

Abstract
Previous studies on the self-assembly of capsid protein CA of human immunodeficiency virus type 1 (HIV-1) in vitro have provided important insights on the structure and assembly of the mature HIV-1 capsid. However, CA polymerization in vitro was previously observed to occur only at very high ionic strength. Here, we have analyzed the effects on CA assembly in vitro of adding unrelated, inert macromolecules (crowding agents), aimed at mimicking the crowded (very high macromolecular effective concentration) environment within the HIV-1 virion. Crowding agents induced fast and efficient polymerization of CA even at low (close to physiological) ionic strength. The hollow cylinders thus assembled were indistinguishable in shape and dimensions from those formed in dilute protein solutions at high ionic strength. However, two important differences were noted: (i) disassembly by dilution of the capsid-like particles was undetectable at very high ionic strength, but occurred rapidly at low ionic strength in the presence of a crowding agent, and (ii) a variant CA from a presumed infectious HIV-1 with mutations at the CA dimerization interface was unable to assemble at any ionic strength in the absence of a crowding agent; in contrast, this mutation allowed efficient assembly, even at low ionic strength, when a crowding agent was used. The use of a low ionic strength and inert macromolecules to mimic the crowded environment inside the HIV-1 virion may lead to a better in vitro evaluation of the effects of conditions, mutations or/and other molecules, including potential antiviral compounds, on HIV-1 capsid assembly, stability and disassembly.
The immature human immunodeficiency virus type 1 (HIV-1) particle contains, underneath the viral membrane, a spherical shell formed by about 4,000 to 5,000 copies of the polyprotein Gag. As the virion buds from the cell, or after budding, Gag is cleaved by the viral protease releasing the matrix (MA), capsid (CA), and nucleocapsid (NC) polypeptides. This event triggers a major structural rearrangement, termed maturation, which involves condensation of NC and the viral RNA, and assembly of CA into a cone-shaped shell (the mature capsid) surrounding the NC-RNA complex (reviewed in references 13, 17, 38, and 39). Maturation involves dissociation of the Gag shell; recent experimental evidence shows that formation of the mature capsid does not proceed through condensation of the CA portion of the Gag lattice. Instead, it requires a de novo CA assembly process occurring at a very high protein concentration in the confined space inside the extracellular virion (1-3).
The study of the HIV-1 mature capsid structure, assembly and disassembly has been greatly facilitated by the capacity of CA to self-assemble in vitro. When incubated with RNA at pH 8, the polyprotein CA-NC polymerizes into hollow tubular structures at low (0.1 M) NaCl concentrations (4), or a mixture of conical and cylindrical structures at high (0.5 M) salt concentrations (15). In the absence of RNA, CA-NC can also form tubes and cones, but only at very high ionic strength (15). Although there is evidence that the RNA does not play a structural role in the assembled particle, the molecular basis for the polymerization-enhancing effect of either RNA or a high ionic strength is unclear. The capsid protein CA alone can also self-assemble into cylindrical and, in some cases, conical structures in vitro although, again, a very high ionic strength (1 to 2.25 M NaCl) had to be used for achieving polymerization, even at the highest protein concentrations tested (9, 19, 20, 25, 26, 40).
Cryo-electron microscopy revealed that the cylindrical CA structures assembled in vitro are composed of helical arrays of CA hexamers (26). Similar arrays of CA hexamers were observed in images of authentic cylindrical cores isolated from HIV-1 virions (3), indicating that CA forms similar assemblies in vitro and in vivo. Moreover, electron diffraction patterns obtained with conical or cylindrical viral capsids shared common reflections, indicating that both cones and cylinders utilize the same CA lattice and, therefore, that the cylinders formed in vitro are reasonable models for mature capsid assembly in vivo (3). Mutational analyses are consistent with the idea that in vitro cylinder assembly accurately mimics some, though not all, structural aspects of mature capsid assembly in vivo (16, 41).
The availability of an in vitro capsid self-assembly system using CA only is proving invaluable also for the dissection of the effects of experimental conditions, mutations in CA, and/or the presence of other molecules, including potential antiviral drugs, on HIV-1 capsid assembly, stability or disassembly (for examples, see references 5, 8, 10, 16, 24, 25, 29, 37, and 40). However, the relatively low CA concentrations used and, perhaps even more importantly, the very high ionic strength needed for polymerization in vitro, are clearly nonphysiological. A very high ionic strength could alter or distort some effects of physical or chemical agents or mutations. In addition, the most recent determinations (1, 2) indicate that the total CA (monomer) concentration inside the virion would reach 7 to 8 mM, more than one or two orders of magnitude higher than the concentrations used in in vitro assays. Moreover, the presence of very high concentrations of CA and several other proteins and of the viral RNA means that the interior of the viral particle is a molecularly crowded environment, similar in this regard to any subcellular compartment within a living cell.
Cellular compartments contain macromolecules at such high (about 50 to 400 g/liter) total concentrations that the macromolecular species occupy a large fraction of the total internal volume. Such media are referred to as “crowded.” In such conditions, the non-specific steric repulsions may significantly influence specific biochemical and biophysical processes through an increase in the chemical activity (“effective concentration”) of the macromolecules involved, and/or other physicochemical effects. Crowding can be mimicked in vitro by adding high concentrations of inert natural or synthetic polymers, termed crowding agents, to the system analyzed. Experimental work using crowding agents, as well as theoretical studies, has demonstrated substantial (order of magnitude) effects of crowding on a wide range of processes, especially the thermodynamics and kinetics of protein assembly (reviewed in references 11, 21, 30, 32, 33, 36, and 43).
In this work, we have analyzed the effects of crowding agents, aimed at mimicking the crowded environment within the HIV-1 virion, on the in vitro assembly and disassembly of nonmutant and mutant CA from HIV-1. The results extend the analyses of in vitro CA assembly carried out so far to conditions much closer to those prevailing inside the HIV virion.
MATERIALS AND METHODS
Plasmids and site-directed mutagenesis.
Construction of the recombinant expression plasmids containing the nucleotide sequences of the full-length CA and of its C-terminal domain (CA-C) corresponding to the HIV-1 strain BH10 has been previously described (5, 28). Mutagenesis of both CA and CA-C was carried out using the QuikChange Site-directed Mutagenesis kit (Stratagene). The mutations introduced were confirmed by sequencing the entire CA- or CA-C-coding regions.
Protein expression and purification.
The nonmutant and mutant CA protein and isolated CA-C domain were expressed in Escherichia coli BL21(DE3) and cell extracts obtained using procedures already described (5, 28). The CA protein was purified using a modification of a previously published procedure (25). Briefly, the CA protein in clarified cell extracts was precipitated with ammonium sulfate to 25% saturation and purified by ion-exchange chromatography in Q-Sepharose (Amersham Biotech). The fractions containing CA were mixed, dialyzed against a buffer containing 50 mM sodium phosphate (pH 8.0) and 150 mM NaCl, applied to a 2.6-cm by 53-cm Superdex 75 (Amersham Biotech) column, and eluted with the same buffer. Fractions containing CA were pooled and dialyzed against 50 mM sodium phosphate (pH 8.0) and kept frozen at −70°C until use. The isolated CA-C domain was purified from clarified cell extracts as described previously (28) by using ammonium sulfate precipitation, ion-exchange chromatography in SP-Sepharose, affinity chromatography in heparin-Sepharose and size exclusion chromatography in Superdex 75 (all media from Amersham Biotech). Purified nonmutant and mutant CA and CA-C proteins were run in overloaded SDS-PAGE gels and found to be free of contaminants. The CA concentrations are given in this work as total protein monomer concentrations and were determined by UV-spectrophotometry (27), using molar extinction coefficients at 280 nm of 33,254 M−1cm−1 or 8,530 M−1cm−1 for CA or CA-C, respectively.
Kinetic analysis of CA polymerization in vitro.
Assembly reactions were carried out at different ionic strengths (150 mM to 2.25 M NaCl), pHs (7.0 to 7.8) and protein concentrations (5 to 600 μM), either in the absence or presence of crowding agents (either Ficoll 70 or dextran 10, Amersham Biosciences), with average molar masses of 70,000 and 10,000, respectively, at final concentrations of up to 250 g/liter. The assays in the absence of crowding agents were carried out essentially as previously described (25). A volume of CA solution at the appropriate concentration in 50 mM sodium phosphate buffer was introduced into a spectrophotometer cuvette (10-mm by 2-mm internal section), and the assembly reaction was triggered by adding a concentrated solution of NaCl or/and a crowding agent in 50 mM sodium phosphate buffer at the specified pH to get the final concentrations desired for each component in a final volume of 500 μl, followed by rapid mixing by repeated inversion of the cuvette. The pH of the final reaction mixture was checked. The time-dependent increase in the optical density at 350 nm as a measure of the light scattered by the assembled particles was monitored at 25°C using a Shimadzu UV-1603 spectrophotometer, with data points generally collected every six seconds. When very high protein concentrations were used, the turbidity value was far too high to be directly measured. In this case, samples were taken at defined intervals, immediately diluted 50- or 100-fold in the same reaction buffer, and rapidly measured in a spectrophotometer. In control assays, it was observed that the optical density decreased as a function of the time elapsed between dilution and the actual measurement. Thus, the measurements were taken at a very short time after dilution, when no significant decrease in the optical density value had yet occurred. In all polymerization conditions tested, the time-dependent increase in optical density fitted very well the empirical Hill function,
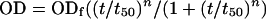 |
(1) |
where OD is the optical density at incubation time t, ODf is the optical density at infinite time, t50 is the time at which the OD is equal to one-half the ODf, and n is a cooperativity parameter (22).
For the determination of the CA critical concentration, the reaction was left to proceed until completion, the polymers formed were collected by centrifugation in a microcentrifuge at 12,000 rpm for 15 min, resuspended in 500 μl of 50 mM sodium phosphate buffer, pH 8.0, containing 3 M guanidinium hydrochloride (Pierce), and the protein concentration determined by spectrophotometry (27), using an extinction coefficient of 33,704 M−1 cm−1. The amount of polymer formed was plotted against the total concentration of protein CA in the assay. Linear extrapolation of the data gave the critical concentration of CA (the minimum concentration needed for polymerization to occur) in the conditions of the assay.
For inhibition assays, CA was mixed with the appropriate amounts of the isolated CA-C domain (previously dialyzed against the buffer used for full-length CA), and incubated for 30 min before triggering the reaction as indicated above.
Kinetic analysis of CA disassembly in vitro.
CA polymers assembled as described above were diluted 10-fold in the reaction buffer while preserving the pH, ionic strength and, if present, the crowding agent concentration. The kinetics of particle dissociation triggered by dilution was determined at 25°C by following the decrease in the optical density at 350 nm, as a measure of the remaining light-scattering particles. Data points were collected generally every 6 s. The time-dependent decrease in optical density at 350 nm was fitted to single- or double-exponential functions. The data fitted very well a double-exponential decay,
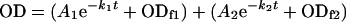 |
(2) |
where OD is the optical density at 350 nm at time t, A1 and A2 are the amplitudes, k1 and k2 are the dissociation rate constants, ODf1 and ODf2 are the optical density values at infinite time, and the subscripts 1 and 2 respectively correspond to the two postulated reactions.
Electron microscopy.
The CA polymers assembled as described above were used undiluted, or, when very high CA and crowding agent concentrations were used for polymerization, diluted 1/5 in reaction buffer. The polymers were deposited on ionized Formvar/carbon-coated copper grids, fixed for 30 s, negatively stained with 2% (wt/vol) uranyl acetate (Fluka) for 30 s (or 60 s if Ficoll was present in the sample), dried, and visualized in JEM-1010 or JEM-1200 (JEOL) electron microscopes at magnifications of ×50,000 or ×80,000. The dimensions of the tubular structures formed correspond to an average of 20 measurements on the micrographs obtained.
Fluorescence spectroscopy.
A Cary Eclipse (Varian) luminescence spectrophotometer, equipped with a temperature-control unit, was used to obtain intrinsic Trp fluorescence spectra at 25°C as previously described in detail (28), except that an excitation wavelength of 295 nm was used.
Analytical gel filtration and analytical ultracentrifugation.
The dimerization equilibrium of isolated CA-C carrying different mutations was investigated by analytical gel filtration exactly as previously described for nonmutant CA-C and other mutants (6). Sedimentation equilibrium experiments of nonmutant and mutant CA-C were performed at 20°C in an Optima XL-A (Beckman) analytical ultracentrifuge equipped with UV-visible optics, using an An50Ti rotor. CA-C samples (75 μl) at different protein concentrations in 25 mM sodium phosphate (pH 7.3), 150 mM NaCl were applied and centrifuged at 20,000 and 30,000 rpm. Baseline offsets were determined afterwards by high-speed sedimentation at 50,000 rpm. Whole-cell apparent weight average molecular weights of CA-C were determined by fitting the equation that describes the radial distribution of concentration gradients of single sedimenting species at sedimentation equilibrium to the experimental data using the program EQASSOC (see reference 31). The partial specific volume of CA-C was calculated from the amino acid composition of CA-C, using the public domain program SEDNTERP.
RESULTS
Effect of crowding agents on assembly of the HIV-1 capsid protein in vitro.
Conditions for efficient assembly of CA of HIV-1 strain BH10 in an in vitro polymerization kinetics assay (25) were first explored in the absence of any crowding agent, using protein (monomer) concentrations in the 1 to 100 μM range at different pHs and ionic strengths (Fig. 1, Table 1, and data not shown). The time-dependent formation of light-scattering CA polymers fitted well the Hill function (equation 1). At close to physiologic ionic strength (150 mM NaCl) no assembly was detected, even after many hours of incubation at high protein concentrations. At an ionic strength as high as 1.75 M NaCl, assembly was still very slow; at 2.25 M NaCl, assembly was efficient and relatively fast (Fig. 1A and Table 1). The assembly rate was also pH dependent, being severalfold higher at pH 7.8 than at pH 7.0 (Fig. 1A and Table 1), and both the rate and the amount of polymer formed increased as expected with the concentration of CA (Fig. 1B and Table 1). According to the empirical Hill analysis (equation 1), CA polymerization was clearly cooperative, with a best-fit cooperativity parameter substantially larger than 1 (Table 1). At 2.25 M NaCl and pH 7.8 the critical CA concentration was about 5.6 μM (Fig. 1C). Transmission electron microscopy images of the polymeric structures formed revealed the presence of hollow cylinders of about 44 ± 6 nm (external diameter) (Fig. 2A). The above results, when directly comparable, are similar to those obtained previously (25) for CA of another HIV variant (strain NY5) which differed from the strain BH10 CA used here in several amino acid residues located in the N- or C-terminal domains.
FIG. 1.

In vitro polymerization of CA at different salt and protein concentrations and pHs, in the absence of a crowding agent. (A) The assay conditions were: circles, 150 mM NaCl, 66 μM CA, pH 7.8; squares, 1.75 M NaCl, 15 μM CA, pH 7.8; triangles, 2.25 M NaCl, 15 μM CA, pH 7.8; diamonds, 2.25 M NaCl, 44 μM CA, pH 7.8; inverted triangles, 2.25 M NaCl, 44 μM CA, pH 7.0. (B) The assay conditions were: pH 7.8, 2.25 M NaCl and the following CA concentrations: circles, 5 μM; squares, 10 μM; triangles, 15 μM; inverted triangles, 22 μM; diamonds, 44 μM. For clarity, in panels A and B only one in each 30 data points are represented (one in each five points for the first minutes at 22 μM or 44 μM CA). Continuous lines represent the best fit to equation 1. (C) amount of CA polymers formed in the experiment shown in panel B, as a function of the total CA concentration. Linear extrapolation of the data yielded the CA critical concentration.
TABLE 1.
Values of kinetic parameters for CA polymerization in vitro in different conditions, in either the absence or presence of molecular crowding agents
| Variable | pH | [NaCl] (M) | [CA]a (μM) | [Crowder] (g/liter) | t50b (min) | nb | Linear ratec (OD/min) |
|---|---|---|---|---|---|---|---|
| pH | 7.0 | 2.25 | 44 | 0 | 8.7 ± 0.02 | 2.8 ± 0.02 | 0.17 |
| 7.8 | 2.25 | 44 | 0 | 1.3 ± 0.01 | 3.1 ± 0.03 | 1.14 | |
| [NaCl] | 7.8 | 0.15 | 66 | 0 | —d | — | — |
| 7.8 | 0.75 | 15 | 0 | — | — | — | |
| 7.8 | 2.25 | 15 | 0 | 27.8 ± 0.02 | 5.3 ± 0.01 | 0.04 | |
| [CA] | 7.8 | 2.25 | 5 | 0 | — | — | — |
| 7.8 | 2.25 | 10 | 0 | — | — | — | |
| 7.8 | 2.25 | 15 | 0 | 27.8 ± 0.02 | 5.3 ± 0.01 | 0.04 | |
| 7.8 | 2.25 | 22 | 0 | 7.5 ± 0.01 | 4.4 ± 0.01 | 0.18 | |
| 7.8 | 2.25 | 44 | 0 | 1.3 ± 0.01 | 3.1 ± 0.03 | 1.14 | |
| [Crowder] | 7.8 | 2.25 | 15 | 0 | 27.8 ± 0.02 | 5.3 ± 0.01 | 0.04 |
| 7.8 | 2.25 | 15 | 25 Ficoll | 13.1 ± 0.01 | 4.7 ± 0.01 | 0.08 | |
| 7.8 | 2.25 | 15 | 50 Ficoll | 7.1 ± 0.01 | 4.6 ± 0.02 | 0.14 | |
| 7.8 | 2.25 | 15 | 100 Ficoll | 2.3 ± 0.02 | 2.6 ± 0.04 | 0.21 | |
| 7.8 | 2.25 | 15 | 100 dextran | 4.5 ± 0.01 | 3.1 ± 0.02 | 0.12 | |
| Mutation and crowder | |||||||
| High salt | 7.8 | 2.25 | 15 nonmutated | 0 | 27.8 ± 0.02 | 5.3 ± 0.01 | 0.04 |
| 7.8 | 2.25 | 15 mutant | 0 | — | — | — | |
| 7.8 | 2.25 | 15 nonmutated | 100 Ficoll | 2.3 ± 0.02 | 2.6 ± 0.04 | 0.21 | |
| 7.8 | 2.25 | 15 mutant | 100 Ficoll | 11.8 ± 0.02 | 3.2 ± 0.02 | 0.07 | |
| Low salt | 7.8 | 0.15 | 600 nonmutated | 0 | — | — | — |
| 7.8 | 0.15 | 600 mutant | 0 | — | — | — | |
| 7.8 | 0.15 | 600 nonmutated | 250 Ficoll | 1.1 ± 0.2 | 1.9 ± 0.6 | 8.6 | |
| 7.8 | 0.15 | 600 mutant | 250 Ficoll | 2.1 ± 0.4 | 1.7 ± 0.6 | 5.4 |
Total CA (monomer) concentration.
t50 is the time at which the optical density (OD) is equal to one-half the optical density extrapolated at infinite time and n is the cooperativity parameter. The values given were obtained by fitting the data to equation 1. The fitting errors are indicated.
The linear polymerization rate is the average increase in optical density per minute for the approximately linear part of the polymerization curve.
—, no significant polymerization observed.
FIG. 2.

Visualization by electron microscopy of CA polymers assembled in vitro. (A) Nonmutated CA polymers formed at 2.25 M NaCl in the absence of a crowding agent. (B) Nonmutated CA polymers formed in the experiment shown in Fig. 4 at 150 mM NaCl in the presence of 250 g/liter Ficoll as a crowding agent. Cylinders (generally forming bundles, which are not shown in the picture) were clearly dominant, but conical structures with about 100 nm length and 40 nm maximum width were also observed. (C) Mutant V181A/E180D CA polymers formed in the experiment shown in Fig. 9A at 150 mM NaCl in the presence of 250 g/liter Ficoll as a crowding agent. The scale bars represent a length of 100 nm.
The effects of two different crowding agents, Ficoll 70 and dextran 10, on the kinetics of CA polymerization were then investigated (Fig. 3 and Table 1), using first a relatively low CA concentration (15 μM), in conditions highly favorable for in vitro assembly (pH 7.8 and 2.25 M NaCl). In the absence of a crowding agent, polymerization was relatively slow. The addition of increasing concentrations of Ficoll accelerated CA polymerization dramatically (Fig. 3A and Table 1). The addition of dextran as a crowding agent instead of Ficoll had an equally remarkable polymerization-enhancing effect (Fig. 3A and Table 1). The critical concentration was reduced from about 5.6 μM in the absence of crowding agent to 3.1 μM or 2.3 μM in the presence of 100 g/liter Ficoll or dextran, respectively (Fig. 3B).
FIG. 3.

In vitro polymerization of CA in the presence of crowding agents. (A) the assay conditions were as follows: pH 7.8, 2.25 NaCl, 15 μM CA. The following crowding agent concentrations were used: circles, no crowding agent was added; triangles, 25 g/liter Ficoll; squares, 50 g/liter Ficoll; inverted triangles, 100 g/liter Ficoll; diamonds, 100 g/liter dextran. For clarity, only 1 in each 20 data points is represented (one in each five points for the first minutes at 50 g/liter or 100 g/liter Ficoll). Continuous lines represent the best fit to equation 1. (B) Relationship between the t50 obtained in the experiment shown in panel A and the Ficoll concentration. (C) Amount of CA polymers formed in the experiment shown in panel A, in the presence of either Ficoll (circles) or dextran (squares) at 100 g/liter, as a function of the total CA concentration. Linear extrapolation of the data gave the CA critical concentration.
The dramatic increase in the polymerization rate in the presence of molecular crowding agents led us to evaluate the possibility of achieving rapid and efficient CA assembly in vitro at very high effective CA concentration and a low (physiologic) ionic strength, conditions that would approach those found inside the authentic HIV-1 virion. At 150 mM NaCl and a high (600 μM) CA concentration, no significant assembly was detected even after 65 h of incubation in the absence of a crowding agent. In sharp contrast, addition of 250 g/liter Ficoll 70 reproducibly led to massive, efficient polymerization of CA in less than 2 min (Fig. 4 and Table 1). Transmission electron microscopy analysis revealed the formation of cylindrical structures (Fig. 2B) with a diameter of 37 ± 9 nm that were indistinguishable in shape and dimensions from those obtained in standard conditions (i.e., at very high ionic strength and relatively low CA concentrations, in the absence of crowding agents) (Fig. 2A). Conical structures with dimensions approaching those of authentic mature capsids of HIV-1 were also observed (Fig. 2B).
FIG. 4.

In vitro polymerization of CA at ionic strength and CA effective concentrations that approach those inside the HIV-1 virion. The sample contained 150 mM NaCl and 600 μM CA, and 250 g/liter Ficoll as a crowding agent (circles), or no crowding agent (squares). At specified times small aliquots were taken, rapidly diluted 100-fold and their optical density at 350 nm was determined immediately. The ordinate values indicate the corrected optical densities corresponding to the nondiluted sample. The continuous line represents the best fit to equation 1. The high viscosity of the crowded solution made rapid and accurate dilution difficult, which led to some dispersion in the values obtained at high polymer concentrations (longer times). However, the result and the values of the parameters obtained (Table 1) were reproducible.
Disassembly of capsid-like particles formed at low ionic strength in the presence of a crowding agent.
In vitro assembly of CA at very high ionic strength in the absence of crowding agents may be essentially irreversible, as the polymers formed at 2.25 M NaCl and 600 μM CA (or lower protein concentrations) dissociated very slowly, or not at all, when the protein was diluted 10-fold while preserving the pH and ionic strength (Fig. 5A). A small gradual decrease in turbidity observed over a period of many minutes was due to sedimentation of the particles in the absence of stirring (Fig. 5A and data not shown). We then tested the reversibility of CA polymerization at low ionic strength and very high effective CA concentrations in the presence of a crowding agent. The polymers formed at 150 mM NaCl, 250 g/liter Ficoll, and 600 μM CA were rapidly diluted ten-fold while preserving the pH, ionic strength, and Ficoll concentration, and the kinetics of particle dissociation triggered by dilution was determined (Fig. 5B). A rapid dissociation process was reproducibly observed by following the reduction in light scattering. This process did not fit the single exponential decay expected for a simple dissociation of the polymers into small (non-light-scattering) subunits. In contrast, fitting to two exponential decays (equation 2) was extremely good (Fig. 5B) and yielded similar amplitudes, and rate constants which differed by one order of magnitude (Table 2) (see Discussion). The experimental values of the scattered light at the end of the reaction and the fitted ordinate values at infinite time for the two exponential decays were not zero, consistent with the expected displacement of the chemical equilibrium, without complete dissociation of all of the light-scattering particles. As another test for equilibrium conditions, polymerization of soluble CA was carried out under the same conditions (including low-salt [150 mM] and CA [about 60 μM] concentrations) used in the disassembly assay described above. Polymerization proceeded very slowly and yielded low amounts of light-scattering particles as expected, even after one or more hours of incubation. Longer incubation times sometimes led to further substantial increases in scattered light that were, however, not reproducible (data not shown). These observations, taken together, suggest that the assembly of CA in vitro at physiologic ionic strength in macromolecular crowding conditions may be reversible, and open the possibility for a thermodynamic characterization of the process (under study).
FIG. 5.

In vitro disassembly of capsid-like particles. Particles were assembled using 600 μM CA in either 2.25 M NaCl in the absence of a crowding agent (panel A), or in 150 mM NaCl in the presence of 250 g/liter Ficoll (panel B). Immediately before the start of the measurements, the particles were diluted 10-fold in the same buffer used for assembly. For clarity, only one in each 20 data points is represented (one in four points for times shorter than 7 min in panel B). The decay in optical density as a function of time after dilution is indicated by circles. Incubation times longer than those shown led to substantial sedimentation of the particles. In the experiment shown in panel A, the sample was stirred at 60 min after dilution; the decay in optical density as a function of time after stirring is indicated by inverted triangles. The similarity between the two curves shows that the very slight decrease in turbidity is mainly due to sedimentation, and not to particle dissociation. In the experiment shown in panel B, the continuous lines represent the best fits to a single exponential decay (nonmatching curve) and to a double exponential decay (equation 2) (matching curve). The experiment was repeated, and quantitatively similar results were obtained.
TABLE 2.
Kinetic analysis of the disassembly of capsid-like particlesa
| CAb | A1 | k1 (min−1) | A2 | k2 (min−1) |
|---|---|---|---|---|
| Nonmutated (1/10), avg | 0.63 ± 0.05 | 0.46 ± 0.05 | 0.37 ± 0.05 | 0.028 ± 0.001 |
| Nonmutated (1/10) | 0.62 ± 0.003 | 0.43 ± 0.007 | 0.38 ± 0.002 | 0.029 ± 0.0003 |
| V181A/E180D (1/10) | 0.68 ± 0.003 | 0.44 ± 0.004 | 0.32 ± 0.003 | 0.068 ± 0.0006 |
| V181A/E180D (1/100) | 0.83 ± 0.002 | 0.44 ± 0.002 | 0.17 ± 0.001 | 0.045 ± 0.0004 |
Fitting values for the relative amplitudes A1 and A2 (total amplitude = 1) and the dissociation rate constants k1 and k2 for the two exponential processes 1 and 2 are indicated (see text).
At t = 0, the samples containing assembled particles were diluted 1/10 or 1/100 as indicated. In the first file, average values and experimental standard deviations obtained in four independent experiments are indicated. In the remaining files, values obtained in representative experiments and fitting errors are indicated.
Effect of molecular crowding agents on assembly of a variant of CA with an extremely reduced dimerization affinity.
CA dimerizes in solution through an interface that is structurally and functionally contained entirely within the C-terminal domain, CA-C (14). Homodimerization of CA is critical for assembly of the immature HIV-1 particle and of the mature capsid (for examples, see references 7, 14, 35, 40, and 41) through the noncovalent linking of CA hexamers (26). As a part of a thorough thermodynamic analysis of the CA dimerization interface (6) we had observed that mutation of a valine (CA residue 181) to alanine at the energetic epitope within the dimerization interface led to essentially monomeric protein in solution, even at very high (mM) protein concentrations. Remarkably, a presumably infectious HIV-1 variant that had alanine at CA position 181 had been identified by sequencing cDNA Gag clones obtained from RNA extracted from virions purified from plasma of HIV-infected patients (42). One possibility to explain the presence of mutation V181A in CA from a presumably infectious virion was that the drastic negative effect of this mutation on CA association could be neutralized by a compensatory mutation at the dimerization interface. The only other difference in the entire CA-C sequence between this variant and the HIV strain we use as a reference (BH10) is the presence of an aspartate instead of a glutamate at the neighboring position 180. Isolated CA-C is correctly folded and dimerizes with the same affinity as the full-length protein (14). Thus, we introduced mutations V181A and the presumed compensatory mutation E180D into isolated CA-C, and tested the effect of these mutations on dimerization.
Both CA-C mutant V181A and the double mutant V181A/E180D were found essentially monomeric in solution, even at high protein concentrations, as shown by intrinsic Trp fluorescence, analytical gel filtration and analytical ultracentrifugation. The intrinsic tryptophan fluorescence emission spectrum of both mutants at 200 μM showed a maximum emission wavelength of about 350 nm (Fig. 6A), corresponding to a monomeric state with the only Trp, which is located at the dimerization interface (14), completely exposed to solvent (28). Analytical gel filtration also showed that both V181A (6) and V181A/E180D (not shown) are mostly in monomeric form, even at mM concentrations. The same was observed using analytical ultracentrifugation (Fig. 6B and data not shown). Given the extremely low dimerization affinity of these mutants, the association equilibrium constant could not be rigorously determined using either frontal analytical gel filtration or analytical ultracentrifugation. However, a rough estimation using zonal analytical gel filtration yielded similar, extremely low association constants for the two CA-C mutants (not shown). From these observations we conclude that mutation E180D does not compensate, or even alleviate, the extreme reduction in CA dimerization affinity caused by mutation V181A.
FIG. 6.

Fluorescence spectrum and sedimentation equilibrium of CA-C mutant V181A. (A) Trp intrinsic fluorescence emission spectra of CA-C obtained at 200 μM protein (monomer) concentration. Continuous line, nonmutated CA-C; dashed line, mutant V181A; dotted line, mutant V181A/E180D. (B) Sedimentation equilibrium gradients (30,000 rpm, 20°C) of nonmutated CA-C (left panel) or mutant V181A (right panel) determined by analytical ultracentrifugation. The protein (monomer) concentration was 20 μM. Circles, experimental data points; lines, theoretical gradients corresponding to monomer or dimer, as indicated.
The effects of mutations V181A and V181A/E180D were also tested on CA polymerization in vitro. The isolated CA-C domain is able to efficiently inhibit the in vitro assembly of full-length CA in standard conditions (i.e., at a high ionic strength in the absence of crowding agents; 25), and mutations that either decrease or increase the CA-C dimerization affinity have a correspondingly lower or higher inhibitory activity on CA polymerization (5). Consistent with their very low homodimerization affinity, both V181A and V181A/E180D CA-C mutants were unable to inhibit the polymerization of nonmutated CA in those conditions, even at relatively high CA-C concentrations (Fig. 7).
FIG. 7.

Effect of CA-C mutants V181A and V181A/E180D on the polymerization of CA in vitro. The conditions were as follows: pH 7.8, 2.25 M NaCl, 44 μM CA, and no isolated CA-C (circles), 88 μM nonmutated CA-C (triangles), 88 μM CA-C mutant V181A (inverted triangles), or 88 μM CA-C mutant V181A/E180D (squares). For clarity, only one in each 30 data points is represented (one in five points for the shorter times when CA-C mutants were present). Continuous lines represent the best fit to equation 1.
The above results provided evidence that mutation V181A, alone or accompanied by mutation E180D, has a most severe negative effect on CA-C and CA association in the standard conditions used. To directly evaluate the capacity of the CA mutant V181A/E180D to assemble into polymeric structures in vitro, a full-length CA that carried both mutations was constructed, purified and analyzed for polymerization in vitro, using at first 15 μM CA and 2.25 M NaCl, either in the absence or presence of Ficoll 70. As expected from the results described above, the CA double mutant was completely unable to polymerize in the absence of Ficoll, even after a long incubation time. In sharp contrast, the same mutant polymerized rapidly and efficiently in the presence of the crowding agent (Fig. 8), at a rate that was only somewhat lower than that of nonmutated CA (Table 1). Use of different CA concentrations showed that the critical concentration in the absence of a crowding agent was substantially higher for the double mutant (14.3 μM) than for nonmutated CA (5.6 μM). In contrast, in the presence of Ficoll at 100 g/liter, the critical concentration for the double mutant was greatly reduced (to 4.7 μM), and approached that obtained with nonmutated CA in the same conditions (3.1 μM).
FIG. 8.

In vitro polymerization of CA mutant V181A/E180D. The conditions were: pH 7.8, 2.25 M NaCl, 15 μM nonmutated CA (circles) or CA mutant V181A/E180D (squares) in the absence (open symbols) or presence (closed symbols) of 100 g/liter Ficoll as a crowding agent. For clarity, only one in each five data points is represented. Continuous lines represent the best fit to equation 1.
Finally, the ability of CA mutant V181A/E180D to polymerize into tubular structures was compared with that of nonmutated CA using the highest possible CA and crowding agent concentrations at 150 mM NaCl, conditions that mimicked the very high effective CA concentration and low ionic strength inside the HIV-1 virion. The results (Fig. 9A) confirmed that the CA mutant was able to polymerize with high efficiency and a rate that approached that of nonmutated CA (Table 1). Analysis of the polymeric structures formed by the double mutant (Fig. 2C) showed them to be hollow cylinders. These showed a tendency to form bundles, like those formed by nonmutated CA in the same conditions (not shown). However, the mutant cylinders, with a diameter of 12 ± 2 nm, were much thinner than the nonmutated cylinders (Fig. 2). The former were also consistently longer. As observed for nonmutated CA, a ten-fold dilution triggered a rapid disassembly of the mutant tubular structures, with a kinetics that fitted very well a double-exponential decay (Fig. 9B). As expected for first order reactions, the values of the kinetic parameters (Table 2) did not depend on the initial polymer concentration. The values obtained for the mutant were very similar to those obtained for nonmutated CA except for the dissociation rate constant of the slower exponential decay, which was about twofold larger for the mutant, consistent with a destabilizing effect of the mutation on this process.
FIG. 9.

In vitro polymerization of CA mutant V181A/E180D at ionic strength and CA effective concentrations that approach those inside the HIV-1 virion. (A) The sample contained 150 mM NaCl and 600 μM CA, and 250 g/liter Ficoll as a crowding agent (circles), or no crowding agent (squares). The experiment was carried out as described in the legend to Fig. 4, except that a 50-fold dilution was used. As in the experiment shown in Fig. 4, the high viscosity of the solution led to some dispersion in the values obtained. The continuous line represents the best fit to equation 1. (B) Disassembly of the mutant V181A/E180D capsid-like particles. Immediately before the start of the measurements, the particles formed in the experiment shown in panel A were diluted 10-fold in the same buffer used for assembly. For clarity, only one in each 7 or 20 data points is represented for times shorter or longer than 7 min, respectively. The continuous lines represent the best fits to a single exponential decay (non-matching curve) and to a double exponential decay (equation 2) (matching curve). The experiment was repeated, and similar results were obtained.
DISCUSSION
Recent determinations (1, 2) indicate that a typical HIV-1 virion may contain, in an internal volume of about 980,000 nm3, about 4,000 to 5,000 molecules of CA, MA, NC, and p6, 250 molecules of reverse transcriptase, integrase, and protease, and many molecules of viral proteins Vpr (700 molecules), Nef, Vif, and cellular proteins, including cyclophyllin A (500 molecules), actin, etc., as well as two genomic RNA molecules. Thus, the CA concentration inside the virion would be as high as 7 to 8 mM, and the total macromolecular concentration would reach 500 to 600 g/liter, which even surpasses those found in cellular compartments. Such crowded conditions would involve a substantial solute volume exclusion effect that may alter the energetics and dynamics of macromolecular reactions. Regarding the de novo CA assembly process occurring inside the extracellular virion during maturation (1-3), the most obvious consequence of the crowded conditions inside the HIV-1 particle is that, from pure excluded volume effects, the chemical activity of CA may be at least ten times its actual concentration (32), or over 50 mM. Such effective concentration is two orders of magnitude above the highest CA concentrations used in assembly assays in vitro. This fact is made even more relevant by considering the weak affinity of CA to hexamerize through its N-terminal domain (34), or dimerize through its C-terminal domain (14). Thus, it may be hardly surprising that, when nearly physiological pH and ionic strength but relatively low CA concentrations in non-crowded solutions were used in vitro, CA polymerization proceeded extremely slowly, or not at all.
Addition of high concentrations of an unrelated inert macromolecule, acting as a crowding agent, allowed us to drastically accelerate the rate of CA polymerization in vitro. The effect was most dramatic at close to physiological ionic strength (150 mM NaCl) and high CA concentration (600 μM), where assembly of CA was insignificant even after three days in the absence of a crowding agent, but completed in less than 2 min in the presence of Ficoll at 250 g/liter. The enhancement effect was not specific, as it occurred irrespective of either Ficoll or dextran being used as a crowder. In addition to a dramatic enhancement of the assembly rate at any protein concentration or ionic strength tested, the presence of the crowding agent caused substantial bundling of the tubular structures formed, an entropically driven effect already described for filamentous proteins in macromolecular crowding conditions (23). Interestingly, CA can form cylinders also in the crowded environment inside live E. coli cells, where evidence of bundling can be appreciated (Fig. 7E in reference 18).
As seen in electron micrographs, the hollow cylinders formed at low ionic strength and high CA effective concentration in the presence of a crowding agent are not significantly different from those formed at very high ionic strength in the absence of a crowding agent, which is consistent with the idea that the underlying structural organization is the same in both cases. This result was not unexpected, as recent analyses indicated that the cylindrical and conical assemblies formed at high ionic strength and authentic cylindrical cores isolated from HIV-1 virions are structurally similar and may share, along with the conical HIV-1 capsids, the same CA lattice (3, 26). However, the capsids formed in physiological conditions could present some differences in local conformation, intersubunit interactions (3, 16, 41), or the kinetics and thermodynamics of assembly, disassembly, or stability. Two aspects in which conditions much closer to those inside the HIV-1 virion led to different results are discussed next.
Firstly, the extremely high CA effective concentrations in vivo may allow assembly of very low-affinity variants. A double mutation (V181A/E180D), presumed to be present in a replicating HIV-1 variant, drastically reduced CA-C dimerization in solution and CA polymerization in standard (high ionic strength) in vitro assays. By simply increasing the chemical activity of mutant CA to values much closer to those that would occur in vivo, efficient polymerization at a rate that approached that of nonmutated CA was achieved. However, that CA mutants with very low dimerization affinity may polymerize in conditions close to those found in the HIV-1 virion does not mean that such mutations would be biologically irrelevant. The cylindrical structures formed by mutant V181A/E180D are unnaturally thin, which suggests that some difference in the protein-protein interfaces involved led to aberrant assembly. This mutant showed also an increased rate of disassembly, and such variations could severely affect the biological fitness of the virus. In addition, for assembly to take place in vivo an immature virion must first be produced; any mutation that impairs Gag assembly would block the subsequent process of core assembly, while it may have no influence on CA polymerization in vitro. Mutation V181A/E180D was detected in a molecular clone obtained from RNA extracted from HIV-1 virions purified from plasma of a patient with a high viral load; thus, this mutation would probably be present in an actively replicating virus (42). However, it must be stressed that, until proven by introducing the V181A/E180D mutation into an infectious proviral clone, we cannot be certain that this mutation does not abolish HIV-1 infectivity. Whatever the case, the results just discussed do emphasize the importance of considering the influence of solvent volume exclusion effects and the extremely high CA effective concentration inside the virion when analyzing the effect of mutations on assembly of the HIV-1 mature capsid.
Secondly, the relatively low ionic strength inside the virion may facilitate capsid dissociation. The very high ionic strength in standard CA polymerization assays could directly modify the strength of protein association because of an intensification of the hydrophobic effect, displacement of protein-bound solvent molecules and/or a drastic screening of charge-charge interactions. For example, electrostatic repulsions between residues located at the CA intersubunit interfaces may help to preserve a low affinity of dimerization (5) and hexamerization (8). A high ionic strength would effectively screen those repulsive interactions, and could be thus predicted to increase the stability of the HIV-1 capsid. Accordingly, we found that capsid-like particles formed at a very high ionic strength (2.25 M NaCl) dissociated very slowly upon dilution in the same buffer, while those formed in the presence of a crowding agent at low ionic strength (150 mM NaCl) dissociated very rapidly. These results indicate that the capsid-like particles formed in vitro in close to physiological conditions (low ionic strength and high CA effective concentration) are kinetically much less stable than those formed at very high (non-physiological) ionic strength. This observation is consistent with observations that indicate a low stability of authentic HIV-1 cores (reference 12 and references therein), and actually supports the view that maturation of the HIV-1 virion is necessary for the formation of an unstable capsid poised for dissociation and release of the viral genome by simple “dissolution” in the cytosol, after the loss of the viral membrane during infection of the host cell (34).
The reduction in light scattering upon dilution of the nonmutated and mutant polymers assembled at low ionic strength and high effective protein concentrations fitted extremely well a double-exponential decay that yielded similar amplitudes, and rate constants which differed by about one order of magnitude. The presence of the double mutation had no effect on the relative amplitudes of the two processes or in the rate constant of the faster decay, but led to a twofold increase in the rate constant of the slower decay. This is consistent with the expected destabilizing role of the intersubunit interactions lost in the mutant due to truncation of the valine side chain within the CA dimerization interface (6), and supports the possibility that the slower exponential decay of turbidity is reporting the actual disassembly of capsid-like particles into small, non-scattering subunits (like CA dimers). The faster exponential decay could be due to (i) disaggregation of the bundles of cylinders, formed under macromolecular crowding conditions, into nonaggregated cylinders that may contribute less to light scattering or (ii) disassembly of the capsid-like particles through a medium-sized intermediate that would contribute roughly half the total light scattering. To evaluate the first possibility, particle disassembly was induced by dilution and aliquots were taken at different times, rapidly fixed and stained on grids and visualized by electron microscopy. At a short time after dilution, most tubules observed were relatively long, and all of them were bundled. From the rate constants, we calculated that, if the faster process corresponds to dissociation of bundles and the slower process corresponds to disassembly of individual tubules, at 5 min after dilution about 9/10 of the bundles should have disappeared, while about 9/10 of the tubules should still be present in free form. In fact, at about 5 min, we observed many shortened individual tubules, but also many bundles, many of them formed by tubules of shortened length. Bundles were still observed at 20 min. This preliminary experiment does not support the first possibility advanced to explain the biphasic kinetics, while it is not inconsistent with the second possibility. Though further analyses are required, the observed reversibility of the assembly process by simple protein dilution opens the possibility of carrying out quantitative thermodynamic analysis of the assembly/disassembly equilibrium and kinetic analysis of disassembly of capsid-like particles of HIV-1, including the identification of possible intermediates.
In conclusion, the use of macromolecular crowding agents allows the reversible assembly of CA into capsid-like particles at low ionic strength and high protein effective concentrations in vitro. Differences in the effect of mutations on the assembly reaction and on the kinetic stability of the particles formed were observed when the ionic strength and CA effective concentration used in in vitro polymerization assays approached those found inside the HIV-1 virion. Thus, the use of such conditions may allow a better in vitro evaluation of the effects of physical or chemical agents, mutations in CA or/and other molecules, including potential antiviral compounds, on HIV-1 capsid assembly.
Acknowledgments
We acknowledge M. Jiménez, C. Alfonso, and M. T. Rejas for assistance with electron microscopy and analytical ultracentrifugation.
M.D.A. is a recipient of a research fellowship from Comunidad Autónoma de Madrid. This work was supported by grants from the Ministerio de Ciencia y Tecnología (BIO2003-04445) and Comunidad Autónoma de Madrid (GR/SAL/0027/2004) to M.G.M., by grant BMC2002-04617-C02 to G.R., and by an institutional grant from Fundación Ramón Areces.
REFERENCES
- 1.Benjamin, J., B. K. Ganser-Pornillos, W. F. Tivol, W. I. Sundquist, and G. J. Jensen. 2005. Three-dimensional structure of HIV-1 virus-like particles by electron cryotomography. J. Mol. Biol. 346:577-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Briggs, J. A. G., M. N. Simon, I. Gross, H.-G. Kräusslich, S. D. Fuller, V. M. Vogt, and M. C. Johnson. 2004. The stoichiometry of gag protein in HIV-1. Nat. Struct. Mol. Biol. 11:672-675. [DOI] [PubMed] [Google Scholar]
- 3.Briggs, J. A. G., T. Wilk, R. Welker, H.-G. Kräusslich, and S. D. Fuller. 2003. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 22:1707-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell, S., and V. M. Vogt. 1995. Self-assembly in vitro of purified CA-NC proteins from Rous Sarcoma Virus and human immunodeficiency virus type 1. J. Virol. 69:6487-6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.del Álamo, M., and M. G. Mateu. 2005. Electrostatic repulsion, compensatory mutations, and long-range non-additive effects at the dimerization interface of the HIV capsid protein. J. Mol. Biol. 345:893-906. [DOI] [PubMed] [Google Scholar]
- 6.del Álamo, M., J. L. Neira, and M. G. Mateu. 2003. Thermodynamic dissection of a low-affinity protein-protein interface involved in human immunodeficiency virus assembly. J. Biol. Chem. 278:27923-27929. [DOI] [PubMed] [Google Scholar]
- 7.Dorfman, T., A. Bukovsky, A. Ohagen, S. Höglund, and H. G. Göttlinger. 1994. Functional domains of the capsid protein of human immunodeficiency virus type 1. J. Virol. 68:8180-8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Douglas, C. C., D. Thomas, J. Lanman, and P. E. Prevelige, Jr. 2004. Investigation of N-terminal domain charged residues on the assembly and stability of HIV-1 CA. Biochemistry 43:10435-10441. [DOI] [PubMed] [Google Scholar]
- 9.Ehrlich, L. S., B. E. Agresta, and C. A. Carter. 1992. Assembly of recombinant human immunodeficiency virus type 1 capsid protein in vitro. J. Virol. 66:4874-4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ehrlich, L. S., T. Liu, S. Scarlata, B. Chu, and C. A. Carter. 2001. HIV-1 capsid protein forms spherical (immature-like) and tubular (mature-like) particles in vitro: structure switching by pH-induced conformational changes. Biophys. J. 81:586-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellis, R. J. 2001. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 26:597-604. [DOI] [PubMed] [Google Scholar]
- 12.Forshey, B. M., U. von Schwedler, W. I. Sundquist, and C. Aiken. 2002. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 76:5667-5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frankel, A. D., and J. A. Young. 1998. HIV-1: fifteen proteins and an RNA. Annu. Rev. Biochem. 67:1-25. [DOI] [PubMed] [Google Scholar]
- 14.Gamble, T. R., S. Yoo, F. F. Vajdos, U. K. von Schwedler, D. K. Worthylake, H. Wang, J. P. McCutcheon, W. I. Sundquist, and C. P. Hill. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278:849-853. [DOI] [PubMed] [Google Scholar]
- 15.Ganser, B. K., S. Li, V. Y. Klishko, J. T. Finch, and W. I. Sundquist. 1999. Assembly and analysis of conical models for the HIV-1 core. Science 283: 80-83. [DOI] [PubMed] [Google Scholar]
- 16.Ganser-Pornillos, B. K., U. K. von Schwedler, K. M. Stray, C. Aiken, and W. I. Sundquist. 2004. Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 78:2545-2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Göttlinger, H. G. 2001. The HIV assembly machine. AIDS 15:S13-S20. [DOI] [PubMed] [Google Scholar]
- 18.Gross, I., H. Hohenberg, C. Huckhagel, and H.-G. Kräusslich. 1998. N-terminal extension of human immunodeficiency virus capsid protein converts the in vitro assembly phenotype from tubular to spherical particles. J. Virol. 72:4798-4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gross, I., H. Hohenberg, and H.-G. Kräusslich. 1997. In vitro assembly properties of purified bacterially expressed capsid proteins of human immunodeficiency virus. Eur. J. Biochem. 249:592-600. [DOI] [PubMed] [Google Scholar]
- 20.Gross, I., H. Hohenberg, T. Wilk, K. Wiegers, M. Grattinger, B. Müller, S. Fuller, and H.-G. Kräusslich. 2000. A conformational switch controlling HIV-1 morphogenesis. EMBO J. 19:103-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall, D., and A. P. Minton. 2003. Macromolecular crowding: qualitative and semiquantitative successes, quantitative challenges. Biochim. Biophys. Acta 1649:127-139. [DOI] [PubMed] [Google Scholar]
- 22.Hatters, D. M., A. P. Minton, and G. J. Howlett. 2002. Macromolecular crowding accelerates amyloid formation by human apolipoprotein C-II. J. Biol. Chem. 277:7824-7830. [DOI] [PubMed] [Google Scholar]
- 23.Herzfeld, J. 1996. Entropically driven order in crowded solutions: from liquid crystals to cell biology. Acc. Chem. Res. 29:31-37. [DOI] [PubMed] [Google Scholar]
- 24.Lanman, J., T. T. Lam, S. Barnes, M. Sakalian, M. R. Emmett, A. G. Marshall, and P. E. Prevelige, Jr. 2003. Identification of novel interactions in HIV-1 capsid protein assembly by high-resolution mass spectrometry. J. Mol. Biol. 325:759-772. [DOI] [PubMed] [Google Scholar]
- 25.Lanman, J., J. Sexton, M. Sakalian, and P. E. Prevelige, Jr. 2002. Kinetic analysis of the role of intersubunit interactions in human immunodeficiency virus type 1 capsid protein assembly in vitro. J. Virol. 76:6900-6908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li, S., C. P. Hill, W. I. Sundquist, and J. T. Finch. 2000. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407:409-413. [DOI] [PubMed] [Google Scholar]
- 27.Mach, H., D. B. Volkin, C. J. Burke, and C. R. Middaugh. 1995. Ultraviolet absorption spectroscopy, p. 91-114. In B.A. Shirley (ed.), Protein folding and stability: theory and practice. Humana Press, Totowa, N.J. [DOI] [PubMed]
- 28.Mateu, M. G. 2002. Conformational stability of dimeric and monomeric forms of the C-terminal domain of human immunodeficiency virus-1 capsid protein. J. Mol. Biol. 318:519-531. [DOI] [PubMed] [Google Scholar]
- 29.Mayo, K., D. Huseby, J. McDermott, B. Arvidson, L. Finlay, and E. Barklis. 2003. Retrovirus capsid protein assembly arrangements. J. Mol. Biol. 325:225-237. [DOI] [PubMed] [Google Scholar]
- 30.Minton, A. P. 1983. The effect of volume occupancy upon the thermodynamic activity of proteins: some biochemical consequences. Mol. Cell Biochem. 55:119-140. [DOI] [PubMed] [Google Scholar]
- 31.Minton, A. P. 1994. Conservation of signal: a new algorithm for the elimination of the reference concentration as an independently variable parameter in the analysis of sedimentation equilibrium, p. 81-93. In T. M. Schuster and T. M. Laue (ed.), Modern analytical ultracentrifugation. Birkhauser, Boston, Ma.
- 32.Minton, A. P. 1998. Molecular crowding: analysis of effects of high concentrations of inert cosolutes on biochemical equilibria and rates in terms of volume exclusion. Methods Enzymol. 295:127-149. [DOI] [PubMed] [Google Scholar]
- 33.Minton, A. P. 2000. Implications of macromolecular crowding for protein assembly. Curr. Opin. Struct. Biol. 10:34-39. [DOI] [PubMed] [Google Scholar]
- 34.Mortuza, G. B., L. F. Haire, A. Stevens, S. J. Smerdon, J. P. Stoye, and I. A. Taylor. 2004. High-resolution structure of a retroviral capsid hexameric amino-terminal domain. Nature 431:481-485. [DOI] [PubMed] [Google Scholar]
- 35.Reicin, A. S., S. Paik, R. D. Berkowitz, J. Luban, I. Lowy, and S. P. Goff. 1995. Linker insertion mutations in the human immunodeficiency virus type 1 gag gene: effects on virion particle assembly, release and infectivity. J. Virol. 69:642-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rivas, G., F. Ferrone, and J. Herzfeld. 2004. Life in a crowded world. EMBO Rep. 5:23-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang, C., E. Loeliger, I. Kinde, S. Kyere, K. Mayo, E. Barklis, Y. Sun, M. Huang, and M. F. Summers. 2003. Antiviral inhibition of the HIV-1 capsid protein. J. Mol. Biol. 327:1013-1020. [DOI] [PubMed] [Google Scholar]
- 38.Turner, B. G., and M. F. Summers. 1999. Structural biology of HIV. J. Mol. Biol. 285:1-32. [DOI] [PubMed] [Google Scholar]
- 39.Vogt, V. M. 1997. Retroviral virions and genomes, p. 27-70. In J. M. Coffin, S. H. Hughes, and H. E. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [PubMed]
- 40.von Schwedler, U. K., T. L. Stemmler, V. Y. Klishko, S. Li, K. H. Albertine, D. R. Davis, and W. I. Sundquist. 1998. Proteolytic refolding of the HIV-1 capsid protein amino terminus facilitates viral core assembly. EMBO J. 17:1555-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.von Schwedler, U. K., K. M. Stray, J. E. Garrus, and W. I. Sundquist. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 77:5439-5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshimura, F. K., K. Diem, G. H. Learn, Jr., S. Riddell, and L. Corey. 1996. Intrapatient sequence variation of the gag gene of human immunodeficiency virus type 1 plasma virions. J. Virol. 70:8879-8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zimmerman, S. B., and A. P. Minton. 1993. Macromolecular crowding: biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct. 22:27-65. [DOI] [PubMed] [Google Scholar]