

Abstract
Kaposi's sarcoma (KS)-associated herpesvirus (KSHV) is the infectious cause of Kaposi's sarcoma and is also associated with two B-cell lymphoproliferative diseases, primary effusion lymphoma and the plasmablastic form of multicentric Castleman's disease. KSHV is also found in the B-cell fraction of peripheral blood mononucleocytes of some KS patients. Despite in vivo infection of B cells and the ability of KSHV to infect many cell types in culture, to date B cells in culture have been resistant to KSHV infection. However, as shown here, the lack of infection is not due to the inability of B cells to support latent KSHV infection. When KSHV DNA is introduced into B cells, the virus is maintained as an episome and can establish and maintain latency over the course of months. As in all primary effusion lymphoma cell lines, there is a low level of spontaneous lytic replication in latently infected BJAB cells. Importantly, viral gene expression is similar to that of primary effusion lymphoma cell lines. Furthermore, the virus can be reactivated to higher levels with specific stimuli and transmitted to other cells, indicating that this is a productive infection. Thus B cells in culture are capable of establishing, maintaining, and reactivating from latency. These studies provide a controlled system to analyze how KSHV alters B cells during KSHV latency and reactivation.
Kaposi's sarcoma (KS)-associated herpesvirus (KSHV; also known as human herpesvirus 8) is present in virtually all KS tumors (17, 18, 25, 37), and many epidemiological studies have found that KSHV is necessary for KS (24, 47). The KS tumor is predominately made up of spindle cells, cells of endothelial origin that express markers of lymphatic endothelium (12, 29). KSHV is found in most if not all the spindle cells in nodule KS tumors. KSHV is also found in B cells in the peripheral blood of KS patients (3).
Soon after the discovery of KSHV in the spindle cells of KS tumors, KSHV was found associated with two B-cell lymphoproliferative disorders. KSHV is found in all primary effusion lymphomas (PELs) and is also present in AIDS-related or plasmablastic multicentric Castleman's disease (13, 15, 49). PELs are rare lymphomas mostly found in AIDS patients. While the lymphoma cells have lost many B-cell-associated antigens, they have rearranged immunoglobulin domains and a number of B-cell markers (4, 10, 11, 22, 30, 39, 52). A number of PEL cells have been isolated and grown in culture (5, 8, 9, 14, 44). The first PEL cells isolated contained both KSHV and Epstein-Barr virus (EBV) (14). However, many EBV-negative PELs have been identified, while KSHV-negative PELs have not been found, indicating that KSHV is the more likely agent, but EBV could facilitate the process of lymphoma formation.
Like all herpesviruses, KSHV has a latent life cycle and a lytic life cycle. During latency, only a few regions are transcriptionally active. The latency-associated nuclear antigen (LANA) locus carries three latent genes from one promoter, coding for LANA, a viral cyclin, and a viral FLICE inhibitory protein (21, 42). Another active region during latency is the kaposin locus that encodes a family of kaposin proteins from a series of noncanonical CUG codons (46). One of the four viral interferon regulatory factor homologs is also expressed during latency in some PEL lines but not in latently infected KS spindle cells (45). As with other characterized herpesviruses, lytic replication occurs in a cascade fashion with immediate-early (IE), early, and late gene synthesis. Lytic replication can be activated in vitro by a number of stimuli. Phorbol esters as well as sodium butyrate have been shown to induce higher levels of lytic replication in PEL lines (36, 44, 55). Small increases have been seen following treatment with gamma interferon and hypoxia (16, 20, 41). In other infected cell types, reactivation of high percentages of cells has been induced with overexpression of open reading frame 50 (ORF 50 or RTA), a KSHV gene that is both necessary and sufficient to activate lytic replication (35, 51).
In both KS tumors and in PELs, the virus is found predominantly in the latent state (44, 50). In KS tumors, approximately 98% of the spindle cells solely express transcripts consistent with latent infection (50, 56). However, in all of these tumors, a low percentage of cells (1 to 3%) express transcripts and viral antigens consistent with lytic replication (50). In cultured PELs, the same is true, though the percentage of lytic replication changes from one PEL line to anther. For example, in the BCBL-1 line the rate of lytic replication is around 2 to 4%, while in the BC-1 line nearly 1% of cells are undergoing lytic replication, and in the JSC-1 line there is a much higher percentage of cells undergoing lytic replication (9, 14, 44).
Virus from induced PEL lines has been used to infect many cell types in culture. Virus isolated from induced PEL cell supernatants was first shown to infect many cell types at very low levels (19, 23, 38, 43). A number of groups have shown infection of endothelial cells, the cell type that KS spindle cells are derived from. The first example showed low-level infection of bone marrow microvascular endothelial cells. Subsequent studies showed low-level infection of primary dermal microvascular endothelial cells (DMVECs) and the same cells immortalized with the E6 and E7 genes of papillomavirus (19, 38). In both primary and immortalized DMVECs the initial low level of infection increased over time until most of the culture maintained latent infection, although with the primary cells constant addition of fresh cells was necessary to maintain the culture. Subsequently it was found that high-level initial infection could be achieved in a TERT-immortalized dermal microvascular endothelial cell line (TIME cells) (32). By 24 h, greater than 90% of the TIME cells could be latently infected. Using the conditions used for TIME cells, it was found that primary dermal microvascular endothelial cells could also be infected to high levels as well as a large number of other cell types, including human foreskin fibroblasts, 293 cells (human kidney epithelial cells), HeLa cells (cervical carcinoma cells), CV-1 (green monkey kidney cells), SLK cells (spindle cells), 3T3 cells (murine fibroblasts), and CHO cells (Chinese hamster ovary cells) (6). Interestingly, cultured B-cell lines cannot be infected. In one study, over 13 cell lines were tested for susceptibility to KSHV and all but B-cell and T-cell lines could be infected (6). Infection of B cells in culture was achieved in an earlier study where they were able to infect B cells from EBV-infected patients (but not from EBV-negative patients), but the levels of infection were extremely low (31). All other attempts to infect B-cell lines or primary human B cells beyond detection by PCR have been unsuccessful (6, 7, 43).
KSHV clearly infects B cells in vivo, as demonstrated by the presence of virus in the peripheral blood mononuclear cells of KS patients and can cause B-cell-related diseases like PEL and multicentric Castleman's disease (13, 15, 49). The question remains why the virus, which can infect many cell types in vitro, is unable to infect B cells in culture. To address this question, we attempted to infect BJAB cells in culture. We created a recombinant virus that expresses both green fluorescent protein (GFP) and the puromycin resistance gene, which allows simple detection and powerful selection of recombinant infected cells. As observed with wild-type virus, we were unable to infect BJAB to any detectable extent. However, when naked viral DNA was introduced into BJAB cells, latency was established and could be maintained when kept under selection. The pattern of viral gene expression was similar to that in PEL cell lines. Virus can be successfully reactivated from the infected BJAB cells and passaged to endothelial cells. These studies indicate that BJAB cells are capable of establishing, maintaining, and reactivating from KSHV latency and thus provide a controlled system for examining the effects of KSHV infection on B cells in culture.
MATERIALS AND METHODS
Cells and media.
BCBL-1 cells have been previously described (44). BJAB cells are KSHV- and EBV-negative B-cell lymphoma cells. BCBL-1 cells and BJAB cells were carried in RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum, penicillin, streptomycin, glutamine, and β-mercaptoethanol. TIME cells are hTERT-immortalized dermal microvascular endothelial cells (32, 53). TIME cells were maintained in an EBM-2 medium bullet kit with supplements (Cambrex). African green monkey kidney (Vero) cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, penicillin, streptomycin, and glutamine.
Construction of recombinant KSHV.
For the construction of the GFP-puro recombinant virus, pBluescript II SK+(Stratagene) was digested with SpeI and then blunted and ligated to destroy the SpeI site in the polylinker (pBSIIspe−). Lambda 102 (33) was digested with BamHI and XhoI, and the 2.5-kb fragment containing the genomic locus surrounding ORF 11 and K2 was cloned into the pBSIIspe− vector to make the SpeI site between the poly(A) signal of ORF 11 and K2, a unique site in the resulting plasmid, pKS11K2. pBSII-eGFP-puro plasmid (a kind gift from Mark Pritchard) was digested with SpeI, and the 2.2-kb fragment containing the eGFP-puromycin fusion gene under the control of the cytomegalovirus (CMV) IE promoter was cloned into the SpeI site in plasmid pKS11K2, creating pKSGFP. pKSGFP was then electroporated into BCBL-1 cells. Twenty-four hours posttransfection, BCBL-1 cells were selected with puromycin. After selection for 1 month, all cells expressed GFP and were puromycin resistant. Recombinant virus was harvested from these cells as previously described (32). African green monkey kidney (Vero) cells were then infected with recombinant KSHV. Forty-eight hours post-KSHV infection, medium was replaced with fresh medium containing 10 μg of puromycin per ml. The culture medium was replaced with fresh puromycin-containing medium every day to remove dead cells until green colonies were visible. The purification of recombinant KSHV virus was done as previously described (54). Briefly, supernatant from KSHV-Vero cells induced with a defective adenovirus expressing the KSHV ORF 50 gene (a kind gift of Don Ganem) and 3 mM sodium butyrate were used to infect new Vero cells at a low multiplicity of infection (MOI). The MOI was determined empirically by determining the dilution of virus stock that yielded infection of less than 1% of the cells as determined by visualization of GFP. Single GFP-expressing colonies were isolated with a cloning cylinder. Each colony was tested for the purity of recombinant virus by PCR and/or Southern analysis. Vero cells carrying a higher percentage of recombinant KSHV were induced, and supernatant was used to infect new Vero cells. Single green colonies were isolated and screened. This procedure was repeated five times, and a KSHV-Vero cell line was established and used in the subsequent study.
Viruses and induction.
KSHV was harvested as previously described (32). For infections, the viral pellet was resuspended in the indicated medium and used to inoculate corresponding cell cultures for 2 h. The cell monolayer was then washed once and overlaid with fresh complete medium. For Vero cell inductions, the cell monolayer was infected with Adeno-50 recombinant virus at a density of approximately 4,000 particles per cell for 2 h. Adenovirus was preincubated with 1 μg/ml polylysine (Sigma) in medium for 100 min at room temperature (34). After infection, the cell monolayer was washed three times and overlaid with fresh complete medium containing 3 mM sodium butyrate. For BJAB cell inductions, cells were pelleted and the cell pellet was resuspended with 0.5 ml of polylysine-treated Adeno-50 virus inoculum. Cells were infected with Adeno-50 virus at a density of 4,000 to 5,000 particles per cell for 2 h. After infection, the cells were washed three times and resuspended in fresh complete medium containing 2 mM sodium butyrate. BJAB cells were also induced with phorbol 12-myristate-13-acetate (TPA) at a final concentration of 20 ng/ml.
To quantify the induction rate using TPA or Adeno-50 with sodium butyrate, four separate inductions on different days from two different KSHV-BJAB isolates were performed and the rate of reactivation was quantified by immunofluorescent antibody (IFA) staining for ORF 59 as described below. To quantify the infection rates from the supernatants of induced KSHV-BJAB cells, virus was harvested from the supernatant of 25 × 106 cells, concentrated, and used to infect TIME cells. The number of cells that were GFP positive was enumerated for each condition. This was done for three separate inductions from two different KSHV-BJAB isolates.
Episomal DNA preparation and electroporation into BJAB cells.
KSHV episomes were isolated from KSHV-Vero cells (5 × 106) by an alkaline lysis procedure as previously described (48). KSHV episomal DNA was then transfected into 2 × 107 BJAB cells via electroporation (Bio-Rad Gene Pulser II; 210 V, 960 μF). Transfected cells were resuspended in 20 ml of culture medium. At 2 days posttransfection, medium was replaced with fresh medium containing 10 μg of puromycin per ml. Half of the culture medium was replaced with fresh puromycin-containing medium every 5 days.
Immunofluorescence assay.
Immunofluorescence assays were done as previously described (32). The primary antibodies used were an anti-LANA peptide rabbit polyclonal antiserum (a kind gift from A. Polson and D. Ganem) and mouse monoclonal antibodies that recognize ORF 59 (Advanced Biotechnologies Inc.). The primary antibodies were diluted 1:1,000, and secondary antibodies—anti-rabbit Alexa fluor 488 and anti-mouse Alexa fluor 594 (Molecular Probes)—were diluted 1:1,000.
Gardella gel electrophoresis.
The Gardella gel assay was performed as previously described (26). Uninduced and induced BJAB cells latently infected with KSHV were washed twice in phosphate-buffered saline. The cell pellets (1 × 106 cells) were resuspended in loading buffer containing 5% Ficoll and 40 μg/ml RNase A in Tris-borate-EDTA and then loaded onto the gels. The gels were run at 40 V for 4 h and then at 120 V for an additional 22 h in Tris-borate-EDTA at 4°C. The gels were then dried, rehydrated in 0.5 M NaOH-0.15 M NaCl buffer, and directly probed with 32P-radiolabeled DNA corresponding to KSHV sequences.
Southern and Northern blot analyses.
Virion DNA was purified from BCBL-1 and KSHV-BJAB supernatant as previously described (33). Purified virion DNA was digested with HincII and subjected to Southern blot analysis as previously described (33). The blot was probed with 32P-radiolabeled K2 ORF and eGFP, respectively. Total RNA was extracted from 2.5 × 107 uninduced and induced BCBL-1 or KSHV-BJAB cells with RNA-Bee RNA isolation reagents (Tel-Test, Friendswood, Tex.). mRNA was then extracted with an Oligotex direct mRNA kit, following the manufacturer's instructions (QIAGEN). For Northern blot hybridization, 2 μg of mRNA from each sample was separated in a 1% agarose gel containing 18% formaldehyde and transferred to a nylon membrane, which was then hybridized with 32P-radiolabeled KSHV-specific probes.
RESULTS
Purification of recombinant KSHV containing an eGFP-puromycin fusion protein.
In order to examine infection of BJAB cells further, we made a recombinant KSHV that expresses an eGFP-puromycin resistance fusion protein, KSHV-GFP1 (Fig. 1A). We inserted the GFP-puro marker into a plasmid between the polyadenylation signals of ORF 11 and K2 as described in Materials and Methods section. The plasmid was transfected into BCBL-1 cells and selected for puromycin resistance. After 1 month, the entire culture was puromycin resistant and GFP positive by fluorescence (L. Chen and M. Lagunoff, data not shown). The BCBL-1 cells were induced with TPA, and virus in the supernatant was used to infect Vero cells at a very low multiplicity of infection. This was done to decrease the likelihood that multiple virus particles would enter a single cell. After selection, nearly 100% of the Vero cells expressed GFP (Fig. 1C). The GFP-expressing Vero cells were induced and used to infect a fresh batch of Vero cells again at a low MOI as described previously (54). This time the GFP-positive, puromycin-resistant cells were single-cell cloned and screened for pure recombinant virus. After a few rounds of selection and purification, virus from induced Vero cells was harvested and DNA was isolated, digested, and subjected to Southern blot analysis. As seen in Fig. 1B, a probe recognizing the region around viral interleukin 6 (vIL-6) recognized a significantly slower-migrating species indicative of the GFP-puromycin insertion. A band of the same size was also recognized by a GFP probe, demonstrating that the slower migration was indeed due to the insertion of GFP-puromycin. This recombinant virus will be referred to as KSHV-GFP1
FIG. 1.

Creation of KSHV-GFP1 recombinant virus. (A) Strategy for generation of KSHV-GFP1 recombinant virus. The structure of the first 30 kb of the KSHV genome is shown at the top. Viral genes are indicated as open boxes. The selective marker GFP-puro under the control of the CMV IE promoter was integrated between the ORF 11 and ORF K2 genes in the KSHV genome by homologous recombination in BCBL-1 cells with the recombination plasmid pKSGFP. Genomic structure of the resulting reconstituted virus KSHV-GFP1 is shown below. (B) Southern analysis of virion DNA isolated from induced BCBL-1 and KSHV-Vero supernatants. The probes used were the full-length vIL-6 gene (left panel), or the enhanced green fluorescence protein gene (right panel). (C) Photomicrograph of Vero cells infected with KSHV-GFP1 and selected with puromycin. (Left column) Phase. (Right column) Fluorescence for GFP.
KSHV does not infect BJAB cells.
The recombinant KSHV expressing the eGFP-puromycin fusion protein can be directly visualized for GFP expression and selected with puromycin to detect very low levels of infection. We used purified KSHV-GFP1 virus to infect different cells. We found that we could reactivate the KSHV-GFP1 to high levels similar to those of wild-type virus, and we could efficiently infect TIME and Vero cells with purified recombinant virus as determined by both GFP expression and immunofluorescence staining with an anti-LANA antibody (data not shown). In contrast to TIME and Vero cells, we could not detect a single GFP-positive cell after infection of BJAB cells under many different conditions. We also attempted to select BJAB cells with puromycin after exposure to large amounts of KSHV, but we could not grow out any cells after 1 week in culture. Thus, even with very sensitive methods to identify infected cells, we could not successfully detect any KSHV infection of BJAB cells. Although KSHV-GFP1 could infect Vero cells and TIME cells, we wanted to ensure that the defect in BJAB infection was not due to defects in our recombinant virus. We obtained another recombinant virus, rKSHV.219 (generously provided by Jeff Vieira), that contains the puromycin resistance gene and genes coding for GFP and red fluorescent protein, all under different promoters and all inserted between K9 and ORF 57 (54). We were unable to isolate any infected BJAB cells with this recombinant KSHV (data not shown).
Establishment and maintenance of KSHV latency in B cells.
We wanted to determine if we could overcome the lack of infection by directly introducing KSHV DNA into BJAB cells. Episomal DNA was isolated from Vero cells latently infected with the KSHV-GFP1 recombinant using the method of Simpson (48). We introduced the DNA into BJAB cells by electroporation. Forty-eight hours after electroporation, a low percentage of cells expressed GFP. We then used puromycin to select for BJAB cells transfected with the KSHV-GFP1 recombinant. After 10 days of selection, 100% of the live cells expressed GFP (Fig. 2). To determine if this was virally expressed GFP, we also stained the selected cells with antibody to LANA (Fig. 2). All of the GFP-positive cells also reacted with the LANA antibody, showing the typical speckled pattern seen previously in many different infected cell types. The untransfected BJAB controls did not stain with the LANA antibody. Immunofluorescence with an anti-ORF59 antibody, a marker of lytic replication, was also performed to determine the extent of spontaneous lytic replication in the KSHV-BJAB cells. Slightly less than 0.1% of the cells stained positive with the anti-ORF59 antibody, indicating that there is a very low but detectable level of spontaneous reactivation. After over 2 months in culture with selection, the BJAB cells were still GFP positive and maintained LANA expression as determined by IFA (Fig. 2). Thus the BJAB cells transfected with KSHV DNA could establish and maintain latent gene expression. We have transfected BJAB cells on multiple occasions with KSHV DNA from different sources and have been able to rapidly establish latency in the cells every time (data not shown).
FIG. 2.

Photomicrographs of BJAB cells and KSHV-BJAB cells 2 weeks and 2 months after puromycin selection, showing phase (left panel), eGFP fluorescence (middle-left panel), 4′,6′-diamidino-2-phenylindole (DAPI) (middle-right panel), and LANA red fluorescence (right panel).
To further prove that KSHV latency was indeed established, infected cell samples were run on a Gardella gel to determine if the latent virus in the BJABs was maintained as a circular episome. BJAB cells and uninduced and induced KSHV-BJAB and BCBL-1 cells were lysed in the well and electrophoresed overnight on an agarose Gardella gel to separate large linear and circular DNA molecules (26). As a control for linear KSHV DNA, KSHV virions were loaded in a control lane. The gel was subsequently probed with radiolabeled KSHV sequences. As seen in Fig. 3, the predominant species in uninduced KSHV-BJAB and BCBL-1 cells that reacts with KSHV sequences migrates much slower than the linear virion DNA, indicating that the KSHV-infected BJAB cells maintain episomal circular KSHV DNA, as occurs in all herpesvirus latent phases. There are significant amounts of linear KSHV DNA in the uninduced BCBL-1 cells indicative of the 3 to 5% of cells undergoing spontaneous reactivation. There is little or no detectable linear KSHV in the BAJB cells (Fig. 3). This was expected due to the much lower levels of spontaneous lytic reactivation in the KSHV-BJAB cells as compared to BCBL-1 cells.
FIG. 3.
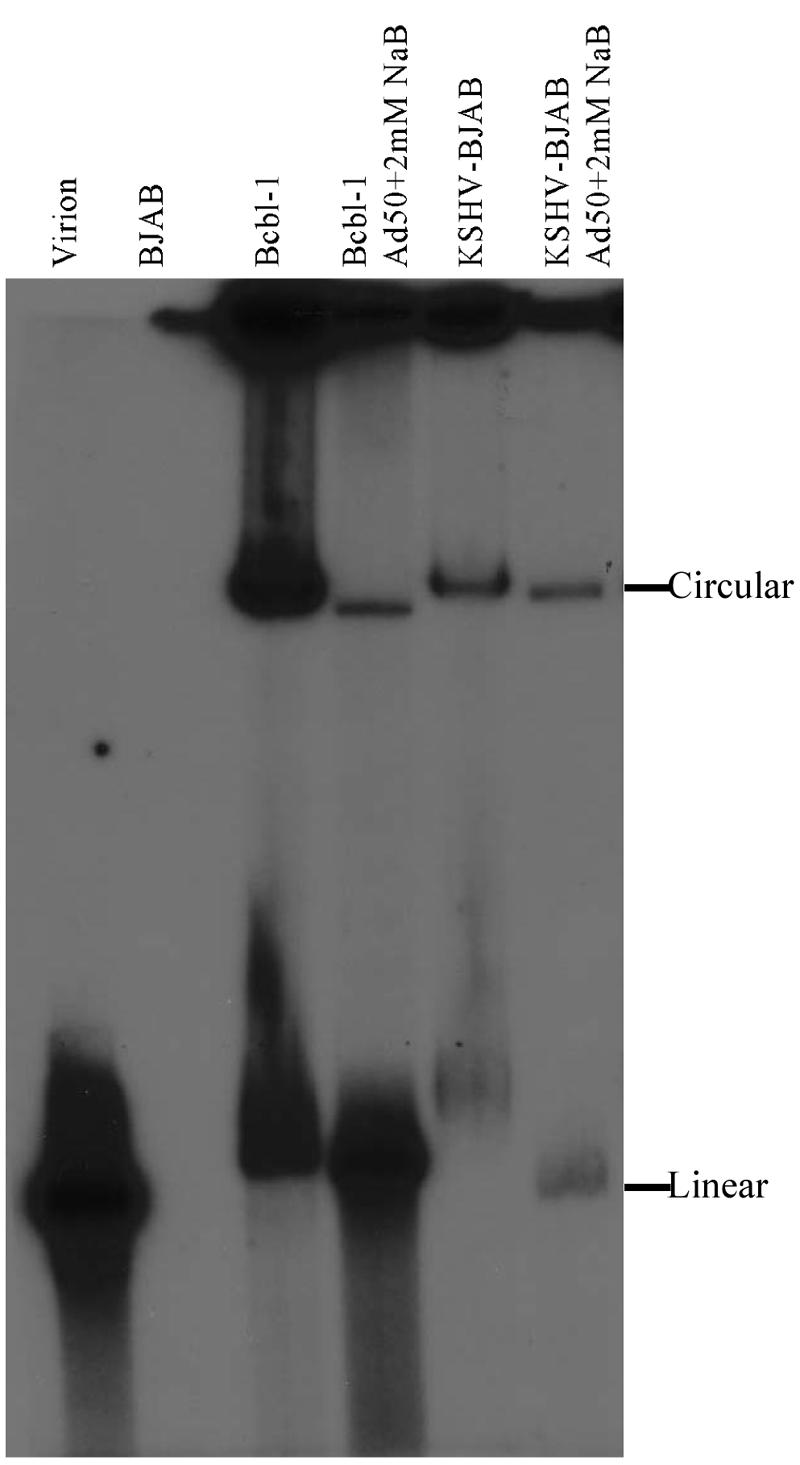
KSHV is episomal in KSHV-BJAB cells. BCBL-1 and KSHV-BJAB cells were induced with Adeno-50 (Ad50) and 2 mM sodium butyrate (NaB). Forty-eight hours after induction, uninduced and induced cells were examined by Gardella gel electrophoresis to detect KSHV episomes. KSHV virion was used as a control. 32P-labeled full-length vIL-6 was used as a probe.
In TIME cells and other cell types, the KSHV latent episome is lost from a large percentage of the cells (27, 32). If we expanded the KSHV-BJAB cells in the absence of puromycin selection, we found that the episome is lost in a significant percentage of the cells over the course of 1 month (data not shown). Two weeks after removal of the selection, approximately 70% of the cells were still GFP positive and stained positively for with the anti-LANA antibody (data not shown). After 1 month in culture without selection, approximately 50 to 60% of the cells remained GFP positive and expressed LANA (not shown).
Reactivation of latency in B cells.
To determine if the transfected BJAB cells established and maintained a productive latent infection, specific stimuli were used to reactivate the virus from the infected cells. Before induction, slightly less than 0.1% of KSHV-BJAB cells stained positive for ORF59, a marker of lytic replication. After induction with TPA, there was a fivefold increase in the number of cells staining positive for ORF 59 as compared to matched uninduced cells (Fig. 4). A 25- to 30-fold increase in ORF 59-positive cells was seen when cells were induced with a combination of sodium butyrate and superinfection with an adenovirus expressing ORF 50 (Fig. 4).
FIG. 4.

Photomicrographs of KSHV-BJAB cells 48 h postinduction showing 4′,6′-diamidino-2-phenylindole (DAPI) (left panel), eGFP fluorescence (middle panel), and ORF59 red fluorescence (right panel). BJAB cells (upper panel) were included as a negative control. Quantification is described in the text. Ad50, Adeno-50; NaB, sodium butyrate.
The induction of lytic replication resulted in an increase in the production of infectious virus. Seventy-two hours after different induction protocols of KSHV-BJAB cells, virus in the supernatant from each sample was used to infect TIME cells. Viral infection rates were determined by measuring the number of cells expressing GFP, indicative of the recombinant KSHV infection. As seen in Fig. 5, we could not detect GFP expression when supernatants from uninduced KSHV-BJAB cells were used as the inoculum. Virus isolated from supernatants of KSHV-BJAB induced with TPA yields on average 10 GFP-positive cells per 25 × 106 cells induced, while virus isolated from the supernatants of KSHV-BJAB induced with an adenovirus expressing ORF 50 and sodium butyrate yielded on average 3,000 GFP-positive cells per 25 × 106 cells induced (numbers are the average of three separate experiments). Interestingly, while consistent with the trend of lytic antigen IFA numbers, a fivefold increase in the number of cells staining with ORF 59 yields much more than a fivefold increase in infectious virus produced.
FIG. 5.

Recombinant KSHV virus reconstituted from induced KSHV-BJAB cells can be transmitted to hTERT-immortalized microvascular endothelial (TIME) cells. Phase (upper panel) and eGFP fluorescence (lower panel) are shown. Three days postinduction, supernatant was briefly centrifuged to remove cell debris and then filtered through a 0.45-μm-pore filter. The resulting supernatant was used to infect TIME cells on a 12-well plate. Twenty-four hours postinfection, phase and fluorescence pictures were taken. Supernatant from BJAB cells was used to infect TIME cells (left panel) as a negative control. TIME cells infected with supernatants from uninduced (middle-left panel), TPA-induced (middle-right panel) and Adeno-50 (Ad50) plus 2 mM sodium butyrate (NaB)-induced (right panel) KSHV-BJAB cells are shown. Quantification is described in the text.
Viral gene expression in B cells.
To determine if KSHV-BJAB cells have similar gene expression patterns as PEL cells, Northern blot analysis was done to probe for the expression of representative latent and lytic genes. Polyadenylated RNA was isolated from uninduced and Adeno-50 plus 2 mM sodium butyrate-induced KSHV-BJAB cells and BCBL-1 cells. The RNA was then electrophoresed on a Northern gel and probed with sequences to the viral cyclin that recognize all the transcripts from the LANA locus. As seen in Fig. 6A, there is little or no increase in the two major transcripts from the LANA locus after Adeno-50 plus 2 mM sodium butyrate induction of KSHV-BJAB cells, while there is a slight induction in the same transcripts in BCBL-1 cells. However, when the same blot was probed with sequences to the PAN RNA (also known as T1.1 or Nut-1), there was a massive increase in RNA in both the BCBL-1 cells and the KSHV-BJAB cells, indicative of the induction of lytic replication. Interestingly, in the KSHV-BJAB cells, the PAN RNA could not be detected in uninduced cells but was easily detected following lytic induction (Fig. 6A). We probed a similar Northern blot with vIL-6 sequences and obtained similar results to the Northern blot probed with the PAN probe; it was not detectable prior to induction but was induced to high levels upon induction (Fig. 6B). Thus in the KSHV-BJAB cells and BCBL-1 cells, the latent transcripts are not strongly increased upon induction while as expected lytic gene expression is greatly increased following TPA or Adeno-50 induction. Importantly, as seen with immunofluorescence with the ORF 59 antibodies, there is a much lower level of spontaneous lytic reactivation in the KSHV-BJAB cells. Importantly, since in KSHV-BJAB cells, vIL-6 cannot be detected until after induction of lytic replication, the insertion of the GFP-puromycin resistance gene downstream of the polyadenylation signal of vIL-6 does not alter its transcription pattern.
FIG. 6.

Northern blot analysis of uninduced and induced KSHV-BJAB cells. BCBL-1 cells and KSHV-BJAB cells were induced by Adeno-50 (Ad50) plus 2 mM sodium butyrate (NaB). Forty-eight hours postinduction, mRNA was extracted and analyzed. BJAB cells were included as a negative control. Uninduced and induced BCBL-1 cells were included as a positive control. (A) Northern blot probed with radiolabeled sequences containing viral cyclin (vCyclin), PAN RNA, and GAPDH consecutively. (B) Northern blot probed with vIL-6 and GAPDH consecutively.
DISCUSSION
A major conundrum of KSHV is that in vivo the virus clearly infects and establishes latency in B cells, but B cells tested in culture are refractory to KSHV infection (6, 28, 43). This report demonstrates the first example of rapid establishment of high-level KSHV infection in cultured B cells. While we were unable to directly infect BJAB cells with wild-type or recombinant virus, we were able to establish and maintain latent KSHV infection via direct introduction of viral DNA. While the efficiency of establishment in the BJAB cells seems relatively low, this is more likely due to the methods of transduction and not the difficulty in establishing latency upon introduction of naked viral DNA. Less than 1% of the transduced BJAB cells expressed GFP at 48 h. However, under the conditions we used, only a very low percentage of BJAB cells are transduced by electroporation of a small plasmid. Large plasmids are notoriously harder to transduce than smaller plasmids, and electroporation of a 165-kb plasmid would be even less efficient. Furthermore, the episome extracts contain large amounts of chromosomal DNA contaminants; thus much of what we transduce is chromosomal DNA and would interfere with transfection of the viral episome. Therefore, the fact that we can easily grow large numbers of KSHV-infected BJAB cells after only 1 week of selection indicates that the efficiency of establishment of latency is probably relatively high.
While there was only a very low level of spontaneous lytic replication in the KSHV-BJAB cells, lytic replication could be induced in the infected BJAB cells and virus produced from induced cells could infect other cell types. Importantly, latent infection of BJAB cells had similar viral gene expression pattern as the primary effusion lymphoma cell line BCBL-1, with the noted exception that there are much lower levels of spontaneous lytic replication. Because of the lower levels of spontaneous lytic replication, lytic transcripts could not be detected without the induction of lytic replication with drugs or ORF 50 expression.
This report demonstrates that B cells can support latent KSHV infection. Thus the block in the establishment of infection in B cells in culture is not due to the inability of these cells to express viral genes. One possible reason for the lack of infection is that the virus is unable to bind or enter B cells in culture. It may be possible that when cultured, primary B cells and B-cell lines lose a surface marker necessary for KSHV binding or entry. Akula et al. identified one receptor for KSHV as the α3β1 integrin (1). The same group also showed that BJAB cells have high levels of this integrin (1). If this integrin can act as the receptor in B cells, this would indicate that the block to viral infection is postentry. Studies from Akula et al. showed by electron microscopy that when they infected BJAB cells KSHV viral particles could enter the cells via endocytosis (2). However, it is not clear if the virus escapes the endosome or if this is not a productive route of infection. Interestingly, the same cells also had low levels of the αvβ3 integrin, which has also been proposed to behave as a KSHV receptor (1; T. Rose, personal communication). Studies are under way to determine where the block in replication of KSHV occurs in BJAB cells. If it is the case that KSHV binds and enters BJAB cells, then the block to infection could be at a number of postentry points: escape from the endosome, transport to the nuclear membrane, DNA extrusion across the nuclear membrane, or DNA circularization. Importantly, the studies here show that if the viral DNA gains access to the nucleus, it is competent to establish latency. Furthermore, the latent state can be maintained for longer than 3 months, and KSHV can be efficiently reactivated from latency and used to infect other cell types.
Like other cells infected in culture, the infected BJAB cells slowly lose the viral episome when passaged regularly. It was previously shown that TIME cells, 293 cells, SLK cells, and human foreskin fibroblast cells lose the viral episome when split regularly (27, 32). Primary dermal microvascular cells also lose the episome slowly when split regularly (27; P. A. Carroll and M. Lagunoff, unpublished). When split only once a week or less, primary dermal microvascular endothelial cells can maintain the episome, indicating that the low level of spontaneous lytic replication in the cells is enough to reinfect the cells that lose the episome when they divide or simply that when there is less cell division cells lose the virus more slowly (40). However, if the cells are split often, there is not enough virus in the supernatant and the cells slowly lose the episome. In SLK cells, while the episome is lost in the bulk of the population, a low percentage of cells maintain LANA expression, indicating that the virus is never completely lost from the bulk culture. BJAB cells transfected with KSHV and then selected for 100% of the cells to have the episome lose the episome when the selection medium is removed, albeit more slowly than other cell types (Chen and Lagunoff unpublished). Grundhoff et al. found that when they expressed both the latent origin of replication and LANA, the viral protein that maintains the episome in BJAB cells, the plasmid was lost at the same rate in culture as in other cells (27). Interestingly, when KS tumor spindle cells are isolated, they also lose the episome when cultured ex vivo. This is in stark contrast to PEL cells. These lymphomas of B-cell origin maintain the KSHV episome with no apparent loss of episome even when constantly passaged for over 1 year (Lagunoff, unpublished). Perhaps it is due to the much higher levels of spontaneous lytic reactivation seen in PEL cell lines as compared to our KSHV-BJAB cells, or perhaps KSHV-induced lymphomas acquire other mutations that allow the better maintenance of KSHV. The acquired mutations would have to be cellular mutations as the origin of the recombinant virus we used to infect BJAB cells is the primary effusion lymphoma line, BCBL-1.
The establishment of latent KSHV infection by introduction of naked KSHV DNA provides a simple, tractable model to study the effects of KSHV infection on B cells in culture. Previous analysis of KSHV in B cells relied on analysis of primary effusion lymphomas that were isolated as human tumors already infected with KSHV. Thus, there are no proper controls for uninfected PEL lines. Therefore, analysis of PELs compared them only to other types of B-cell lymphomas, never to a matched uninfected control. The studies presented here provide a system to analyze the changes in EBV-negative B cells following establishment of latency. Recent advances in our ability to create recombinant viruses have led to the need for a method to examine recombinants in different cell types. As demonstrated in this paper, we are now able to analyze KSHV mutants in B cells. This system will allow us to begin to understand how specific viral genes affect B cells as well as other relevant cell types like endothelial cells. Furthermore, since we can establish and maintain KSHV latency in B cells, we can now dissect the block to B-cell infection in culture.
Acknowledgments
We thank Almira Punjabi for critically reading the manuscript.
M.L. is supported by the PEW scholars program in the biological sciences sponsored by the PEW charitable trust and by a grant (5RO1CA97934-03) from the National Cancer Institute.
REFERENCES
- 1.Akula, S. M., N. P. Pramod, F. Z. Wang, and B. Chandran. 2002. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 108:407-419. [DOI] [PubMed] [Google Scholar]
- 2.Akula, S. M., F. Z. Wang, J. Vieira, and B. Chandran. 2001. Human herpesvirus 8 interaction with target cells involves heparan sulfate. Virology 282:245-255. [DOI] [PubMed] [Google Scholar]
- 3.Ambroziak, J. A., D. J. Blackbourn, B. G. Herndier, R. G. Glogau, J. H. Gullett, A. R. McDonald, E. T. Lennette, and J. A. Levy. 1995. Herpes-like sequences in HIV-infected and uninfected Kaposi's sarcoma patients. Science 268:582-583. [DOI] [PubMed] [Google Scholar]
- 4.Ansari, M. Q., D. B. Dawson, R. Nador, C. Rutherford, N. R. Schneider, M. J. Latimer, L. Picker, D. M. Knowles, and R. W. McKenna. 1996. Primary body cavity-based AIDS-related lymphomas. Am. J. Clin. Pathol. 105:221-229. [DOI] [PubMed] [Google Scholar]
- 5.Arvanitakis, L., E. A. Mesri, R. G. Nador, J. W. Said, A. S. Asch, D. M. Knowles, and E. Cesarman. 1996. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood 88:2648-2654. [PubMed] [Google Scholar]
- 6.Bechtel, J. T., Y. Liang, J. Hvidding, and D. Ganem. 2003. Host range of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 77:6474-6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blackbourn, D. J., E. Lennette, B. Klencke, A. Moses, B. Chandran, M. Weinstein, R. G. Glogau, M. H. Witte, D. L. Way, T. Kutzkey, B. Herndier, and J. A. Levy. 2000. The restricted cellular host range of human herpesvirus 8. AIDS 14:1123-1133. [DOI] [PubMed] [Google Scholar]
- 8.Boshoff, C., S. J. Gao, L. E. Healy, S. Matthews, A. J. Thomas, L. Coignet, R. A. Warnke, J. A. Strauchen, E. Matutes, O. W. Kamel, P. S. Moore, R. A. Weiss, and Y. Chang. 1998. Establishing a KSHV+ cell line (BCP-1) from peripheral blood and characterizing its growth in Nod/SCID mice. Blood 91:1671-1679. [PubMed] [Google Scholar]
- 9.Cannon, J. S., D. Ciufo, A. L. Hawkins, C. A. Griffin, M. J. Borowitz, G. S. Hayward, and R. F. Ambinder. 2000. A new primary effusion lymphoma-derived cell line yields a highly infectious Kaposi's sarcoma herpesvirus-containing supernatant. J. Virol. 74:10187-10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capello, D., G. Gaidano, M. Gallicchio, A. Gloghini, E. Medico, D. Vivenza, D. Buonaiuto, L. Fassone, G. C. Avanzi, G. Saglio, M. Prat, and A. Carbone. 2000. The tyrosine kinase receptor met and its ligand HGF are co-expressed and functionally active in HHV-8 positive primary effusion lymphoma. Leukemia 14:285-291. [DOI] [PubMed] [Google Scholar]
- 11.Carbone, A., A. Gloghini, M. R. Cozzi, D. Capello, A. Steffan, P. Monini, L. De Marco, and G. Gaidano. 2000. Expression of MUM1/IRF4 selectively clusters with primary effusion lymphoma among lymphomatous effusions: implications for disease histogenesis and pathogenesis. Br. J. Haematol. 111:247-257. [DOI] [PubMed] [Google Scholar]
- 12.Carroll, P. A., E. Brazeau, and M. Lagunoff. 2004. Kaposi's sarcoma-associated herpesvirus infection of blood endothelial cells induces lymphatic differentiation. Virology 328:7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cesarman, E., Y. Chang, P. S. Moore, J. W. Said, and D. M. Knowles. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186-1191. [DOI] [PubMed] [Google Scholar]
- 14.Cesarman, E., P. S. Moore, P. H. Rao, G. Inghirami, D. M. Knowles, and Y. Chang. 1995. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood 86:2708-2714. [PubMed] [Google Scholar]
- 15.Cesarman, E., R. G. Nador, K. Aozasa, G. Delsol, J. W. Said, and D. M. Knowles. 1996. Kaposi's sarcoma-associated herpesvirus in non-AIDS related lymphomas occurring in body cavities. Am. J. Pathol. 149:53-57. [PMC free article] [PubMed] [Google Scholar]
- 16.Chang, J., R. Renne, D. Dittmer, and D. Ganem. 2000. Inflammatory cytokines and the reactivation of Kaposi's sarcoma-associated herpesvirus lytic replication. Virology 266:17-25. [DOI] [PubMed] [Google Scholar]
- 17.Chang, Y., E. Cesarman, M. S. Pessin, F. Lee, J. Culpepper, D. M. Knowles, and P. S. Moore. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865-1869. [DOI] [PubMed] [Google Scholar]
- 18.Chuck, S., R. M. Grant, E. Katongole-Mbidde, M. Conant, and D. Ganem. 1996. Frequent presence of a novel herpesvirus genome in lesions of human immunodeficiency virus-negative Kaposi's sarcoma. J. Infect. Dis. 173:248-251. [DOI] [PubMed] [Google Scholar]
- 19.Ciufo, D. M., J. S. Cannon, L. J. Poole, F. Y. Wu, P. Murray, R. F. Ambinder, and G. S. Hayward. 2001. Spindle cell conversion by Kaposi's sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J. Virol. 75:5614-5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis, D. A., A. S. Rinderknecht, J. P. Zoeteweij, Y. Aoki, E. L. Read-Connole, G. Tosato, A. Blauvelt, and R. Yarchoan. 2001. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood 97:3244-3250. [DOI] [PubMed] [Google Scholar]
- 21.Dittmer, D., M. Lagunoff, R. Renne, K. Staskus, A. Haase, and D. Ganem. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 72:8309-8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drexler, H. G., C. C. Uphoff, G. Gaidano, and A. Carbone. 1998. Lymphoma cell lines: in vitro models for the study of HHV-8+ primary effusion lymphomas (body cavity-based lymphomas). Leukemia 12:1507-1517. [DOI] [PubMed] [Google Scholar]
- 23.Flore, O., S. Rafii, S. Ely, J. J. O'Leary, E. M. Hyjek, and E. Cesarman. 1998. Transformation of primary human endothelial cells by Kaposi's sarcoma-associated herpesvirus. Nature 394:588-592. [DOI] [PubMed] [Google Scholar]
- 24.Ganem, D. 1997. KSHV and Kaposi's sarcoma: the end of the beginning? Cell 91:157-160. [DOI] [PubMed] [Google Scholar]
- 25.Gao, S. J., L. Kingsley, M. Li, W. Zheng, C. Parravicini, J. Ziegler, R. Newton, C. R. Rinaldo, A. Saah, J. Phair, R. Detels, Y. Chang, and P. S. Moore. 1996. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi's sarcoma. Nat. Med. 2:925-928. [DOI] [PubMed] [Google Scholar]
- 26.Gardella, T., P. Medveczky, T. Sairenji, and C. Mulder. 1984. Detection of circular and linear herpesvirus DNA molecules in mammalian cells by gel electrophoresis. J. Virol. 50:248-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grundhoff, A., and D. Ganem. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Investig. 113:124-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henry, M., A. Uthman, A. Geusau, A. Rieger, L. Furci, A. Lazzarin, P. Lusso, and E. Tschachler. 1999. Infection of circulating CD34+ cells by HHV-8 in patients with Kaposi's sarcoma. J. Investig. Dermatol. 113:613-616. [DOI] [PubMed] [Google Scholar]
- 29.Hong, Y. K., K. Foreman, J. W. Shin, S. Hirakawa, C. L. Curry, D. R. Sage, T. Libermann, B. J. Dezube, J. D. Fingeroth, and M. Detmar. 2004. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 36:683-685. [DOI] [PubMed] [Google Scholar]
- 30.Jenner, R. G., K. Maillard, N. Cattini, R. A. Weiss, C. Boshoff, R. Wooster, and P. Kellam. 2003. Kaposi's sarcoma-associated herpesvirus-infected primary effusion lymphoma has a plasma cell gene expression profile. Proc. Natl. Acad. Sci. USA 100:10399-10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kliche, S., E. Kremmer, W. Hammerschmidt, U. Koszinowski, and J. Haas. 1998. Persistent infection of Epstein-Barr virus-positive B lymphocytes by human herpesvirus 8. J. Virol. 72:8143-8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lagunoff, M., J. Bechtel, E. Venetsanakos, A.-M. Roy, N. Abbey, B. Herndier, M. McMahon, and D. Ganem. 2002. De novo infection and serial transmission of Kaposi's sarcoma-associated herpesvirus in cultured endothelial cells. J. Virol. 76:2440-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lagunoff, M., and D. Ganem. 1997. The structure and coding organization of the genomic termini of Kaposi's sarcoma-associated herpesvirus. Virology 236:147-154. [DOI] [PubMed] [Google Scholar]
- 34.Liang, Y., and D. Ganem. 2003. Lytic but not latent infection by Kaposi's sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc. Natl. Acad. Sci. USA 100:8490-8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lukac, D. M., J. R. Kirshner, and D. Ganem. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller, G., L. Heston, E. Grogan, L. Gradoville, M. Rigsby, R. Sun, D. Shedd, V. M. Kushnaryov, S. Grossberg, and Y. Chang. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71:314-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore, P. S., and Y. Chang. 1995. Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. N. Engl. J. Med. 332:1181-1185. [DOI] [PubMed] [Google Scholar]
- 38.Moses, A. V., K. N. Fish, R. Ruhl, P. P. Smith, J. G. Strussenberg, L. Zhu, B. Chandran, and J. A. Nelson. 1999. Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J. Virol. 73:6892-6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nador, R. G., E. Cesarman, A. Chadburn, D. B. Dawson, M. Q. Ansari, J. Sald, and D. M. Knowles. 1996. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood 88:645-656. [PubMed] [Google Scholar]
- 40.Poole, L. J., Y. Yu, P. S. Kim, Q.-Z. Zheng, J. Pevsner, and G. S. Hayward. 2002. Altered patterns of cellular gene expression in dermal microvascular endothelial cells infected with Kaposi's sarcoma-associated herpesvirus. J. Virol. 76:3395-3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pozharskaya, V. P., L. L. Weakland, and M. K. Offermann. 2004. Inhibition of infectious human herpesvirus 8 production by gamma interferon and alpha interferon in BCBL-1 cells. J. Gen. Virol. 85:2779-2787. [DOI] [PubMed] [Google Scholar]
- 42.Rainbow, L., G. M. Platt, G. R. Simpson, R. Sarid, S.-J. Gao, H. Stoiber, C. S. Herrington, P. S. Moore, and T. F. Schulz. 1997. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) is encoded by orf73 and is a component of the latency-associated nuclear antigen. J. Virol. 71:5915-5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Renne, R., D. Blackbourn, D. Whitby, J. Levy, and D. Ganem. 1998. Limited transmission of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 72:5182-5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Renne, R., W. Zhong, B. Herndier, M. McGrath, N. Abbey, D. Kedes, and D. Ganem. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342-346. [DOI] [PubMed] [Google Scholar]
- 45.Rivas, C., A.-E. Thlick, C. Parravicini, P. S. Moore, and Y. Chang. 2001. Kaposi's sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 75:429-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sadler, R., L. Wu, B. Forghani, R. Renne, W. Zhong, B. Herndier, and D. Ganem. 1999. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi's sarcoma-associated herpesvirus. J. Virol. 73:5722-5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schalling, M., M. Ekman, E. E. Kaaya, A. Linde, and P. Biberfeld. 1995. A role for a new herpes virus (KSHV) in different forms of Kaposi's sarcoma. Nat. Med. 1:707-708. [DOI] [PubMed] [Google Scholar]
- 48.Simpson, K., and C. Huxley. 1996. A shuttle system for transfer of YACs between yeast and mammalian cells. Nucleic Acids Res. 24:4693-4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soulier, J., L. Grollet, E. Oksenhendler, P. Cacoub, D. Cazals-Hatem, P. Babinet, M. F. d'Agay, J. P. Clauvel, M. Raphael, L. Degos et al. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276-1280. [PubMed] [Google Scholar]
- 50.Staskus, K. A., W. Zhong, K. Gebhard, B. Herndier, H. Wang, R. Renne, J. Beneke, J. Pudney, D. J. Anderson, D. Ganem, and A. T. Haase. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71:715-719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suscovich, T. J., M. Paulose-Murphy, J. D. Harlow, Y. Chen, S. Y. Thomas, T. J. Mellott, B. D. Walker, D. T. Scadden, S. Zeichner, and C. Brander. 2004. Defective immune function of primary effusion lymphoma cells is associated with distinct KSHV gene expression profiles. Leuk. Lymphoma 45:1223-1238. [DOI] [PubMed] [Google Scholar]
- 53.Venetsanakos, E., A. Mirza, C. Fanton, S. R. Romanov, T. Tlsty, and M. McMahon. 2002. Induction of tubulogenesis in telomerase-immortalized human microvascular endothelial cells by glioblastoma cells. Exp. Cell Res. 273:21-33. [DOI] [PubMed] [Google Scholar]
- 54.Vieira, J., and P. M. O'Hearn. 2004. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology 325:225-240. [DOI] [PubMed] [Google Scholar]
- 55.Yu, Y., J. B. Black, C. S. Goldsmith, P. J. Browning, K. Bhalla, and M. K. Offermann. 1999. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J. Gen. Virol. 80:83-90. [DOI] [PubMed] [Google Scholar]
- 56.Zhong, W., H. Wang, B. Herndier, and D. Ganem. 1996. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. USA 93:6641-6646. [DOI] [PMC free article] [PubMed] [Google Scholar]