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 200 | (0.6 ± 0.01) × 104 | (1.2 ± 0.1) × 10−3 |
| NVP | 3,300 | (0.03 ± 0.005) × 104 | (1 ± 0.2) × 10−3 | 3,300 | (0.03 ± 0.005) × 104 | (1 ± 0.2) × 10−3 | n.d. | n.d. | n.d. |
Enzyme-substrate complexes and kinetic constants are as defined in Fig. 1. Numbers are the mean values of three independent experiments and (standard deviations). n.d., not determined; EFV, efavirenz; NVP, nevirapine; n.a., not applicable (the affinity of EFV for the RTK103N and RTK103N/TP complexes was too low to be measured).
A high potency of inhibition towards Lys103Asn mutant RT correlates with preferential association of IASs with the unliganded form of the enzyme.
As reported in Table 1, both RS1202 and RS1588 retained high activity against the Lys103Asn mutant RT. Remarkably, RS1588 inhibited the mutated enzyme with the same potency as the wild type. Thus, the mechanism of inhibition by RS1202 and RS1588 towards the Lys103Asn mutant was investigated. The calculated kinetic parameters for the different enzymatic forms are listed in Table 2. Both RS1202 and RS1588 preferentially bound to the unliganded form of the Lys103Asn mutant, as in the case of wild-type RT. However, their association rates (kon) were decreased by the Lys103Asn mutation, in accord with previous structural and enzymatic studies which showed that this mutation increased the thermodynamic barrier for drug binding. Remarkably, RS1202 and RS1588 showed a slower dissociation rate (koff) from the mutant enzyme with respect to wild-type RT, thus explaining their high inhibition potencies (Ki). Compared with the NNRTI nevirapine, which does not discriminate among the different enzymatic forms, or efavirenz, which selectively targets the ternary RT/TP/dNTP complex, it could be seen that both IASs showed more favorable interaction parameters. These results suggested that drugs which selectively target the unliganded form of the RT might be less sensitive to the steric barrier imposed by the Lys103Asn mutation.
DISCUSSION
The results of our investigation of the mechanism of action of selected IAS derivatives showed that the nature of the substituents on the drug pharmacophore can influence the selective interaction of the drug with different mechanistic forms of the viral RT. Similar properties have been already observed for other NNRTIs, such as efavirenz, the pyrrolobenzoxazepinone derivative PPO464, and the carboxanilide derivatives UC38 and UC84 (10, 13, 15), suggesting that a preferential complementarity exists between the structure of the inhibitor molecule and the different spatial conformations of the NNBS.
The structure-function relationships of our IAS derivatives are schematically drawn in Fig. 4. The compound RS1866 {4,5-difluoro-3-[(3,5-dimethylphenyl)sulfonyl]-1H-indole-2-carboxyamide} showed a fully noncompetitive mode of action, being able to bind to all of the different reaction intermediates (RT, RT/TP, and RT/TP/dNTP complexes) (Fig. 1A) with equal affinities. Changing the nature of the alkyl group to give the compound RS1588 {5-chloro-3-[(3,5-dimethylphenyl)sulfonyl]-1 H-indole-2-carboxyamide} also modified the mechanism of inhibition, rendering the drug more selective for the free enzyme with respect to the binary and ternary complexes. Elimination of the 3,5-methyl groups gave the compound RS1202 [5-chloro-3-(phenylsulfonyl)-1H-indole-2-carboxyamide], which was very selective for the free enzyme, like RS1588, but also showed a partially competitive mechanism of action towards the nucleic acid substrate. On the other hand, introducing an alaninamide moiety into compound RS1588 to give its derivative RS1980 (N-{3-[(3,5-dimethylphenyl)sulfonyl]-5-chloro-1H-indole-2-carbonyl}alaninamide) made the drug capable of discriminating against the RT/TP/dNTP ternary complex, with a partially competitive mode of action towards the dNTP substrate.
FIG. 4.

Structure-activity relationships for the IAS derivatives used in this study. The different substituents on the common IAS scaffold are highlighted with circles. Arrows indicate the structural relationships among the different compounds. For each compound, the mechanism of action is indicated below its structure. For details, see the Discussion.
Drug binding studies showed that the main determinant for drug selectivity was the inhibitor association rate, suggesting that the structural reorganization of the NNBS upon formation of the binary and ternary complexes of RT with its substrates introduced some steric and/or thermodynamic barriers which were differentially “sensed” by the substituents on the drug molecule, depending on their spatial arrangement.
This inherent flexibility of the IAS derivatives, and their consequent ability to selectively target specific enzymatic forms, also had some consequences for their interaction with NNRTI-resistant mutated RT enzymes, such as Lys103Asn and Tyr181Ile. In fact, as can be seen from Table 1, the compounds which selectively targeted the free enzyme, that is, RS1588 and RS1202, were also the least affected by the Lys103Asn mutation. On the other hand, those compounds which did not discriminate between the free enzyme and the binary RT/TP complex, that is, RS1866 and RS1980, showed a loss of activity of about 75- to 150-fold. The RS1866 derivative was the most affected by the Tyr181Ile mutation, but the other compounds also showed significant losses of potency against this mutant.
When the mechanism of inhibition of the Lys103Asn mutant by RS1202 and RS1588 was investigated, both compounds were shown to selectively target the unliganded form of the mutant RT, as in the case of the wild-type enzyme, but the association rates (kon) of both drugs were reduced. This is consistent with the well-known effect of the Lys103Asn mutation, which introduces a steric barrier to drug binding (12). On the other hand, RS1202 and RS1588 showed slower dissociation rates (koff) from the Lys103Asn mutant than from wild-type RT. By comparison, the NNRTIs efavirenz and nevirapine suffered from similar reductions in their kon values but no compensatory decrease in their koff rates. Thus, our results suggest that drugs selectively targeting the unliganded form of the enzyme might efficiently overcome the steric barrier introduced by the Lys103Asn mutation by decreasing their association rates from the mutated enzyme, as we have shown in the case of RS1202 and RS1588. This observation might help in the design of more active compounds.
Several hypotheses have been made, based on kinetic, cross-linking, and structural studies, to explain the mechanism of inhibition of the chemical step by NNRTIs (11, 19, 26, 27). Binding of an NNRTI leads to displacement of the β12-β13 hairpin, which has direct interaction with the nucleic acid substrate. Thus, it is possible that this alteration in the position of the nucleic acid relative to the polymerase active site is responsible for NNRTI inhibition. Another possible mechanism can be hypothesized from the observation that the NNBS includes residues (Tyr181 and Tyr188) in the β9-β10 hairpin, which contains two of the three active-site aspartic acids (Asp185 and Asp186). There are also contacts between some NNRTIs and the β6 strand that carries the third active-site aspartate (Asp110). Again, even a moderate shift in the positions of the active-site residues could interfere with the chemical step of polymerization. Finally, it has been suggested that the fingers subdomain of RT may not adopt a fully closed conformation if both a dNTP and an NNRTI are bound to the enzyme. Since proper closure of the fingers is important for the positioning of the dNTP and the primer relative to the polymerase active site, preventing the proper closure of the fingers subdomain would interfere with the chemical step of DNA synthesis.
Different NNRTIs might act through one or more of these mechanisms; however, all of the NNRTIs studied so far do not interfere with the binding of either the dNTP or the nucleic acid substrate. On the other hand, the IAS derivatives described here showed different mechanisms of action, being ableto discriminate between different mechanistic forms of theviral RT and being partially competitive towards either the nucleic acid or the dNTP substrate. These observations further reinforce the notion that the interaction of IASs with the NNBS of HIV-1 RT is novel and different from the other NNRTIs (18).
It thus appears that the IAS derivatives shown here are very sensitive to the structural modifications occurring at the NNBS upon complexation of the RT with its substrates and that their sensitivity can be modulated through small modifications of the drug molecule.
Acknowledgments
We thank S. H. Hughes (NCI-Frederick Cancer Research and Development Center) for the coexpression vectors pUC12N/p66(His)/p51 with the wild-type or the mutant forms of HIV-1 RT.
This work was supported by the Italian Ministero della Salute, Istituto Superiore di Sanità, Fourth National Research Program on AIDS (R.S., M.A., and R.R.), Fifth National Research Program on AIDS (grant 40F.78 to S.S.), Italian MIUR-Cofin 2002 and 2004 (R.S. and M.A.), Istituto Pasteur-Fondazione Cenci Bolognetti (R.S.), and the EU FP6 Research Project LSHB-CT-2003-503480-TRIoH (G.M.). U.H. is supported by the University of Zürich.
REFERENCES
- 1.Bahar, I., B. Erman, R. L. Jernigan, A. R. Atilgan, and D. G. Covell. 1999. Collective motions in HIV-1 reverse transcriptase: examination of flexibility and enzyme function. J. Mol. Biol. 285:1023-1037. [DOI] [PubMed] [Google Scholar]
- 2.Baldanti, F., S. Paolucci, G. Maga, N. Labo, U. Hubscher, A. Y. Skoblov, L. Victorova, S. Spadari, L. Minoli, and G. Gerna. 2003. Nevirapine-selected mutations Tyr181Ile/C of HIV-1 reverse transcriptase confer cross-resistance to stavudine. AIDS 17:1568-1570. [DOI] [PubMed] [Google Scholar]
- 3.Basavapathruni, A., C. M. Bailey, and K. S. Anderson. 2004. Defining a molecular mechanism of synergy between nucleoside and nonnucleoside AIDS drugs. J. Biol. Chem. 279:6221-6224. (First published 13 January 2004.) [DOI] [PubMed] [Google Scholar]
- 4.Blanca, G., F. Baldanti, S. Paolucci, A. Y. Skoblov, L. Victorova, U. Hubscher, G. Gerna, S. Spadari, and G. Maga. 2003. Nevirapine resistance mutation at codon 181 of the HIV-1 reverse transcriptase confers stavudine resistance by increasing nucleotide substrate discrimination and phosphorolytic activity. J. Biol. Chem. 278:15469-15472. (First published 24 February 2003.) [DOI] [PubMed] [Google Scholar]
- 5.Buckheit, R. W., Jr., E. L. White, V. Fliakas-Boltz, J. Russell, T. L. Stup, T. L. Kinjerski, M. C. Osterling, A. Weigand, and J. P. Bader. 1999. Unique anti-human immunodeficiency virus activities of the nonnucleoside reverse transcriptase inhibitors calanolide A, costatolide, and dihydrocostatolide. Antimicrob. Agents Chemother. 43:1827-1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campiani, G., A. Ramunno, G. Maga, V. Nacci, C. Fattorusso, B. Catalanotti, E. Morelli, and E. Novellino. 2002. Non-nucleoside HIV-1 reverse transcriptase (RT) inhibitors: past, present, and future perspectives. Curr. Pharm. Des. 8:615-657. [DOI] [PubMed] [Google Scholar]
- 7.De Clercq, E. 2002. Highlights in the development of new antiviral agents. Mini Rev. Med. Chem. 2:163-175. [DOI] [PubMed] [Google Scholar]
- 8.De Clercq, E. 2002. New anti-HIV agents and targets. Med. Res. Rev. 22:531-565. [DOI] [PubMed] [Google Scholar]
- 9.Dixon, M., and E. C. Webb. 1979. Enzymes, 3rd ed. Longman, London, United Kingdom.
- 10.Fletcher, R. S., K. Syed, S. Mithani, G. I. Dmitrienko, and M. A. Parniak. 1995. Carboxanilide derivative non-nucleoside inhibitors of HIV-1 reverse transcriptase interact with different mechanistic forms of the enzyme. Biochemistry 34:4346-4353. [DOI] [PubMed] [Google Scholar]
- 11.Madrid, M., J. A. Lukin, J. D. Madura, J. Ding, and E. Arnold. 2001. Molecular dynamics of HIV-1 reverse transcriptase indicates increased flexibility upon DNA binding. Proteins 45:176-182. [DOI] [PubMed] [Google Scholar]
- 12.Maga, G., M. Amacker, N. Ruel, U. Hubscher, and S. Spadari. 1997. Resistance to nevirapine of HIV-1 reverse transcriptase mutants: loss of stabilizing interactions and thermodynamic or steric barriers are induced by different single amino acid substitutions. J. Mol. Biol. 274:738-747. [DOI] [PubMed] [Google Scholar]
- 13.Maga, G., A. Ramunno, V. Nacci, G. A. Locatelli, S. Spadari, I. Fiorini, F. Baldanti, S. Paolucci, M. Zavattoni, A. Bergamini, B. Galletti, S. Muck, U. Hubscher, G. Giorgi, G. Guiso, S. Caccia, and G. Campiani. 2001. The stereoselective targeting of a specific enzyme-substrate complex is the molecular mechanism for the synergic inhibition of HIV-1 reverse transcriptase by (R)-(-)-PPO464: a novel generation of nonnucleoside inhibitors. J. Biol. Chem. 276:44653-44662. (First published 25 September 2001.) [DOI] [PubMed] [Google Scholar]
- 14.Maga, G., and S. Spadari. 2002. Combinations against combinations: associations of anti-HIV 1 reverse transcriptase drugs challenged by constellations of drug resistance mutations. Curr. Drug Metab. 3:73-95. [DOI] [PubMed] [Google Scholar]
- 15.Maga, G., D. Ubiali, R. Salvetti, M. Pregnolato, and S. Spadari. 2000. Selective interaction of the human immunodeficiency virus type 1 reverse transcriptase nonnucleoside inhibitor efavirenz and its thio-substituted analog with different enzyme-substrate complexes. Antimicrob. Agents Chemother. 44:1186-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reference deleted.
- 17.Reference deleted.
- 18.Ragno, R., M. Artico, G. De Martino, G. La Regina, A. Coluccia, A. Di Pasquali, and R. Silvestri. 2005. Docking and 3-D QSAR studies on indolyl aryl sulfones. Binding mode exploration at the HIV-1 reverse transcriptase non-nucleoside binding site and design of highly active N-(2-hydroxyethyl)carboxamide and N-(2-hydroxyethyl)carbohydrazide derivatives. J. Med. Chem. 48:213-223. [DOI] [PubMed] [Google Scholar]
- 19.Ren, J., R. Esnouf, E. Garman, D. Somers, C. Ross, I. Kirby, J. Keeling, G. Darby, Y. Jones, D. Stuart, and D. Stammers. 1995. High resolution structures of HIV-1 RT from four RT-inhibitor complexes. Nat. Struct. Biol. 2:293-302. [DOI] [PubMed] [Google Scholar]
- 20.Selmi, B., J. Deval, K. Alvarez, J. Boretto, S. Sarfati, C. Guerreiro, and B. Canard. 2003. The Y181C substitution in 3′-azido-3′-deoxythymidine-resistant human immunodeficiency virus, type 1, reverse transcriptase suppresses the ATP-mediated repair of the 3′-azido-3′-deoxythymidine 5′-monophosphate-terminated primer. J. Biol. Chem. 278:40464-40472. [DOI] [PubMed] [Google Scholar]
- 21.Silvestri, R., M. Artico, G. De Martino, G. La Regina, R. Loddo, M. La Colla, M. Mura, and P. La Colla. 2004. Simple, short peptide derivatives of a sulfonylindolecarboxamide (L-737,126) active in vitro against HIV-1 wild type and variants carrying non-nucleoside reverse transcriptase inhibitor resistance mutations. J. Med. Chem. 47:3892-3896. [DOI] [PubMed] [Google Scholar]
- 22.Silvestri, R., M. Artico, G. La Regina, G. De Martino, M. La Colla, R. Loddo, and P. La Colla. 2004. Anti-HIV-1 activity of pyrryl aryl sulfone (PAS) derivatives: synthesis and SAR studies of novel esters and amides at the position 2 of the pyrrole nucleus. Farmaco 59:201-210. [DOI] [PubMed] [Google Scholar]
- 23.Silvestri, R., G. De Martino, G. La Regina, M. Artico, S. Massa, L. Vargiu, M. Mura, A. G. Loi, T. Marceddu, and P. La Colla. 2003. Novel indolyl aryl sulfones active against HIV-1 carrying NNRTI resistance mutations: synthesis and SAR studies. J. Med. Chem. 46:2482-2493. [DOI] [PubMed] [Google Scholar]
- 24.Smith, M. B., M. L. Lamb, J. Tirado-Rives, W. L. Jorgensen, C. J. Michejda, S. K. Ruby, and R. H. Smith, Jr. 2000. Monte Carlo calculations on HIV-1 reverse transcriptase complexed with the non-nucleoside inhibitor 8-Cl TIBO: contribution of the L100I and Y181C variants to protein stability and biological activity. Protein Eng. 13:413-421. [DOI] [PubMed] [Google Scholar]
- 25.Smith, M. B., S. Ruby, S. Horouzhenko, B. Buckingham, J. Richardson, I. Puleri, E. Potts, W. L. Jorgensen, E. Arnold, W. Zhang, S. H. Hughes, C. J. Michejda, and R. H. Smith, Jr. 2003. HIV-1 reverse transcriptase variants: molecular modeling of Y181C, V106A, L100I, and Lys103Asn mutations with nonnucleoside inhibitors using Monte Carlo simulations in combination with a linear response method. Drug Des. Discov. 18:151-163. [DOI] [PubMed] [Google Scholar]
- 26.Spence, R., K. S. Anderson, and K. A. Johnson. 1996. HIV-1 reverse transcriptase resistance to nonnucleoside inhibitors. Biochemistry 35:1054-1063. [DOI] [PubMed] [Google Scholar]
- 27.Spence, R. A., W. M. Kati, K. S. Anderson, and K. A. Johnson. 1995. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science 267:988-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Temiz, N. A., and I. Bahar. 2002. Inhibitor binding alters the directions of domain motions in HIV-1 reverse transcriptase. Proteins 49:61-70. [DOI] [PubMed] [Google Scholar]
- 29.Vella, S., and L. Palmisano. 2000. Antiretroviral therapy: state of the HAART. Antivir. Res. 45:1-7. [DOI] [PubMed] [Google Scholar]
- 30.Williams, T. M., T. M. Ciccarone, S. C. MacTough, C. S. Rooney, S. K. Balani, J. H. Condra, E. A. Emini, M. E. Goldman, W. J. Greenlee, L. R. Kauffman, et al. 1993. 5-Chloro-3-(phenylsulfonyl)indole-2-carboxamide: a novel, non-nucleoside inhibitor of HIV-1 reverse transcriptase. J. Med. Chem. 36:1291-1294. [DOI] [PubMed] [Google Scholar]