

Abstract
This study was designed to describe the population pharmacokinetics of rifapentine (RFP) and 25-desacetyl RFP in a South African pulmonary tuberculosis patient population. Special reference was made to studying the influence of previous exposure to rifampin (RIF) and the variability in pharmacokinetic parameters between patients and between occasions and the influence of different covariates. Patients were included in the study if they had been receiving first-line antimycobacterial therapy (rifampin, isoniazid, pyrazinamide, and ethambutol) for not less than 4 weeks and not more than 6 weeks and were divided into three RFP dosage groups based on weight: 600 mg, <45 kg; 750 mg, 46 to 55 kg; and 900 mg, >55 kg. Participants received a single oral dose of RFP together with concomitant antimycobacterial agents, excluding RIF, on study days 1 and 5 after they ingested a soup-based meal. The RFP and 25-desacetyl RFP concentration-time data were analyzed by nonlinear mixed-effect modeling using NONMEM. The pharmacokinetics of the parent drug were modeled separately, and the individual pharmacokinetic parameters were used as inputs for the 25-desacetyl RFP pharmacokinetic model. A one-compartment disposition model was found to best describe the data for both the parent and the metabolite, and the metabolite was assumed to be formed only from the central compartment of the parent drug. Prior treatment with RIF did not alter the pharmacokinetics of RFP but appeared to increase the excretion of 25-desacetyl RFP in a nonlinear fashion. The RFP oral clearance and volume of distribution were found to increase by 0.049 liter/h and 0.691 liter, respectively, with a 1-kg increase from the median weight of 50 kg. The oral clearance of 25-desacetyl RFP was found to be 35% lower in female patients. The model developed here describes the population pharmacokinetics of RFP and its primary metabolite in tuberculosis patients and includes the effects of prior administration with RIF and covariate factors.
Rifapentine (RFP) is a member of the rifamycin family and is currently registered in the United States for the treatment of tuberculosis in human immunodeficiency virus (HIV)-negative patients with noncavitary tuberculosis (TB) and with a sputum sample which is negative for acid-fast bacilli by smear at 2 months of therapy (2). Treatment guidelines recommend dosing RFP (600 mg orally) together with the companion first-line antimycobacterial drug isoniazid (INH) once weekly, following the first 8 weeks of treatment with rifampin (RIF), INH, pyrazinamide (PZA), and ethambutol (EMB), to complete 6 months of therapy.
The primary metabolic pathways for RFP are deacetylation and nonenzymatic hydrolysis. This results in one primary enzymatic metabolite, 25-desacetyl RFP, and two secondary nonenzymatic metabolites, 3-formyl RFP and 3-formyldesacetyl RFP (18). The primary route of elimination for the rifamycins is via biliary excretion with enterohepatic recirculation, although gastrointestinal secretion and renal clearance also play a role (1, 4, 18). RFP is an inducer of cytochrome P450 (CYP) 3A4 and CYP2C8/9 at the same order of magnitude as RIF (1, 3, 8), although it does not possess the same autoinductive properties (10, 11).
The pharmacokinetics of RFP in TB patients have been described previously by using noncompartmental techniques (16, 21). The effects of age (14), sex (13), various degrees of hepatic dysfunction (15), and HIV infection (12) on the pharmacokinetics of RFP have all been investigated in separate studies. The data from those studies were all analyzed by model-independent methods, and only a single covariate factor at a time was investigated. Furthermore, the impact of prior administration of RIF on the pharmacokinetics of RFP has not been investigated in patients.
This study was designed to describe the population pharmacokinetics of RFP and 25-desacetyl RFP in a South African pulmonary TB patient population receiving companion frontline antimycobacterial agents concomitantly. Special reference was made to investigating the influence of previous exposure to RIF and the variability in the pharmacokinetic parameters between patients and between occasions and the influence of different covariates on RFP and 25-desacetyl RFP pharmacokinetics.
MATERIALS AND METHODS
Patient inclusion and exclusion criteria.
Forty-six hospitalized patients diagnosed with either an initial or a secondary episode of pulmonary TB were enrolled in this study at the DP Marais SANTA Centre, a specialist tuberculosis hospital in Cape Town, South Africa. The demographic characteristics of the patients are displayed in Table 1. All patients had been receiving first-line antimycobacterial combination therapy (Rifafour e-200; Aventis, Midrand, South Africa) of RIF (body weight [WT] <50 kg, 480 mg/day; body weight >50 kg, 600 mg/day), INH (body weight <50 kg, 240 mg/day; body weight >50 kg, 300 mg/day), PZA (body weight <50 kg, 1,200 mg/day; body weight >50 kg, 1,500 mg/day), and EMB (body weight <50 kg, 800 mg/day; body weight >50 kg, 1,200 mg/day) for not less than 4 weeks and not more than 6 weeks. None of the patients had known resistance to RIF prior to study initiation or had previously shown hypersensitivity to rifamycin antibiotics.
TABLE 1.
Patient characteristicsa
| Dose group | Mean (SD) age (yr) | Sex (no. of males/ no. of females) | Mean (SD) body wt (kg) | Mean (SD) BMI (kg/m2) | Mean (SD) dose (mg/kg) | No. of patients who were:
|
|||
|---|---|---|---|---|---|---|---|---|---|
| Smokersb | Alcohol users | HIV positive | Previously treated for TB | ||||||
| 600 mg (n = 10) | 35.5 (11.1) | 4/6 | 43.8 (2.48) | 16.5 (1.72) | 13.7 (0.85) | 9 | 8 | 5 | 5 |
| 750 mg (n = 19) | 36.0 (10.2) | 12/7 | 50.0 (2.44) | 17.9 (1.52) | 15 (0.75) | 18 | 13 | 6 | 12 |
| 900 mg (n = 16) | 32.5 (9.7) | 13/3 | 60.5 (5.23) | 21.5 (1.22) | 14.9 (1.22) | 14 | 16 | 3 | 12 |
| All patients | 35.0 (10.0) | 29/16 | 50.0 (7.97) | 19.7 (2.27) | 15.0 (1.04) | 41 | 37 | 14 | 29 |
SD, standard deviation; BMI, body mass index.
Evaluated by direct questioning about smoking tobacco while a hospitalized patient.
Evaluated by direct questioning about alcohol consumption during or prior to admission.
The study protocol was reviewed and approved by the Medicines Control Council of South Africa; the University of Cape Town Research Ethics Committee, South Africa; and the Ethics Review Committee of the SANTA Centre, South Africa. Each subject provided written informed consent before being admitted into the study.
Treatment and sample collection.
Patients were divided into the following three RFP dosage groups based on weight: 600 mg/day for those with body weights of 36 to 45 kg, 750 mg/day for those with body weights of 46 to 55 kg, and 900 mg/day for those with body weights of ≥56 kg. Participants received a single oral dose of RFP (Priftin; Hoechst Marion Roussel, Lainate, Italy) on study days 1 (occasion 1) and 5 (occasion 2), approximately 30 min after they ingested a soup-based meal. Patients continued to receive their concomitant antimycobacterial therapy on study days 1 to 8 at the following doses: subjects weighing <50 kg received 240 mg INH (INH-Betabs; Betabs, Johannesburg, South Africa), 1,200 mg PZA (Pyrazide; Hoechst Marion Roussel, Midrand, South Africa), and 800 mg EMB (Rolab 400, Rolab, Kempton Park, South Africa); subjects weighing >50 kg received 300 mg INH, 1,500 mg PZA, and 1,200 mg EMB. None of the patients received RIF during the study period. Blood samples (4 ml) were collected via an indwelling cannula (Introcan; 1.1 by 32 mm; B. Braun AG, Melsungen, Germany) prior to dosing and at 2, 3, 4, 5, 6, 8, 24, 48, and 72 h following drug administration on both occasions. The samples were stored temporarily in darkness on ice before undergoing centrifugation (3E-1 bench-top centrifuge; Sigma, Osterode am Harz, Germany) at 3,500 rpm for 10 min. The plasma was subsequently harvested into labeled 1.5-ml microcentrifuge tubes (Greiner Bio-One International, Kremsmuenster, Austria) and stored at −80°C until analysis.
Drug quantification.
The simultaneous determination of RFP and 25-desacetyl RFP plasma concentrations were made by using a validated high-pressure liquid chromatography method developed at the Division of Clinical Pharmacology, University of Cape Town, Cape Town, South Africa (16). The standard curve ranges of concentrations in plasma were 6.84 × 10−4 to 3.42 × 10−2 mmol/liter for RFP and 2.99 × 10−4 to 2.39 × 10−2 mmol/liter for 25-desacetyl RFP. The intraday coefficients of variation ranged from 2.8% to 4.4% for RFP and from 4.4% to 5.6% for 25-desacetyl RFP. The interday coefficients of variation for RFP and 25-desacetyl RFP were 2.5 to 4.7% and 4.0 to 6.3%, respectively.
Population pharmacokinetic analysis.
Plasma molar concentration-time data for RFP and 25-desacetyl RFP for all patients were modeled by use of a nonlinear mixed-effects approach with NONMEM (version V, double precision, level 1.1) (20). The first-order conditional estimation method was used to estimate all population pharmacokinetic parameters except for the parent absorption lag time, where the first-order estimation method was employed. The model building was divided into two stages: parent drug model development followed by metabolite model development. Tracking of run information and parameter estimates during the model-building process was managed by using the software utility Census (22).
Single- and multicompartment pharmacokinetic models with linear elimination were fitted to the RFP concentration-time data. The models included first-order absorption with and without a lag time to determine the basic pharmacokinetic structural model. The need for interindividual variability (IIV) was evaluated in all basic structural parameters and was modeled exponentially as in the case for oral clearance (CL/F):
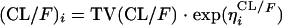 |
(1) |
where (CL/F)i is the oral clearance value for the ith patient and TV(CL/F) is the oral clearance in a typical individual. ηiCL/F is the interindividual variability, which is assumed to be normally distributed around zero and which has a variance (ω2)CL/F, to distinguish the ith patient's clearance from the population mean predicted from the regression model. This was done to avoid negative individual parameter estimates. Residual variability incorporated both additive and proportional error terms. Concentration-time profiles of the patient data displayed a secondary peak for 23 of 45 patients at 6 h. The 6-h sample was the first sample taken after lunch, and the peak could have resulted from RFP reabsorption from the small intestine following the release of bile from the gall bladder. An enterohepatic recirculation model was employed to characterize this. This model incorporated a hypothetical gall bladder compartment whereby drug entered from the central compartment and was later emptied into the absorption compartment. Separate rate constants were estimated for each process, and the time of gall bladder emptying was determined by the time of food intake relative to the time of drug ingestion. The potential effect of prior administration of RIF on RFP pharmacokinetics was also investigated. An empirical model explored changes in RFP clearance due to previous RIF administration with putative induction and the need for separate population CL/F values for the two dose occasions. Furthermore, interoccasional differences (IOV) in the pharmacokinetic parameters were explored and modeled as in the case for the volume of distribution of the central compartment (V/F):
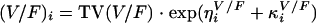 |
(2) |
where κiV/F is the interoccasional variability in the ith individual, which is assumed to be normally distributed around zero and which has a variance of (π2)V/F.
Individual empirical Bayes post hoc estimates were generated from the basic parent model, and individual η values for each pharmacokinetic parameter were plotted against the following covariates to identify potential relationships as well as the shape of the relationship: WT; body mass index; sex; HIV status; new or retreatment TB patient; smoking; alcohol abuse; recreational drug use; hemoglobin level; creatinine clearance; and total protein, albumin, serum alanine aminotransferase, aspartate aminotransferase, and alkaline phosphatase levels. Further testing and selection of covariates was achieved by using a stepwise generalized additive modeling, as implemented in Xpose (7). Covariates identified as being important were first assessed in the basic model by univariate addition and were ranked in descending order according to the change in objective function value (ΔOFV). The variables were then tested by forward stepwise inclusion into the model. Covariates were included in the model at a significance level of 0.05 (ΔOFV = 3.84). When no further covariates could be included at the 5% significance level, a backwards deletion was carried out at the 10% (ΔOFV = 10.83) significance level. Continuous covariates were centered at the median values and were included in the model as exemplified in the case of body weight:
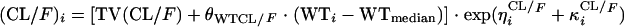 |
3 |
where θWTCL is the change in CL/F for each WT unit and WTi is the ith individual's weight. The individual empirical Bayesian post hoc estimates from the final parent drug model were fixed and served as input for the metabolite model. Single-compartment and multicompartment models with linear and nonlinear elimination were fitted to the metabolite plasma concentration-time data. Presystemic formation of the metabolite via the first-pass effect was investigated and modeled by using a hypothetical metabolite absorption compartment (9). Models that assumed that the metabolite was formed only systemically and models that included the elimination of 25-desacetyl RFP through a second nonhepatic pathway were also tested. Different models were applied to describe a change in metabolite exposure observed between the first and second RFP doses. These included a linear change in the oral clearance of the metabolite (CLM/F) over time (equation 4), an exponential change in CLM/F over time (equation 5), and a saturable elimination model (equation 6).
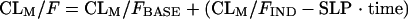 |
(4) |
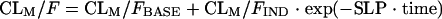 |
(5) |
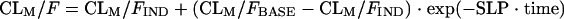 |
(6) |
where CLM/FBASE is the oral clearance in a typical individual at the end of the second RFP dosing occasion, i.e., at the baseline; CLM/FIND is the clearance in a typical individual on the first dosing occasion, i.e., RIF-induced clearance; and SLP is the slope of the decline in CLM/F over the study duration. The significance of covariate relations in the metabolite model were evaluated in the same manner as they were for the parent pharmacokinetic model. Residual variability in the metabolite model incorporated both additive and proportional error terms.
Model evaluation and qualification.
Models were selected by visual inspection of basic goodness-of-fit plots, including plots of the observed data versus population predictions (PREDs) and individual predictions (IPREDs). Plots of individual weighted residuals versus IPREDs and the distribution of weighted residuals over time were assessed. The relative standard errors (RSEs) of the parameters were also compared to measure parameter precision, and the objective function value (OFV) was used to discriminate between hierarchical (nested) models. This discrimination was based on a significance level of 0.05, which corresponds to a decrease in OFV of ≥3.84 (one parameter difference), as the difference in OFV is approximately χ2 distributed.
Model validation for both parent and metabolite were performed by mapping the response surface of the objective function (6) and by bootstrap resampling (5) to confirm parameter stability and sensitivity as well as the robustness of the model. For the former method, individual parameter values were fixed at ±5, 10, 15, 20, 30, 40, and 60% of the population estimate from the final model; and changes in the OFV were plotted against the parameter values. Polynomial equations (fourth order) were fitted to the plotted data. If it is the case that the OFV had a χ2 distribution, the 95% confidence intervals (CIs) for the parameter estimate would correspond to a change in OFV of 3.84. The CIs were compared with those based on the standard errors (SEs) of the NONMEM estimates and were calculated as a point estimate (±1.96 × SE). Parameter estimates were reestimated for each of the 1,000 bootstrap samples. The mean, SE, and 95% CIs were also compared with the NONMEM estimates from the final model.
RESULTS
Parent drug model.
A total of 775 RFP and 756 25-desacetyl RFP concentrations were collected from intensive sampling of 45 patients. The observed concentration-time data are presented in Fig. 1. One patient was withdrawn on the first dosing occasion due to poor venous access. A one-compartment model with first-order absorption and elimination including an absorption lag time was found to be optimal for further modeling of the data. IIV was included on the following structural model parameters: absorption lag time (ALAG), absorption rate constant (ka), V/F, and CL/F. Parameter estimates for RFP pharmacokinetics from the final model are presented in Table 2.
FIG. 1.

Means and standard deviations of observed RFP (solid circle and line) and observed 25-desacetyl RFP (solid triangle and dashed line) concentrations at each time point.
TABLE 2.
Final parameter estimates and comparison of the 95% confidence intervals for RFP population pharmacokinetic parameters estimated by standard errors of the NONMEM final estimates, bootstrapping, and objective function mapping
| Population parameter | NONMEM final estimate
|
Bootstrap resampling
|
Objective function mapping
|
|||
|---|---|---|---|---|---|---|
| Mean | SE | 95% confidence interval | Median | 95% confidence interval | 95% confidence interval | |
| ALAG (h) | 1.45 | 0.08 | 1.31-1.59 | 1.48 | 1.26-1.60 | 1.28-1.53 |
| ka (h−1) | 0.641 | 0.121 | 0.404-0.878 | 0.637 | 0.446-0.978 | 0.506-0.832 |
| CL/F (liters/h) | 2.03 | 0.09 | 1.85-2.21 | 2.03 | 1.86-2.22 | 1.90-2.21 |
| V/F (liters) | 37.8 | 1.6 | 34.7-40.9 | 37.8 | 34.3-44.5 | 35.3-42.0 |
| Additive residual error (mmol/liter) | 3.82 × 10−4 | 5.80 × 10−5 | 2.68 × 10−4-4.96 × 10−4 | 3.74 × 10−4 | 2.49 × 10−4-4.90 × 10−4 | 2.71 × 10−4-5.33 × 10−4 |
| Proportional residual error | 0.144 | 0.012 | 0.121-0.167 | 0.144 | 0.122-0.171 | 0.130-0.164 |
| Effect of WT on CL/F (liter · kg/h) | 0.049 | 0.04 | 0.014-0.084 | 0.050 | 0.029-0.071 | 0.029-0.073 |
| Effect of WT on V/F (liters/kg) | 0.691 | 0.04 | 0.592-0.789 | 0.696 | 0.157-1.235 | 0.386-0.994 |
| ω2 for ALAG | 0.0541 | 0.0211 | 0.0127-0.0955 | 0.0473 | 0.0164-0.1525 | 0.0236-0.1163 |
| ω2 for ka | 0.27 | 0.16 | 0.00-0.58 | 0.23 | 0.00-0.75 | 0.04-0.50 |
| ω2 for CL/F | 0.0485 | 0.0101 | 0.0287-0.0683 | 0.0457 | 0.0249-0.0656 | 0.0265-0.0882 |
| ω2 for V/F | 0.0268 | 0.0130 | 0.0013-0.0523 | 0.0257 | 0.0008-0.1569 | 0.0071-0.0537 |
| π2 for CL/F and V/F | 0.0271 | 0.0099 | 0.0077-0.0464 | 0.0278 | 0.0091-0.0537 | 0.0168-0.0421 |
| π2 for ka | 0.361 | 0.097 | 0.172-0.550 | 0.351 | 0.185-0.560 | 0.228-0.611 |
The secondary RFP peaks observed in some individuals could not be adequately characterized by models that included enterohepatic recirculation due to a lack of proper convergence and overparameterization. The precisions of the parameter estimates and objective function from the empirical enzyme model were not better than those from the model that incorporated only IOV on RFP CL/F (ΔOFV = 48.16), and the latter model was followed further. Addition of IOV terms to ka and V/F further improved the model and provided the best fit to the data. Graphical analysis and stepwise generalized additive modeling identified WT as a possible covariate that influenced both CL/F and V/F. The inclusion of the effect of WT on CL/F provided the biggest drop in OFV (ΔOFV = 22.66). In the forward inclusion step only the influence of WT on V/F produced a significant decrease in OFV (ΔOFV = 15.85), which negated the need for a backward deletion process. A typical individual weighing 50 kg was estimated to have an apparent oral clearance of 2.03 liters/h and a volume of distribution of 37.8 liters. An increase of 0.049 liter/h and 0.691 liter was observed for a 1-kg increase in weight from the median value of 50 kg for CL/F and V/F, respectively.
The PRED RFP concentration and the IPRED RFP concentration described the observed RFP concentrations well (Fig. 2), and no trends were seen in plots of weighted and individual weighted residuals versus IPRED (Fig. 3). The majority of individual weighted residuals were within 2.5 units of perfect agreement and were normally distributed around zero over the duration of the study (Fig. 3).
FIG. 2.

Observed RFP concentrations versus the PRED and IPRE concentrations on a normal scale. The solid line represents the line of identity.
FIG. 3.

IPRE RFP concentrations versus the individual weighted residuals and individual weighted residuals plotted over time.
No local minimum in any of the parameters was found following the objective function mapping. The 95% CIs of the objective function mapping and those of the final model coincided well (Table 2). The results from the bootstrap resampling are presented in Table 2 and concurred with the NONMEM parameter estimates.
Metabolite model.
A one-compartment model with linear elimination was found to be optimal for further modeling of the data. It was not possible to characterize any first-pass formation of the metabolite, nor could the distinction between hepatic and nonhepatic clearance of the parent drug be made. Therefore, it was assumed that all metabolite was formed centrally and that all parent drug was eliminated through formation of the metabolite. Of the various models tested to describe the shape of the change in clearance of the metabolite, the exponential decline-over-time model and the saturated elimination model provided equally good fits of the data. The saturation model was chosen based on the physiological plausibility that the enzymes responsible for the formation of metabolite decline from a maximal induced CLM/F value rather than the “infinite” value assumed in the exponential model. Final population pharmacokinetic parameter estimates are presented in Table 3. Variability between individuals in the parameters describing the pharmacokinetics of 25-desacetyl RFP were 23% for VM/F and 36% for CLM/F. Inclusion of variability terms in the other parameters were not supported by the data.
TABLE 3.
Final parameter estimates and a comparison of the 95% confidence intervals for 25-desacetyl RFP population pharmacokinetic parameters estimated by standard errors of the NONMEM final estimates, bootstrapping, and objective function mapping
| Population parameter | NONMEM final estimate
|
Bootstrap resampling
|
Objective function mapping
|
|||
|---|---|---|---|---|---|---|
| Mean | SE | 95% confidence interval | Median | 95% confidence interval | 95% confidence interval | |
| VM/F (liters) | 11.6 | 0.8 | 10.0-13.2 | 13.2 | 10.1-13.2 | 10.6-13.2 |
| CLM/FBASE (liters/h) | 3.56 | 0.29 | 2.98-4.13 | 3.57 | 2.91-4.15 | 3.14-4.07 |
| CLM/FIND (liters/h) | 21.0 | 1.6 | 13.2-24.2 | 20.5 | 13.2-45.0 | 13.2-34.1 |
| SLP (h−1) | 1.70 | 0.16 | 1.38-2.02 | 1.67 | 1.18-2.56 | 1.22-2.47 |
| Additive residual error (mmol/liter) | 6.3 × 10−4 | 5.6 × 10−5 | 5.2 × 10−4-7.4 × 10−4 | 6.2 × 10−4 | 5.3 × 10−4-7.4 × 10−4 | 5.6 × 10−4-7.1 × 10−4 |
| Proportional residual error | 0.196 | 0.017 | 0.163-0.229 | 0.193 | 0.162-0.225 | 0.169-0.225 |
| Effect of sex on CLM/Fa (liter/h) | 0.647 | 0.078 | 0.495-0.799 | 0.651 | 0.527-0.844 | 0.535-0.784 |
| Effect of weight on VM/Fb (liter/kg) | 0.267 | 0.096 | 0.046-0.079 | 0.268 | 0.102-0.461 | 0.119-0.417 |
| ω2 for CLM/F | 0.131 | 0.035 | 0.062-0.199 | 0.123 | 0.059-0.201 | 0.083-0.211 |
| ω2 for VM/F | 0.0646 | 0.0206 | 0.0242-0.1050 | 0.0585 | 0.0129-0.1510 | 0.0276-0.1229 |
CLM/Fi = (TVCLM · θSEX)·exp(ηiCLM/F)
(VM/Fi) = [TV(VM/F) + θWTVM/F · (WTi − WTmedian)]·exp(ηiVM/F)
Graphical analysis and stepwise generalized additive modeling identified WT and sex as possible covariates that influenced both CLM/F and VM/F. The inclusion of the effect of sex on CLM/F provided the biggest drop in OFV (ΔOFV = 23.98). In the forward inclusion step only the influence of WT on VM/F produced a further significant decrease in OFV (ΔOFV = 11.35). The final 25-desacetyl RFP model therefore included two covariate relations: the combined effects of sex on CLM/F and WT on VM/F. Females had a 35% lower CLM/F than males, and an increase of 0.267 liter was observed for a 1-kg increase in weight from the median value of 50 kg. The CLM/F on day 1 for a male subject weighing 50 kg was estimated to be 6.74 liters/h. This value declined to 3.56 liters/h on study day 8 (Table 3).
The population predicted 25-desacetyl RFP concentration and individual predicted 25-desacetyl RFP concentration described the observed concentrations well (Fig. 4), and no trends were seen in plots of weighted and individual weighted residuals versus IPRED or over time.
FIG. 4.

Observed 25-desactyl RFP concentrations versus PRED and IPRE concentrations on a normal scale. The solid line represents the line of identity.
No local minima were found in any of the parameters following the objective function mapping. The 95% CIs of the objective function mapping and those of the final model coincided well (Table 3). The results from the bootstrap resampling are presented in Table 3 and coincided well with the NONMEM parameter estimates.
DISCUSSION
The pharmacokinetics of RFP in the patient population were best described by using a one-compartment model with an absorption lag time and first-order absorption and elimination. We were not able to characterize fully the enterohepatic recirculation in our model, and this was mostly due to the lack of sufficient sampling following the first postdose meal. These spikes in concentration were, however, carried in the residual error terms, and the estimates of CL/F were not biased as a result. The final model included a positive association between WT and CL/F, with a 0.049-liter/h change in CL/F for each kilogram deviation from the median weight of 50 kg. The time of unbound RFP concentrations (≈5%) above an MIC threshold (ratio free drug concentration/MIC), where the MIC for RFP in drug-sensitive isolates is 0.06 mg/liter, is seen as a determining factor with regard to treatment outcome (2). If 95% protein binding in our population is assumed, all subjects maintained a free drug concentration/MIC ratio ≥1 for up to 48 h on each occasion, and the increased CL/F with increased WT is not seen to have a negative impact on clinical outcomes. The lack of support for the inclusion of a change in CL/F between occasion 1 and occasion 2 supports the findings from the previously published noncompartmental analysis (16) that the prior administration of RFP for a period of between 4 and 6 weeks does not significantly alter the oral clearance of RFP.
Sex differences have been shown to influence RFP pharmacokinetic measures (the maximum concentration in plasma, the area under the concentration-time curve, CL/F, V/F) derived by noncompartmental analysis (16). The results from the population analysis of the parent drug indicate that the differences in weight between individuals correlate better with the observed differences in CL/F and V/F rather than discrete sex differences. Females in this study generally had lower body weights, with only 3 of 16 weighing more than 50 kg but with 13 of 29 men weighing more than 50 kg. This resulted in lower median CL/F and V/F values in the female group and would account for the previously observed differences. Furthermore, coinfection with HIV, prior administration of RIF, smoking, alcohol abuse, and recreational drug use did not significantly affect the pharmacokinetics of RFP. A larger sample size may be required to detect differences within these subgroups, if indeed they exist.
The pharmacokinetics of 25-desacetyl RFP in this study were best described by a one-compartment model with no first-pass formation and a clearance value that declined in a nonlinear fashion over time after the withdrawal of RIF administration. The increased metabolite levels on the second dosing occasion are thought to be related to changes in the elimination of the metabolite, as no significant change in the pharmacokinetics of the parent drug was observed over time. This assumption is based on the fact that in previously published studies (1) prior RIF administration has been shown to increase the capacity of the liver to excrete hydrophobic compounds into the bile. Furthermore, sex was found to have a significant impact on CLM/F, with female patients demonstrating a 35% lower value than male patients. The weight differences between the two sex groups could not account for the lower value. A previous study by Schuetz et al. (19) found that women displayed only one-third to one-half the hepatic P-glycoprotein levels of men, which could account for the disparity between the sexes.
25-Desacetyl RFP has been shown to be active in vitro, with an MIC of 0.25 mg/liter in drug-susceptible isolates of Mycobacterium tuberculosis (17). If it is assumed that protein binding is similar between parent and metabolite (≈95%), then total plasma concentrations ≥5 mg/liter for an extended period of time are required to have an impact on treatment outcome. Only 25% (137of 656) of the plasma samples analyzed had metabolite concentrations above this level. Due to the limited pharmacological activity of the metabolite in our study population, the changes in metabolite kinetics between occasions is therefore not seen to have a negative clinical impact.
A population pharmacokinetic model for RFP and its deacetylated metabolite was developed that characterized the increased CL/F and V/F with increasing WT as well as the lower oral clearance of the metabolite in female patients. Prior treatment with RIF did not alter the pharmacokinetics of the parent drug but appeared to increase the excretion of the metabolite. Parameter estimates were consistent between individuals and between occasions, with low levels of interindividual variability observed for all parameters except the absorption rate constant.
Acknowledgments
This study was funded by the Medical Research Council of South Africa and the Division of Clinical Pharmacology, Department of Medicine, University of Cape Town.
REFERENCES
- 1.Acocella, G. 1978. Clinical pharmacokinetics of rifampicin. Clin. Pharmacokinet. 3:108-127. [DOI] [PubMed] [Google Scholar]
- 2.American Thoracic Society, Centers for Disease Control and Prevention, and Infectious Diseases Society of America. 2003. Official joint statement on the treatment of tuberculosis. Am. J. Respir. Crit. Care Med. 167:603-662. [DOI] [PubMed] [Google Scholar]
- 3.Aventis Pharmaceuticals. 2000. Priftin package insert. Aventis Pharmaceuticals, Midrand, South Africa.
- 4.Battaglia, R., E. Pianezzola, G. Salgarollo, G. Zini, and B. M. Strolin. 1990. Absorption, disposition and preliminary metabolic pathway of 14C-rifabutin in animals and man. J. Antimicrob. Chemother. 26:813-822. [DOI] [PubMed] [Google Scholar]
- 5.Ette, E. I. 1997. Stability and performance of a population pharmacokinetic model. J. Clin. Pharmacol. 37:486-495. [DOI] [PubMed] [Google Scholar]
- 6.Holford, N. H., and K. E. Peace. 1992. Results and validation of a population pharmacodynamic model for cognitive effects in Alzheimer patients treated with tacrine. Proc. Natl. Acad. Sci. USA 89:11471-11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jonsson, E. N., and M. O. Karlsson. 1999. Xpose: an S-PLUS based population pharmacokinetic/ pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed 58:51-64. [DOI] [PubMed] [Google Scholar]
- 8.Kenny, M. T., and B. Strates. 1981. Metabolism and pharmacokinetics of the antibiotic rifampin. Drug Metab. Rev. 12:159-218. [DOI] [PubMed] [Google Scholar]
- 9.Kerbusch, T., U. Wahlby, P. A. Milligan, and M. O. Karlsson. 2003. Population pharmacokinetic modelling of darifenacin and its hydroxylated metabolite using pooled data, incorporating saturable first-pass metabolism, CYP2D6 genotype and formulation-dependent bioavailability. Br J. Clin. Pharmacol. 56:639-652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keung, A., K. Reith, M. G. Eller, K. A. McKenzie, L. Cheng, and S. J. Weir. 1999. Enzyme induction observed in healthy volunteers after repeated administration of rifapentine and its lack of effect on steady-state rifapentine pharmacokinetics: part I. Int. J. Tuberc. Lung Dis. 3:426-436. [PubMed] [Google Scholar]
- 11.Keung, A., M. G. Eller, K. A. McKenzie, and S. J. Weir. 1999. Single and multiple dose pharmacokinetics of rifapentine in man: part II. Int. J. Tuberc. Lung Dis. 3:437-444. [PubMed] [Google Scholar]
- 12.Keung, A. C., R. C. Owens, Jr., M. G. Eller, S. J. Weir, D. P. Nicolau, and C. H. Nightingale. 1999. Pharmacokinetics of rifapentine in subjects seropositive for the human immunodeficiency virus: a phase I study. Antimicrob. Agents Chemother. 43:1230-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keung, A. C., M. G. Eller, and S. J. Weir. 1998. Single-dose pharmacokinetics of rifapentine in women. Pharmacokinet. Biopharm. 26:75-85. [DOI] [PubMed] [Google Scholar]
- 14.Keung, A. C., M. G. Eller, and S. J. Weir. 1998. Single-dose pharmacokinetics of rifapentine in elderly men. Pharm. Res. 15:1286-1291. [DOI] [PubMed] [Google Scholar]
- 15.Keung, A. C., M. G. Eller, and S. J. Weir. 1998. Pharmacokinetics of rifapentine in patients with varying degrees of hepatic dysfunction. J. Clin. Pharmacol. 38:517-524. [DOI] [PubMed] [Google Scholar]
- 16.Langdon, G., J. J. Wilkins, P. J. Smith, and H. McIlleron. 2004. Consecutive-dose pharmacokinetics of rifapentine in patients diagnosed with pulmonary tuberculosis. Int J. Tuberc. Lung Dis. 8:862-867. [PubMed] [Google Scholar]
- 17.Rastogi, N., K. S. Goh, M. Berchel, and A. Bryskier. 2000. Activity of rifapentine and its metabolite 25-O-desacetylrifapentine compared with rifampicin and rifabutin against Mycobacterium tuberculosis, Mycobacterium africanum, Mycobacterium bovis and M. bovis BCG. J. Antimicrob. Chemother. 46:565-570. [DOI] [PubMed] [Google Scholar]
- 18.Reith, K., A. Keung, P. C. Toren, L. Cheng, M. G. Eller, and S. J. Weir. 1998. Disposition and metabolism of 14C-rifapentine in healthy volunteers. Drug Metab. Dispos. 26:732-738. [PubMed] [Google Scholar]
- 19.Schuetz, E. G., A. H. Schinkel, M. V. Relling, and J. D. Schuetz. 1996. P-glycoprotein: a major determinant of rifampicin-inducible expression of cytochrome P4503A in mice and humans. Proc. Natl. Acad. Sci. USA 93:4001-4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheiner, L. B., and S. L. Beal. 1998. NONMEM users guide, part V. University of California, San Francisco.
- 21.Weiner, M., N. Bock, C. A. Peloquin, W. J. Burman, A. Khan, A. Vernon, Z. Zhao, S. Weis, T. R. Sterling, K. Hayden, S. Goldberg, and the Tuberculosis Trials Consortium. 2004. Pharmacokinetics of rifapentine 600, 900 and 1,200 mg during once-weekly tuberculosis therapy. Am. J. Respir. Crit. Care Med. 169:1191-1197. [DOI] [PubMed] [Google Scholar]
- 22.Wilkins, J. J. 2005. NONMEMory: a run management tool for NONMEM. Comput. Methods Programs Biomed. 78:259-267. [DOI] [PubMed] [Google Scholar]