

Abstract
Chromatin rearrangements occur during repair of cyclobutane pyrimidine dimers (CPDs) by nucleotide excision repair (NER). Thereafter, the original structure must be restored to retain normal genomic functions. How NER proceeds through nonnucleosomal chromatin and how open chromatin is reestablished after repair are unknown. We analyzed NER in ribosomal genes (rDNA), which are present in multiple copies but only a fraction are actively transcribed and nonnucleosomal. We show that removal of CPDs is fast in the active rDNA and that chromatin reorganization occurs during NER. Furthermore, chromatin assembles on nonnucleosomal rDNA during the early events of NER but in the absence of DNA repair. The resumption of transcription after removal of CPDs correlates with the reappearance of nonnucleosomal chromatin. To date, only the passage of replication machinery was thought to package ribosomal genes in nucleosomes. In this report, we show that early events after formation of UV photoproducts in DNA also promote chromatin assembly.
Nucleotide excision repair (NER) removes several types of lesions from DNA, including bulky adducts caused by chemicals, inter- or intrastrand cross-links, and the UV photoproducts cis-syn cyclobutane pyrimidine dimer (CPD) and pyrimidine (6-4) pyrimidone (15). In general, transcriptionally active genes are repaired faster than inactive DNA due to preferential removal of DNA lesions from the transcribed strands (TS) (37, 58, 62). This transcription-coupled repair process (or TCR) has been thought to require elongating RNA polymerase II (5, 27). However, recently TCR was found in the active fraction of ribosomal genes (rDNA) in yeast wild-type (wt) cells (7, 35), which are transcribed at a very high rate by RNA polymerase I. Furthermore, strand-specific repair was observed in total rDNA of rad7Δ, rad16Δ, and rad4Δ Saccharomyces cerevisiae strains (61).
The yeast RAD26 gene is the counterpart to the human Cockayne syndrome B (CSB) gene, and its inactivation creates a defect in the TCR of UV lesions (60). Since UV photoproducts present on the TS of active genes block RNA polymerases and arrest transcription (21), it was proposed that the Rad26/CSB proteins may act in the displacement of RNA polymerase II arrested at damaged sites and, subsequently, help in the recruitment of NER proteins to the lesion sites (62). Also, there are indications that Rad26 plays a role in RNA polymerase II-dependent transcription elongation in the absence of DNA damage (28). Thus, the Rad26/CSB proteins may promote RNA polymerase II transcription through damaged DNA bases (29).
Ribosomal genes are localized in the nucleolus, a dense chromatin region composed of rDNA, RNA polymerase I, rRNA, and assembling ribosomes, among other proteins (reviewed in reference 39). The ribosomal genes are present in multiple copies (∼150 in yeast) that are organized in long tandem repeats (55). In most organisms only a portion of rDNA is transcriptionally active (reviewed in references 20 and 32), and the fraction of active rDNA varies markedly among organisms and cell types (e.g., from ∼20% to ∼70%) (8-11). Furthermore, in yeast, the active rDNA population varies according to growth rate, with the highest proportion of active genes (about 50%) present in exponentially growing cells (10, 13). At the chromatin level, inactive rDNA is assembled in arrays of nucleosomes, while canonical nucleosomes are not present on active rDNA (32).
Like transcription and DNA replication, DNA repair is restricted by the structure of chromatin. At present, little is known about how nucleotide excision repair enzymes recognize and remove DNA lesions from nucleosomes, 30-nm fibers, and higher-order structures (49). In an early study it was found that nascent repair patches lack the canonical nucleosome DNase I footprint, implying that the structure of nucleosomes is altered during DNA repair in intact cells (50). Furthermore, it was shown that ligation, the final step in the NER pathway, precedes the formation of canonical nucleosomes onto repaired DNA (51). More recently, it was found that removal of CPDs from the TS of the URA3 and the MET16 active genes is fast and uniform, while removal of CPDs from the nontranscribed strands (NTS) is generally less efficient and is modulated by the position of nucleosomes (14, 57, 66). In addition, it was shown that nucleosome positioning modulates NER at the MET17 promoter (43). On the other hand, analysis of NER at the nucleotide level in a yeast minichromosome showed only a mild correlation between repair and nucleosome positions in the TS of the inactive GAL1::URA3 gene, and this correlation was abolished upon galactose induction of the gene (30). Thus, despite these and other investigations on DNA repair in chromatin, the details of how NER occurs in nucleosomal DNA remain vague. Furthermore, it is possible that the heterogeneity of chromatin structure within the nucleus calls for different requirements of chromatin remodeling during NER (18). An important process occurring during DNA repair, or soon after removal of DNA lesions, is the restoration of chromatin to its original state (31). Current results from in vitro experiments support a mechanism in which NER initiates formation of nucleosomes from the repair site. Additionally, it is proposed that de novo nucleosome assembly coupled to DNA repair involves the histone chaperone CAF-1 (chromatin assembly factor 1) (16, 36). On the other hand, very little is known about the process of NER in active, nonnucleosomal chromatin domains and about restoration during or after DNA repair of nonnucleosomal regions. To gain insight into this process, we have analyzed removal of CPDs and chromatin rearrangements throughout DNA repair in the active fraction of ribosomal genes in yeast. Using three independent assays, psoralen photo-cross-linking, restriction enzyme accessibility, and micrococcal nuclease sensitivity, our experiments demonstrate that an inactive chromatin structure forms onto active genes within 30 min after UV irradiation but not immediately after induction of DNA damage. Thus, formation of the inactive structure is triggered by the presence of UV-induced DNA lesions. Additionally, assembly of chromatin on transcribing rDNA correlates with arrest of transcription by DNA damage. Finally, while active chromatin is restored in NER-competent cells after removal of CPDs, the inactive structure assembled following CPD formation is retained, and transcription does not recover, in rad1Δ and rad14Δ strains, where lesions are not repaired. Thus, although it has been thought that passage of the replication fork is required for packaging ribosomal genes in nucleosomes (33), we show that UV-induced DNA damage either directly or indirectly promotes chromatin assembly.
MATERIALS AND METHODS
Yeast cells and UV irradiation.
S. cerevisiae strain JS311 (RAD+) (53) cells were grown in complete medium (yeast extract, peptone, and dextrose [YEPD]) to early log phase (∼1.2 × 107 cells/ml). Cultures were washed in ice cold phosphate-buffered saline (137 mM NaCl, 2.5 mM KCl, 2 mM KH2PO4, 10 mM Na2HPO4, pH 7.0) and resuspended in the same buffer to a final concentration of 2 × 107 cells/ml. Cell suspensions were irradiated (primary, 254 nm) with a UV dose of 180 J/m2 as measured with a UVX radiometer(Ultra-Violet Products, Upland, Calif.). Then, cells were harvested, resuspended in YEPD, and incubated in the dark at 30°C with continuous shaking for different repair times. The isogenic rad1Δ and rad14Δ mutant strains of JS311 were constructed by PCR-mediated gene disruption as previously described (1).
Nuclei isolation and DNA extraction.
Yeast cells (∼2 × 109) were collected, washed with ice-cold phosphate-buffered saline, suspended in 1.5 ml of nuclei isolation buffer (NIB; 50 mM morpholinepropanesulfonic acid [MOPS], pH 8.0, 150 mM potassium acetate, 2 mM MgCl2, 17% glycerol, 0.5 mM spermine, and 0.15 mM spermidine) and transferred to 15-ml polypropylene tubes containing 1.5 ml of glass beads (425 to 600 μm; Sigma). Cell disruption and nuclei preparation were done as described previously (7). Nuclei were suspended in 0.5 ml of restriction enzyme buffers (New England BioLabs) and digestions were carried out according to manufacturers' recommendations. Alternatively, CaCl2 (2 mM final concentration) was added to 0.2-ml aliquots of nuclei in NIB, together with 30 U of micrococcal nuclease (Worthington), and the nuclear suspensions were incubated for 1 min at 37°C. After extraction, DNA was treated with RNase (BioShop Canada, Inc.), reextracted with phenol, and precipitated with ethanol.
Psoralen cross-linking of nuclei.
Cross-linking of nuclei was done in 24-multiwell plates (uncoated; Falcon). Psoralen (4,5′,8-trimethylpsoralen) (Sigma) stock solution (400 μg/ml) was added at a volume equal to 0.025 times the nuclei suspension volume. After 5 min on ice in the dark, the nuclear suspension was irradiated on ice for 10 min as described previously (7). The irradiation step was repeated twice.
Transcription run-on.
The in vitro transcription run-on (TRO) reaction mixture contained 1 mM ATP, 1 mM CTP, 1 mM GTP, 80 μM UTP, 5 mM magnesium acetate, 90 mM KCl, 3 mM MgCl2, 8% glycerol, 0.1 mM EDTA, pH 8.0, 5 mM dithiothreitol and 100 μCi of [α-32P]UTP (800 Ci/mmol). Nuclei in NIB were thawed on ice, and 100-μl aliquots were added to 100-μl aliquots of TRO reaction mixture. The samples were held on ice for 5 min, and the TRO reactions were initiated by transferring the samples to a 25°C heat block where in vitro transcription was allowed to occur for 10 min. Transcription was stopped by the addition of 1 ml of Trizol (Invitrogen), and RNA was isolated according to the manufacturer.
The purified pSPTmr100 plasmid samples (9) containing a portion of the rDNA transcription unit were denatured in 0.4N NaOH and 10 mM EDTA, pH 8.0, boiled for 3 min, and placed on ice. Using a slot-blot apparatus, DNA was immobilized in triplicate on Hybond N+ membrane (Amersham Pharmacia). Hybridization to radiolabeled RNA from TRO reactions was done at 68°C as described previously (9).
T4 endonuclease digestion, alkaline gel electrophoresis, and Southern blotting.
DNA samples were cleaved specifically at CPD sites by T4 endonuclease V (T4 endo V) (12) as described elsewhere (52). After T4 endo V digestion, DNA strands were separated on 1% alkaline agarose gels (46) and transferred to Hybond N+ membranes (Amersham). Radioactive probes were generated using random primers or strand-specific riboprobes (Promega).
Quantification of CPD yield.
Quantification of CPDs was performed on data from phosphorimages of the Southern blots using ImageQuant software (Molecular Dynamics), and measurement of CPDs in each strand of rDNA was performed as previously described (40).
RESULTS
As outlined in Fig. 1A, yeast cells were harvested at the specified repair times, nuclei were prepared, and one portion of nuclei was digested with EcoRI to release the active fraction of rDNA chromatin (Fig. 1A, left branch). Only active ribosomal genes are digested by EcoRI because nucleosomes are absent from these genes, making them accessible to restriction endonucleases (42). To monitor the separation of active rDNA from inactive rDNA, one aliquot of each sample of EcoRI-digested nuclei was photo-cross-linked with psoralen (Fig. 1A, a). The principle of psoralen cross-linking of chromatin is that active (nonnucleosomal) rDNA binds more psoralen than inactive (nucleosomal) rDNA. Consequently, psoralen cross-linked rDNA fragments from active genes have a slower migration on gels than fragments from inactive genes (reviewed in reference 20). To follow NER in active and inactive ribosomal genes, DNA was isolated from a second aliquot of EcoRI-digested nuclei and treated with T4 endo V, an enzyme that generates strand breaks specifically at CPD sites (Fig. 1A, b). Thereafter, DNA repair was measured by alkaline gel electrophoresis in combination with Southern blotting (reviewed in reference 3). Another portion of nuclei was directly cross-linked with psoralen (Fig. 1A, c) to follow the overall chromatin structure of ribosomal genes during repair. In addition, the presence of histone protein complexes in ribosomal genes during NER was assessed by the accessibility of the restriction enzyme EcoRI (Fig. 1A, e). In order to monitor resumption of transcription during NER, a third portion of the nuclei preparation was incubated under conditions permitting transcription elongation in vitro (TRO) (Fig. 1A, d).
FIG. 1.

(A) Experimental design. (B) Map of the yeast 35S rRNA gene. The rRNA gene, 5′ and 3′ ends, and direction of transcription (wavy arrow) are shown. The short black box represents the probe (∼140 bp) used in this work. The E1 to E7 positions mark the seven EcoRI restriction sites, N1 and N2 indicate the positions of the two NheI restriction sites, and P indicates the position of the PvuII sites.
Restriction enzyme digestions of isolated nuclei selectively release active rDNA chromatin.
At each repair time, nuclei were isolated and digested with EcoRI. Aliquots of nuclei were photoreacted with psoralen, and the DNA was extracted and digested with NheI (Fig. 1A, a). Since EcoRI sites are located within the two NheI sites (Fig. 1B), the complete NheI band (∼4.4 kb) can only originate from rDNA that was not cleaved by EcoRI.
As shown in Fig. 2, EcoRI digestion releases only nonnucleosomal rDNA (EcoRI, lanes 5 to 11, filled circle). On the other hand, intact NheI fragments contain primarily inactive rDNA (NheI, lanes 5 to 11, open circle), where EcoRI accessibility is inhibited by the presence of nucleosomes. As controls, single digests by EcoRI and NheI of DNA isolated from psoralen cross-linked nuclei show the migration of both active and inactive rDNA (Fig. 2, filled and open circles, respectively, in lanes 2 and 14 and lanes 4 and 12). The faint bands between the EcoRI and NheI fragments represent partial EcoRI digests of rDNA chromatin in nuclei (Fig. 1B, small Es). (It is noted that nuclei suspended in restriction enzyme digestion buffers can form small clumps, which decrease the activity of the restriction enzymes and yield partial digestion bands.) We observe that EcoRI accessibility to rDNA chromatin changes during NER, being low at early repair times and increasing at late repair times (Fig. 2B, lanes 7 to 11, EcoRI). This is indicative of a change in chromatin structure during repair.
FIG. 2.

Separation of active and inactive ribosomal gene chromatin. (A) Nuclei were isolated from nonirradiated (lane 5) and irradiated (lanes 6 to 11) cells that were harvested after different repair times. These nuclei were digested with EcoRI before psoralen cross-linking (lanes 5 to 11). The isolated DNA was then digested with NheI, separated on a 1% native agarose gel, blotted, and hybridized with a random primer-labeled probe (Fig. 1B). As controls, genomic DNA was isolated from non-cross-linked cells and digested with either EcoRI (lanes 1 and 15) or NheI (lanes 3 and 13). The presence of active and inactive rDNA chromatin was monitored by digesting nuclei with EcoRI or NheI before psoralen cross-linking and by redigesting the isolated DNA with EcoRI and NheI, respectively (lanes 2 and 14 and lanes 4 and 12). Labels indicate active rDNA (filled circle) and inactive rDNA (open circle). The major bands N1-N2 (NheI; 4,417 bp) and E4-E5 (EcoRI; 2,846 bp) are indicated, together with the products of partial digestions: E3-N2 (4,196 bp), N1-E6 (4,098 bp), E3-E6 (3,877 bp), N1-E5 (3,731 bp), E4-N2 (3,532 bp), E3-E5 (3,510 bp), and E4-E6 (3,213 bp). (B) Quantification of band intensities of active and inactive rDNA chromatin. Intensities for the N1-N2 (NheI) bands were divided by intensities for the E4-E5 (EcoRI) bands. Data show the average of two independent experiments. (Note that since the .25-h time point measurement was taken only once, it was not included.)
NER operates more efficiently in active (nonnucleosomal) rDNA chromatin than in inactive (nucleosomal) rDNA chromatin.
Previously, we examined NER in active and inactive rDNA chromatin (7). In that study yeast cells were irradiated with a dose of 80 J/m2 in the presence of 100 mM hydroxyurea (HU) to prevent replicative DNA synthesis. Since we recently observed that exposure of yeast cells to 100 mM HU for more than 4 h yields changes in the rDNA chromatin (unpublished data), we first examined NER of active and inactive rDNA in cells irradiated in the absence of HU. Higher UV doses (180 J/m2) were used to induce the threshold of photoproducts needed to inhibit DNA synthesis during the repair period (reviewed in reference 23). Arrest of cell division and inhibition of DNA synthesis up to 4 h after UV irradiation at 180 J/m2 were confirmed by cell enumeration and by flow cytometry (data not shown). At these UV doses, the average yields of CPDs for the active fraction (EcoRI fragment, 2.9 kb) were 1.5 ± 0.1 in the TS and 1.2 ± 0.1 in the NTS, and for the inactive fraction (NheI fragment, 4.4 kb) the average yields were 2.4 ± 0.1 in the TS and 2.0 ± 0.2 in the NTS (mean ± 1 standard deviation [SD] of three experiments).
To follow NER separately in the active and the inactive rDNA chromatin, DNA was isolated from EcoRI-digested nuclei and redigested with NheI, before T4 endo V analysis (Fig. 1A, b). Representative results for repair of the NTS and TS of predominantly active (EcoRI band) and inactive (NheI band) rDNA are shown in Fig. 3A and B, respectively, and the percentage of CPDs removed from each strand is shown in Fig. 3D. Fast repair is observed in the TS of active genes (filled circles), confirming the existence in yeast of TCR in RNA polymerase I genes (7). Furthermore, the NTS of active ribosomal genes (open circles) is repaired faster than both strands of the inactive genes, which show similar repair rates (open and filled triangles). This suggests that active rDNA chromatin is more accessible to NER enzymes than inactive rDNA chromatin. In addition, the repair of active rDNA chromatin is initially fast and rapidly levels off during removal of the remaining ∼20% of CPDs, while CPD removal from inactive rDNA chromatin occurs at a more uniform rate throughout the entire repair time (Fig. 3D). We note that previous studies in our laboratory compared repair of total rDNA (containing both active and inactive rDNA) in two different restriction fragments. The time course for CPD removal was found to be very similar for both a 2.9-kb EcoRI fragment (containing ∼0.68 CPDs/TS and ∼0.51 CPDs/NTS) and a 6.4-kb HindIII fragment (containing ∼1.52 CPDs/TS and 1.38 CPDs/NTS) (7). These analyses showed that the average number of CPDs present in the two rDNA fragments do not account for the differences in repair observed between the active and the inactive rDNA fractions (7) (Fig. 3D). In addition, under our experimental conditions the rate of CPD removal for total rDNA is similar to the rate of CPD removal from total genomic DNA (7) and the TS of the RPB2 gene (41).
FIG. 3.

(A) Repair of NTS of active (EcoRI) and inactive (NheI) rDNA in wt cells. Yeast cells were irradiated with 180 J/m2 UV and harvested at the times indicated. DNA was isolated from EcoRI-treated nuclei and digested with NheI. DNA samples were mock treated (−) or treated with T4 endo V (+); −UV indicates nuclei from nonirradiated cells, and 0 to 4 denotes nuclei from irradiated cells harvested after the indicated repair times (in hours). Samples were separated on a 1% alkaline agarose gel, blotted, and hybridized with strand-specific riboprobes (Fig. 1B). Dots point to the partial digestion products described in the legend of Fig. 2. (B) Repair of TS of active (EcoRI) and inactive (NheI) rDNA in wt cells. Experimental procedure and figure legends are as described for panel A. Dots point to the partial digestion products described in the legend of Fig. 2. (C) Repair of both strands of total rDNA in rad1Δ and rad14Δ mutant cells. Following different repair times, total DNA was isolated from rad1Δ and rad14Δ cells and digested with EcoRI. After separation on alkaline agarose gels and blotting as described for panel A, filter membranes were hybridized with random primer-labeled probe (Fig. 1B). Figure legends are as described for panel A. (D) Quantification of phosphorimages. DNA repair is expressed as the percentage of CPDs removed as a function of repair time. For wt cells data are from active rDNA (EcoRI, circles; wt-a) and inactive rDNA (NheI, triangles; wt-i). Solid and open symbols represent data from the TS and NTS, respectively. Data are the means ± 1 SD of three independent experiments. For both rad1Δ and rad14Δ cells, data are from total rDNA (diamonds) and represent the mean of two independent experiments each.
To determine if changes in chromatin structure during NER are directly related to the repair process (see below), we analyzed CPD removal from total rDNA in two NER-deficient strains (rad1Δ and rad14Δ) (Fig. 3C and D, diamonds). As expected, no removal of CPDs occurs in the rDNA locus of these cells.
Chromatin assembly occurs in active ribosomal genes during early events of NER.
As shown in Fig. 2, chromatin rearrangements appear to occur during repair of rDNA chromatin. To gain further insight into this process, nuclei were isolated from wt cells during NER and cross-linked with psoralen (Fig. 1A, c). The DNA was isolated and digested with EcoRI. In Fig. 4A, it is shown that in nonirradiated cells, ribosomal genes are distributed between active and inactive chromatin (lane 2, filled and open circles, respectively), and a similar pattern is found immediately after UV irradiation (lane 3). However, during early repair times (1/2 h and 1 h) the band representing active rDNA chromatin is no longer evident and is replaced by a smear representing a distribution of partially inactivated rDNA chromatin (lanes 4 and 5). At later repair times (2 to 4 h), the original proportions of active and inactive chromatin are restored (lanes 6 and 7). These data indicate that chromatin rearrangement occurs during the process of NER.
FIG. 4.

(A) Chromatin structure of ribosomal genes during NER in wt cells. Nuclei were isolated from nonirradiated (lane 2) or irradiated (lanes 3 to 7) cells, before (lane 3) and during NER (lanes 4 to 7). After cross-linking with psoralen, DNA was extracted from nuclei, digested with EcoRI, and separated on 1% native agarose gels. As a control (C), DNA was isolated from non-cross-linked nuclei and digested with EcoRI (lanes 1 and 8). After blotting, the filter membranes were hybridized with the random primer-labeled probe (Fig. 1B). Labels at right denote active rDNA (filled circle) and inactive rDNA (open circle). (B) Chromatin structure of ribosomal genes during NER in rad1Δ and rad14Δ strains. Experimental procedures and lanes are as described for panel A. (C) Scan profiles of gels shown in panels A and B.
It was proposed that the DNA synthesis step of NER takes place simultaneously with nucleosome assembly onto newly repaired DNA (reviewed in reference 19). This prompted us to investigate whether chromatin assembly onto active rDNA chromatin during NER is a consequence of DNA repair synthesis. To this end, psoralen cross-linking analyses were performed on nuclei from NER-deficient rad1Δ and rad14Δ cells. The results shown in Fig. 4B indicate that both rDNA populations are present in nonirradiated cells (lane 2) and soon after UV irradiation (lane 3). During repair incubation, the band representing active rDNA chromatin (filled circle) disappears into a smear while there is only a small accumulation of signal in the band representing inactive rDNA (Fig. 4B, open circle; compare lanes 2 and 3 with lanes 4 to 7). This is seen more clearly in scans of these gels (Fig. 4C). In contrast to wt cells, the original proportions of active and inactive chromatin are not restored in the repair-deficient cells. These data suggest that chromatin assembly in the active rDNA fraction shortly after irradiation is not a consequence of the DNA repair synthesis step.
Inhibition of RNA polymerase I elongation by UV photoproducts correlates with chromatin assembly, while resumption of transcription correlates with CPD removal and chromatin disassembly.
The presence of UV photoproducts on the TS causes RNA polymerase I to stall (21, 26). Since RNA polymerase I elongation affects the structure of chromatin and vice versa (2, 10), we investigated the relationship between the observed chromatin assembly during NER and the levels of rRNA transcription. Nuclei isolated from wt, rad1Δ, and rad14Δ cells during DNA repair were incubated under conditions allowing transcription elongation (Fig. 1A, d). At the UV dose used in our experiments, ∼3.6 CPDs were introduced in the TS of the 35S rDNA, and the resulting reduction in rDNA transcription is shown in Fig. 5. In wt cells, transcription is reduced to about 50% and gradually recovers as repair proceeds (black bars). In contrast, transcription does not recover in rad1Δ (gray bars) and rad14Δ (hatched bars) cells, being reduced to almost 10% of the nonirradiated level, which reflects a lack of repair of CPDs. The levels of rRNA elongation measured in nuclei isolated soon after irradiation from wt and rad cells corresponds to about 55% and 45%, respectively, of the levels obtained for nuclei from nonirradiated cells (Fig. 5, compare −UV and 0 h). This is expected from the limited elongation by RNA polymerases on damaged transcribed strands of wt, rad1Δ, and rad14Δ cells. While elongating RNA polymerases just upstream of DNA lesions incorporate only low levels of radioactive UTP before encountering a UV photoproduct, RNA polymerases located downstream of damage sites contribute significantly to the signals measured at 0 h repair time in wt and rad cells.
FIG. 5.
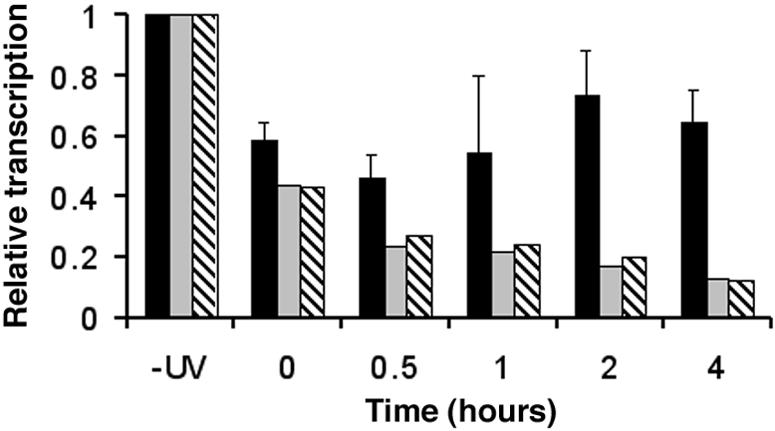
Transcription of ribosomal genes during NER in wt, rad1Δ, and rad14Δ mutant strains. Nuclei were isolated from nonirradiated (−UV) cells and from UV-irradiated cultures at different repair times. After incubation under TRO reaction conditions, the purified radiolabeled RNA was used as a probe to hybridize the membrane-bound rDNA. Membranes were then exposed to phosphorimager screens, and the rDNA signals were normalized to the corresponding signals for the external standard (13). Transcription of the nonirradiated cultures is given an arbitrary value of 1, and transcription of other samples is expressed relative to this value. Black bars, wt strain; gray bars, rad1Δ strain; hatched bars, rad14Δ strain. Data are the means ± 1 SD of three independent experiments for wt and the average of two independent experiments for rad1Δ and rad14Δ cells.
During the first 30 min of repair, most RNA polymerases that are not blocked at a DNA lesion are expected to be at (or near) the end of elongation (34). Therefore, the TRO levels of ribosomal genes measured in nuclei from rad1Δ and rad14Δ cells from 30 min to 1 h are only about 20% of TRO levels measured in nuclei from nonirradiated cells (compare 0.5 to 1 h and −UV). Furthermore, it is believed that transcription initiation does not occur during TRO (34), explaining why transcription levels continue to decrease in rad cells after the 0 h time. Finally, TRO levels obtained from rad1Δ and rad14Δ nuclei after 4 h of repair are close to the background levels of our assays (see also Fig. 6C). In contrast, there is a slow recovery of transcription elongation in wt cells during the first hour of repair (Fig. 5; compare black with gray and hatched bars at 0.5 and 1 h) coincident with removal of CPDs from the TS of active ribosomal genes (Fig. 3D). After 2 to 4 h of repair, about 80% of CPDs are removed from the TS, and rRNA transcription elongation recovers to about 75% of the values measured in nuclei from nonirradiated cells. In addition, after 4 h nonirradiated cells in mid-exponential phase are not arrested and proceed to early stationary phase. In these cells rDNA transcription drops to less than 20% (Fig. 6).
FIG. 6.
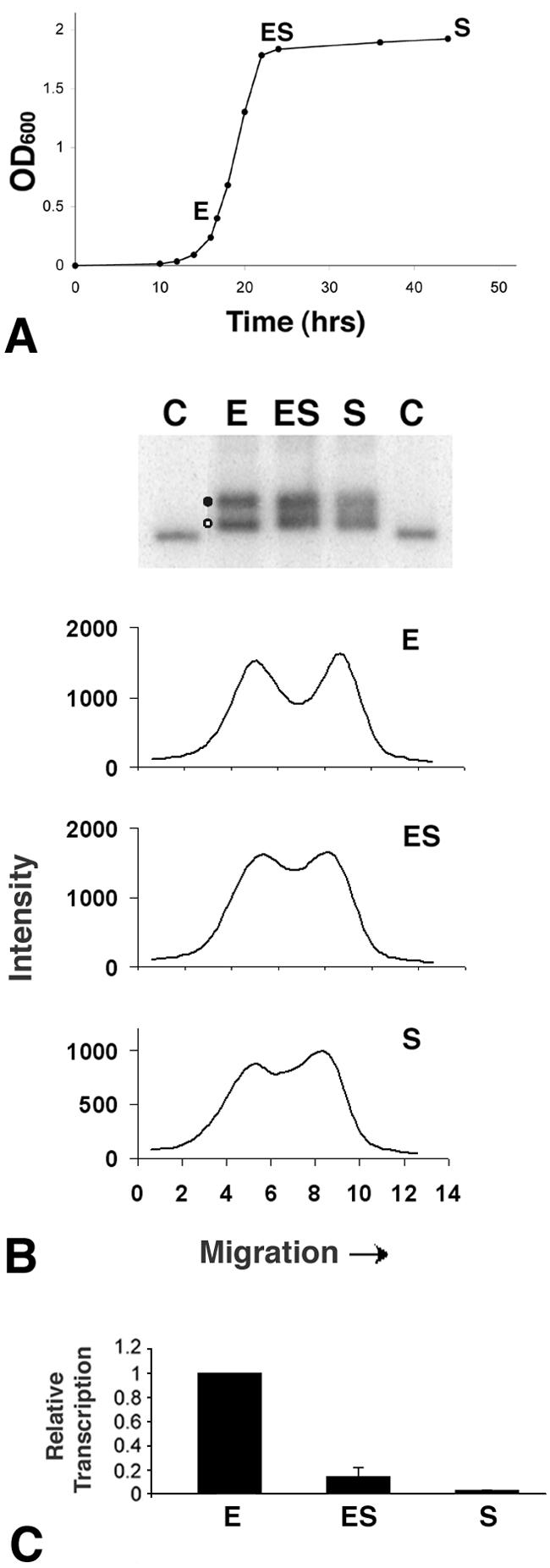
Transcription and chromatin structure of ribosomal genes during cell growth. (A) Cells were grown in YEPD medium and samples for nuclei isolation were harvested during exponential (E) growth (optical density at 600 nm [OD600] of 0.4 × 107 to ∼1.2 × 107 cells/ml), at early stationary phase (ES; 6 h after exponential phase) and at stationary phase (S; 24 h after exponential phase). (B) Chromatin structure of ribosomal genes during cell growth phases. At top, nuclei isolated from cells collected at selected growth stages, as described for panel A, were cross-linked with psoralen. DNA was extracted from nuclei, digested with EcoRI, and separated on 1% native agarose gels (lanes E, ES, and S). As a control (C), DNA was isolated from non-cross-linked nuclei and digested with EcoRI. After blotting, the filter membranes were hybridized with the random primer-labeled probe (Fig. 1B). Labels at left denote active rDNA (filled circle) and inactive rDNA (open circle). Below the blot are three scan profiles of the gels. (C) Transcription of ribosomal genes at different cell growth phases. Nuclei isolated from cells collected at selected growth stages as described for panel A were used for TRO reactions. The purified radiolabeled RNA was employed as a probe to hybridize the membrane-bound rDNA. Membranes were then exposed to phosphorimager screens, and the rDNA signals were normalized to the corresponding signals for the external standard. Transcription of the exponential phase culture is given an arbitrary value of 1, and transcription of the other samples is expressed relative to this value. Data are from the average of three independent experiments.
Decrease in RNA polymerase I elongation in non UV-irradiated cells does not support major chromatin assembly in active ribosomal genes.
As with other organisms, rRNA synthesis in yeast is coupled to growth rate (24; reviewed in reference 64). Since the presence of CPDs in the TS of active rDNA causes inactivation of transcription and assembly of chromatin, we wished to establish whether comparable changes in the structure of chromatin occur when cell growth decreases and rDNA transcription is diminished. Nuclei were isolated from exponentially growing cells having a doubling time of ∼1.5 h, from early stationary cells and from stationary cells (Fig. 6A). Aliquots of nuclei were cross-linked with psoralen, and the results presented in Fig. 6B (upper panel) clearly show that exponentially growing cells contain both nonnucleosomal (filled circle) and nucleosomal (open circle) rDNA. As cells approach stationary phase, the separation of active and inactive rDNA-bands decreases. However, scans of the lanes for early stationary and stationary cells clearly reveal two local maxima (Fig. 6B, lower panel, ES and S, respectively) and the corresponding bands can be deconvoluted into two distinct populations (data not shown). These results differ from the data obtained for wt cells 30 to 60 min after irradiation and for rad1Δ and rad14Δ cells 30 min to 4 h after irradiation, where the local maximum corresponding to the active fraction is not discernible (Fig. 4C).
To analyze rRNA synthesis during cell growth, the TRO assay was performed on aliquots of the same nuclei. The results presented in Fig. 6C show that RNA polymerase I activity decreases as cells leave the exponential growth phase. Thus, despite the drop in transcription in early stationary phase cells, a substantial portion of ribosomal genes is still present in a nonnucleosomal conformation (Fig. 6B) and, as previously shown, the fraction of active versus inactive rDNA repeats does not significantly change (13).
Nucleosome loading onto active ribosomal genes is promoted by the occurrence of UV photoproducts.
As shown above, the presence of photoproducts and the decrease in cellular growth rate reduce rRNA synthesis to about 10 to 20% of the maximal levels. Concomitantly, changes in the structure of rDNA chromatin occurred in both instances, although with significant differences. Since nucleosomal DNA is highly protected from digestion by restriction enzymes (25), this assay was employed to further characterize the structure of rDNA chromatin during NER and cell growth.
Nuclei were isolated from wt, rad1Δ, and rad14Δ cells during NER. In parallel, nuclei were isolated from the wt strain during cell growth. Isolated nuclei were digested with EcoRI prior to DNA purification and gel electrophoresis (Fig. 1A, e), and representative exposures of hybridized filter membranes are shown in Fig. 7 (A and B, upper panels; D, left panel). To correct for possible sample-to-sample variations during DNA extraction, parallel aliquots were spotted in triplicate directly onto membranes (lower panels). The signals were quantified, and plots of the data are shown in Fig. 7C and D. These experiments illustrate that the degree of EcoRI accessibility to rDNA chromatin changes during NER and correlates with the extent of psoralen cross-linking (compare Fig. 7 with Fig. 4). Specifically, the presence of CPDs and/or inactivation of transcription by damaged DNA promote the assembly of nucleosomes on the active portion of ribosomal genes. In contrast to wt cells, where removal of CPDs restores the original EcoRI sensitivity, measurements in rad1Δ and rad14Δ cells show that EcoRI accessibility remains at about 30% of that obtained before UV irradiation (Fig. 7C). Finally, despite the drop of RNA polymerase I transcription as cells reach the stationary phase (Fig. 6C), the sensitivity to EcoRI remains unchanged (Fig. 7D), again substantiating the results obtained by psoralen cross-linking (Fig. 6B).
FIG. 7.

EcoRI accessibility to rDNA chromatin during NER and during cell growth. (A) Chromatin structure of ribosomal genes during NER in wt cells (top gel). Nuclei were isolated from nonirradiated (−UV; lane 2) or irradiated (lanes 3 to 7) cells, before (lane 3) and during (lanes 4 to 7) NER. After EcoRI digestion, DNA was extracted from nuclei and separated on 1% native agarose gels. As a control (C), DNA was isolated from nuclei and digested with EcoRI (lane 8). After being blotted, the filter membranes were hybridized with the random primer-labeled probe (Fig. 1B) to obtain the major band E4-E5 (EcoRI; 2,846 bp). The bands E3-E7 (4,468 bp), E3-E6 (3,877 bp), E4-E7 (3,804 bp), E3-E5 (3,510 bp) and E4-E6 (3,213 bp) are products of partial digestions (Fig. 1B). DNA size-markers (lane M): 5,090, 4,072, 3,054 and 2,036 bp. To correct for gel loading, the amount of total rDNA present in each sample was quantified by spotting aliquots of DNA in triplicate (bottom gel). The membranes were hybridized with the probe shown in Fig. 1B and exposed to phosphorimager screens. The average of each triplicate was used to correct for loading. (B) Chromatin structure of ribosomal genes during NER in rad1Δ strains. Experimental procedure and lanes are as described for panel A. (C) For wt cells, signals for the E4-E5 (EcoRI) bands were quantified, and, after corrections for DNA loading, the rDNA signals were normalized to the −UV samples (given an arbitrary value of 1). The EcoRI sensitivity of the other samples is expressed relative to this value (data are the means ± 1 SD of three experiments). Similar analyses were performed with gels from rad1Δ and rad14 Δ cells. Data show the average of two independent experiments each. (D) Chromatin structure of ribosomal genes during cell growth. Nuclei isolated from cells collected at selected growth stages (Fig. 6A) were digested with EcoRI. DNA was extracted and separated on 1% native agarose gels. As a control (C), DNA was isolated from nuclei and digested with EcoRI. After being blotted, membranes were hybridized with the random primer-labeled probe (Fig. 1B). Lanes are as described for Fig. 6A, and quantification of the gel (right panel) was performed as described for panel C. Data are the means ± 1 SD of three independent experiments.
The chromatin structure of rDNA, as well as the inactive GAL locus, was also analyzed by the micrococcal nuclease (MNase) assay. The coexistence of inactive and active rDNA copies limits the interpretation of biochemical assays such as the standard MNase digestion repeat (32). In fact, MNase digestions of rDNA chromatin produce a nucleosomal ladder (deriving from inactive rDNA) over an undefined smear of fragments with random sizes (deriving from active rDNA) (59). Since after UV irradiation active rDNA sequences are only partially assembled into inactive chromatin (Fig. 4), it is difficult to determine by classical MNase digestion whether the signal of the nucleosomal ladder increases, compared to the same signal obtained from nonirradiated cells. Thus, we compared the MNase sensitivities of fragments from the GAL and rDNA loci having similar lengths. Nuclei were isolated from wt cells that were allowed to repair for different times after UV irradiation. Aliquots of nuclei were treated with MNase, and the DNA was purified as described in Materials and Methods. Equal amounts of DNA were double digested with the restriction enzymes EcoRI and PvuII to release a ∼1.96-kb rDNA fragment (Fig. 1B) and a ∼1.66-kb GAL fragment (Fig. 8A). After gel electrophoresis and blotting, the membranes were first hybridized with a probe specific to the GAL locus (Fig. 8A), and the results are shown in Fig. 8B (upper panel). Then, the same membranes were rehybridized with a probe specific to the rDNA locus (Fig. 1B), and the results are shown in Fig. 8B (lower panel). The signals, quantified and plotted in Fig. 8C, show that no changes in MNase sensitivity occur during NER of the inactive GAL locus. Conversely, a decrease in MNase sensitivity was measured in the rDNA locus at 0.5- and 1-h repair incubation times (Fig. 8C). These data further strengthen the results from the previous experiments showing that chromatin assembly occurs on the nonnucleosomal active rDNA.
FIG. 8.
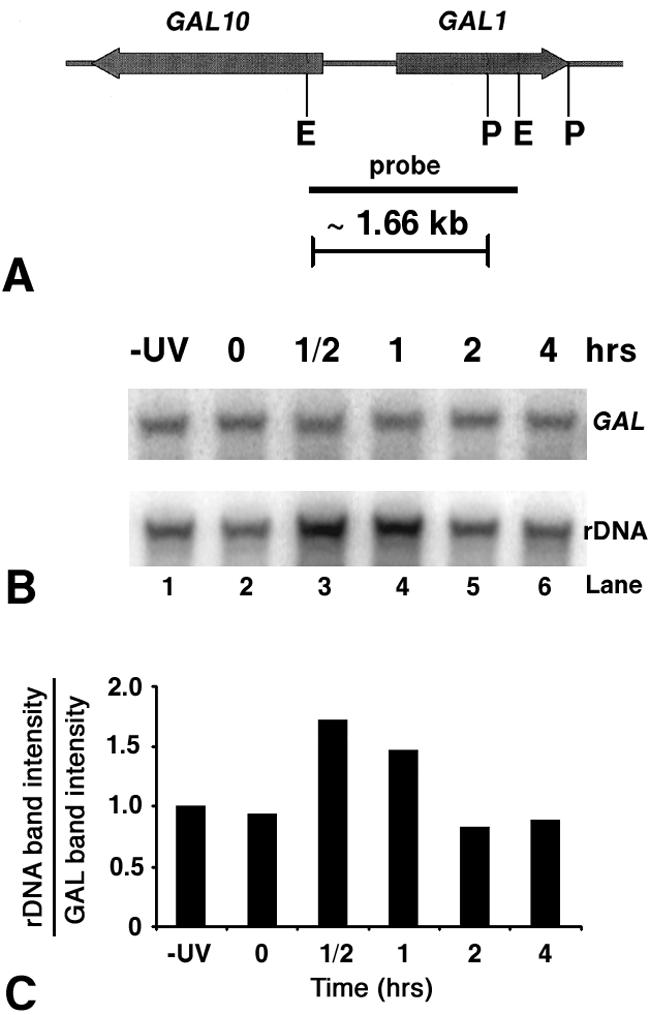
MNase accessibility of rDNA and GAL chromatin during NER in wt cells. (A) Map of the GAl10-GAL1 locus. Thick arrows represent the GAL10 and GAL1 genes and directions of transcription. The solid bar denotes the location of the probe used in this work. The positions of the two EcoRI restriction sites (E) and the two PvuII restriction sites (P) are also shown. (B) MNase sensitivity of the GAL10-GAL1 region and ribosomal genes during NER in wt cells. Nuclei were isolated from nonirradiated (−UV, lane 1) or irradiated (lanes 2 to 6) cells, before (lane 2) and during (lanes 3 to 6) NER. After MNase treatment, DNA was extracted from nuclei, digested with EcoRI and PvuII, and separated on 1% native agarose gels. After being blotted, membranes were first hybridized with a random primer-labeled probe specific for the GAL locus (8A) to obtain the E-P band (∼1.66 kb). Membranes were rehybridized with probe specific for rDNA to obtain the P-E5 band (∼1.96 kb) shown in Fig. 1B. (C) Quantification of intensities of rDNA and GAL bands. Histogram shows the ratio of signals for the rDNA and GAL bands at different repair times. The rDNA/GAL ratios during NER were normalized to the −UV samples (given an arbitrary value of 1). Data are the average of two independent experiments.
DISCUSSION
To investigate the structure of active chromatin during NER, we took advantage of the yeast rDNA locus as a model, where active ribosomal genes are depleted of canonical nucleosomes over the entire coding region (∼6.9 kb) (6, 10). We first showed that repair of CPDs from the NTS of active rDNA is faster than from either strand of inactive rDNA, indicating that active rDNA chromatin is repaired faster than inactive rDNA chromatin. These results prompted us to investigate the mechanisms whereby NER operates in nonnucleosomal active chromatin. Two techniques, psoralen cross-linking and MNase digestion, were employed to study chromatin structure over long stretches of rDNA (∼2 to 3 kb), whereas restriction enzyme accessibility to rDNA chromatin was employed to detect changes in constrained domains (e.g., EcoRI sites). All three methods show that upon irradiation with UV light, chromatin assembly occurs onto active ribosomal genes and in the absence of NER. These results strongly suggest that formation of inactive chromatin is promoted by the presence of damaged DNA. It could be argued that changes in nuclease accessibility (restriction enzymes and MNase) and psoralen cross-linking to damaged active rDNA are caused by the presence of the NER complex. However, these changes are observed in the absence of the damage recognition protein Rad14, known to participate in a very early step of the NER pathway and to be required for targeting the repair complex to the damaged site (44). In addition, we observed these changes in the absence of Rad1 protein, required for the early step of endonuclease cleavage 5′ to the DNA lesion and for the correct assembly of the NER machinery at the damage site (44). Furthermore, the DNase I footprint of the NER complex encompasses only ∼30 nucleotides around the damaged site (63). Since an average yield of less than 1 CPD/kb was introduced in the active rDNA fraction at the UV dose used, it is unlikely that the presence of one NER complex per kilobase decreases MNase sensitivity and causes the major changes in psoralen cross-linking observed in our experiments. Moreover, psoralen cross-linking is inhibited by the presence of nucleosomes but not by the presence of large multiprotein complexes such as those used in transcription and replication nor by the presence of regulatory proteins at promoters, origins of replication, and enhancer DNA elements (54). Finally, from the average number of CPDs introduced in the rDNA sequence in our experiments, there is only a low probability that CPDs form at the EcoRI restriction sites. Yet EcoRI accessibility to the active rDNA genes decreases during the early repair times in wt cells and during the entire repair incubation time in two NER-deficient mutant strains. These results further support the data obtained with the MNase and psoralen cross-linking assays and suggest that inactive chromatin could spread from the site containing a UV lesion into flanking DNA regions, as previously reported for an in vitro repair assay (17).
The three independent methods employed in this work indicate that the presence of UV photoproducts, in the absence of NER, promote chromatin assembly onto active rDNA in vivo. It is possible to consider that minor UV photoproducts, such as thymine glycols (22), might lead to the alternative explanation that oxidative DNA damage and/or base excision repair could cause the inactivation of rDNA chromatin observed in our study. However, it is well established that formation of oxidative DNA damage (such as thymine glycols) following short-wavelength UV irradiation at these doses is negligible (reviewed in reference 4); J. Cadet, personal communication. Finally, it was recently reported that in mammalian cells p53 prevents the accumulation of double-strand DNA breaks at replication forks stalled at UV-induced pyrimidine dimers (56). Thus, it is also possible to consider that DNA ends in double-strand breaks and nicks could be responsible for chromatin assembly onto active rDNA. Indeed, in vitro experiments have shown repair-coupled deposition of nucleosomes at DNA nicks (17, 38). However, yeast cells do not contain p53, and the formation of double-strand breaks induced by UV radiation in our system is expected to be small. For instance, psoralen cross-linking experiments were also performed at very early times after UV irradiation, and the same amount of chromatin assembly observed after 30 min was also measured at ∼7 min after irradiation (data not shown). In addition, the yeast cell cycle is ∼1.5 h, ∼20 to 25% of the cells are in S phase at any given time in log-phase cultures, and the S phase lasts between 20 to 40 min. Consequently, the results of psoralen cross-linking experiments at very short times after irradiation suggest that UV photoproducts, rather than double-strand breaks, initiate chromatin assembly. Thus, although we cannot completely exclude the existence of a small number of double-strand breaks, we think it is much more likely that UV photoproducts alone promote, directly or indirectly (see below), chromatin assembly onto active rDNA in intact yeast cells.
Using the TRO assay, decreased rDNA transcription was measured and correlated with the incidence of CPDs. It is known that the presence of UV photoproducts on the TS of active genes forces RNA polymerase to stall and, in Escherichia coli, the incomplete transcription complex to dissociate (48). Even though it is still unclear whether RNA polymerase II complexes dissociate at damage sites (28, 29; reviewed in reference 58), it was shown that the transcription release factor 2 discharges RNA polymerases I and II stalled at CPDs (21). Therefore, the displacement of RNA polymerase I could also promote the formation of inactive rDNA chromatin. On the other hand, the reduction in rDNA transcription during the transition from exponential cell growth to stationary phase has little effect on the structure of rDNA chromatin. This was previously observed in higher eukaryotes, where nonnucleosomal rDNA is preserved in stationary cells and in metaphase chromosomes and was explained by the presence of nontranscribing (or very slow transcribing) RNA polymerase I that prevents nucleosome formation in rDNA (47, 65; reference 9 and references therein; reviewed in reference 45).
In summary, we have shown that there is a repair-independent chromatin assembly onto active ribosomal genes after UV irradiation and that chromatin disassembly occurs only when CPDs are removed in the NER-competent cells. Although at present we cannot distinguish whether inactive chromatin forms before the arrest of transcription or whether arrest of transcription precedes the formation of inactive chromatin, it is clear that chromatin assembly occurs at very early times after UV irradiation. Interestingly, in wt cells this process takes place before repair of CPDs can be measured and is in line with a new series of observations showing that nucleosomes assemble at, and propagate from, the damaged sites before the repair synthesis step of NER (17, 38). Furthermore, it was recently shown that hyperacetylation of histone H3 and chromatin remodeling occur within minutes at the repressed MFA2 promoter in yeast after UV irradiation and in the absence of NER (67), suggesting that certain chromatin modifications occur independently of NER. Altogether, these findings indicate that nucleosomes may play an active role in NER rather than simply being a passive impediment.
Acknowledgments
We are grateful to L. Gaudreau and B. Leblanc at the Université de Sherbrooke for critical reading of the manuscript and to M. Tremblay for technical help.
The majority of this work was supported by a grant from the Canadian Institutes of Health Research (CIHR) to A.C. D.F. and V.B. were supported by National Institutes of Health grants ES02614 and ES04106 from the National Institute of Environmental Health Sciences (NIEHS) to M.J.S.
The contents of this study are solely the responsibility of the authors and do not represent the official views of the NIEHS.
REFERENCES
- 1.Baker-Brachmann, C., A. Davies, G. J. Cost, E. Caputo, J. Li, P. Hieter, and J. D. Boeke. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14:115-132. [DOI] [PubMed] [Google Scholar]
- 2.Banditt, M., T. Koller, and J. M. Sogo. 1999. Transcriptional activity and chromatin structure of enhancer-deleted rRNA genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 19:4953-4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bohr, V. A., and D. S. Okumoto. 1988. Analysis of pyrimidine dimers in defined genes, p. 347-366. In E. C. Friedberg and P. C. Hanawalt (ed.), DNA repair: a laboratory manual of research procedures. Marcel Dekker, Inc., New York, N.Y.
- 4.Cadet, J., E. Sage, and T. Douki. 2005. Ultraviolet radiation-mediated damage to cellular DNA. Mutat. Res. 571:3-17. [DOI] [PubMed] [Google Scholar]
- 5.Christians, F. C., and P. C. Hanawalt. 1992. Inhibition of transcription and strand-specific DNA repair by alpha-amanitin in Chinese hamster ovary cells. Mutat. Res. 274:93-101. [DOI] [PubMed] [Google Scholar]
- 6.Conconi, A. 2005. The yeast rDNA locus: a model system to study DNA repair in chromatin. DNA Repair 4:897-908. [DOI] [PubMed] [Google Scholar]
- 7.Conconi, A., V. A. Bespalov, and M. J. Smerdon. 2002. Transcription-coupled repair in RNA polymerase I-transcribed genes of yeast. Proc. Natl. Acad. Sci. USA 99:649-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conconi, A., J. M. Sogo, and C. A. Ryan. 1992. Ribosomal gene clusters are uniquely proportioned between open and closed chromatin structures in both tomato leaf cells and exponentially growing suspension cultures. Proc. Natl. Acad. Sci. USA 89:5256-5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conconi, A., R. M. Widmer, T. Koller, and J. M. Sogo. 1989. Two different chromatin structures coexist in ribosomal RNA genes throughout the cell cycle. Cell 57:753-776. [DOI] [PubMed] [Google Scholar]
- 10.Dammann, R., R. Lucchini, T. Koller, and J. M. Sogo. 1993. Chromatin structures and transcription of rDNA in yeast Saccharomyces cerevisiae. Nucleic Acids Res. 21:2331-2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dammann, R., R. Lucchini, T. Koller, and J. M. Sogo. 1995. Transcription in the yeast rDNA locus: distribution of the active gene copies and chromatin structure of their flanking regulatory sequences. Mol. Cell. Biol. 15:5294-5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dodson, M. L., and R. S. Lloyd. 1989. Structure-function studies of the T4 endonuclease V repair enzyme. Mutat. Res. 218:49-65. [DOI] [PubMed] [Google Scholar]
- 13.Fahy, D., A. Conconi, and M. J. Smerdon. 2005. Rapid changes in transcription and chromatin structure of ribosomal genes in yeast during growth phase transitions. Exp. Cell Res. 305:365-373. [DOI] [PubMed] [Google Scholar]
- 14.Ferreiro, J. A., N. G. Powell, N. Karabetsou, N. A. Kent, J. Mellor, and R. Waters. 2004. Cbf1p modulates chromatin structure, transcription and repair at the Saccharomyces cerevisiae MET16 locus. Nucleic Acids Res. 32:1617-1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedberg, E. C., G. C. Walker, and W. Siede. 1995. DNA repair and mutagenesis. ASM Press, Washington, D.C.
- 16.Gaillard, P.-H. L., E. M.-D. Martini, P. D. Kaufman, B. Stillman, E. Moustacchi, and G. Almouzni. 1996. Chromatin assembly coupled to DNA repair: a new role for chromatin assembly factor I. Cell 86:887-896. [DOI] [PubMed] [Google Scholar]
- 17.Gaillard, P.-H. L., J. G. Moggs, D. M. J. Roche, J.-P. Quivy, P. B. Becker, R. D. Wood, and G. Almouzni. 1997. Initiation and bidirectional propagation of chromatin assembly from a target site for nucleotide excision repair. EMBO J. 16:6281-6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gong, F., Y. H. Kwon, and M. J. Smerdon. 2005. Nucleotide excision repair in chromatin and the right of entry. DNA Repair 4:884-896. [DOI] [PubMed] [Google Scholar]
- 19.Green, C. M., and G. Almouzni. 2001. When repair meets chromatin. EMBO Rep. 3:28-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grummt, I., and C. S. Pikaard. 2003. Epigenetic silencing of RNA polymerase I transcription. Nature Rev. Mol. Cell. Biol. 4:641-649. [DOI] [PubMed] [Google Scholar]
- 21.Hara, R., C. P. Selby, M. Liu, D. H. Price, and A. Sancar. 1999. Human transcription release factor 2 dissociates RNA polymerase I and II stalled at a cyclobutane thymine dimer. J. Biol. Chem. 274:24779-24786. [DOI] [PubMed] [Google Scholar]
- 22.Hariharan, P. V., and P. Cerutti. 1977. Formation of products of the 5,6-dihydroxydihydrothymine type by ultraviolet light in HeLa cells. Biochem. 16:2791-2795. [DOI] [PubMed] [Google Scholar]
- 23.Holmquist, G. P., and V. M. Maher. 2002. The bypass of DNA lesions by DNA and RNA polymerases. Mutat. Res. 510:1-7. [DOI] [PubMed] [Google Scholar]
- 24.Kief, D. R., and J. R. Warner. 1981. Coordinate control of syntheses of ribosomal ribonucleic acid and ribosomal proteins during nutritional shift-up in Saccharomyces cerevisiae. Mol. Cell. Biol. 1:1007-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kireeva, M. L., W. Walter, V. Tchernajenko, V. Bondarenko, M. Kashlev, and V. M. Studitsky. 2002. Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol. Cell 9:541-552. [DOI] [PubMed] [Google Scholar]
- 26.Labhart, P., and R. H. Reeder. 1989. High initiation rates at the ribosomal gene promoter do not depend upon spacer transcription. Proc. Natl. Acad. Sci. USA 86:3155-3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leadon, S. A., and D. A. Lawrence. 1991. Preferential repair of DNA damage on the transcribed strand of the human metallothionein gene requires RNA polymerase II. Mutat. Res. 255:67-78. [DOI] [PubMed] [Google Scholar]
- 28.Lee, S.-K., S.-L. Yu, L. Prakash, and S. Prakash. 2001. Requirement for yeast RAD26, a homolog of the human CSB gene, in elongation by RNA polymerase II. Mol. Cell. Biol. 21:8651-8656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee, S.-K., S.-L. Yu, L. Prakash, and S. Prakash. 2002. Yeast RAD26, a homolog of the human CSB gene, functions independently of nucleotide excision repair and base excision repair in promoting transcription through damaged bases. Mol. Cell. Biol. 22:4383-4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li, S., M. Livingstone-Zatchej, R. Gupta, M. Meijer, F. Thoma, and M. J. Smerdon. 1999. Nucleotide excision repair in a constitutive and inducible gene of a yeast minichromosome in intact cells. Nucleic Acids Res. 27:3610-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lieberman, M. W., M. J. Smerdon, T. D. Tlsty, and F. B. Oleson. 1979. The role of chromatin structure in DNA repair in human cells damaged with chemical carcinogens and ultraviolet radiation, p. 345-363. In P. Emmelot and E. Kriek (ed.), Environmental carcinogenesis. Elsevier/North Holland Biomedical Press, Amsterdam, The Netherlands.
- 32.Lucchini, R., and J. M. Sogo. 1998. The dynamic structure of ribosomal RNA gene chromatin, p. 254-276. In M. R. Paule (ed.), Transcription of ribosomal RNA genes by eukaryotic RNA polymerase I. Landes Bioscience, Austin, Tex.
- 33.Lucchini, R., and J. M. Sogo. 1995. Replication of transcriptionally active chromatin. Nature 374:276-280. [DOI] [PubMed] [Google Scholar]
- 34.Marzluff, W. F., and R. C. C. Huang. 1985. Transcription of RNA in isolated nuclei, p. 89-129. In B. D. Hames and S. J. Higgins (ed.), Transcription and translation. IRL Press, Oxford, United Kingdom.
- 35.Meier, A., M. Livingstone-Zatchej, and F. Thoma. 2002. Repair of active and silenced rDNA in yeast. J. Biol. Chem. 277:11845-11852. [DOI] [PubMed] [Google Scholar]
- 36.Mello, J. A., and G. Almouzni. 2001. The ins and outs of nucleosome assembly. Curr. Opin. Gen. Dev. 11:136-141. [DOI] [PubMed] [Google Scholar]
- 37.Mellon, I., G. Spivak, and P. C. Hanawalt. 1987. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell 51:241-249. [DOI] [PubMed] [Google Scholar]
- 38.Moggs, J. G., P. Grandi, J.-P. Quivy, Z. O. Jonsson, U. Hubscher, P. B. Becker, and G. Almouzni. 2000. A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol. Cell. Biol. 20:1206-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moss, T., and V. Y. Stefanovsky. 2002. At the center of eukaryotic life. Cell 109:545-548. [DOI] [PubMed] [Google Scholar]
- 40.Mueller, J. P., and M. J. Smerdon. 1995. Repair of plasmid and genomic DNA in a rad7 delta mutant of yeast. Nucleic Acids Res. 23:3457-3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mueller, J. P., and M. J. Smerdon. 1996. Rad23 is required for transcription-coupled repair and efficient overall repair in Saccharomyces cerevisiae. Mol. Cell. Biol. 16:2361-2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ness, P. J., P. Labhart, E. Banz, T. Koller, and R. W. Parish. 1983. Chromatin structure along the ribosomal DNA of Dictyostelium. Regional differences and changes accompanying cell differentiation. J. Mol. Biol. 166:361-381. [DOI] [PubMed] [Google Scholar]
- 43.Powell, N. G., J. Ferreiro, N. Karabetsou, J. Mellor, and R. Waters. 2003. Transcription, nucleosome positioning and protein binding modulate nucleotide excision repair of the Saccharomyces cerevisiae MET17 promoter. DNA Repair 2:375-386. [DOI] [PubMed] [Google Scholar]
- 44.Prakash, S., and L. Prakash. 2000. Nucleotide excision repair in yeast. Mutat. Res. 451:13-24. [DOI] [PubMed] [Google Scholar]
- 45.Reeder, R. H. 1999. Regulation of RNA polymerase I transcription in yeast and vertebrates. p. 293-327. In K. Moldave (ed.), Progress in nucleic acids research and molecular biology, vol. 62. Academic Press, San Diego. [DOI] [PubMed] [Google Scholar]
- 46.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1982. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 47.Scheer, U., and K. M. Rose. 1984. Localization of RNA polymerase I in interphase cells and mitotic chromosomes by light and electron microscopic immunocytochemistry. Proc. Natl. Acad. Sci. USA 81:1431-1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Selby, C. P., and A. Sancar. 1993. Molecular mechanism of transcription-repair coupling. Science 260:53-58. [DOI] [PubMed] [Google Scholar]
- 49.Smerdon, M. J., and A. Conconi. 1999. Modulation of DNA damage and DNA repair in chromatin, p. 227-255. In K. Moldave (ed.), Progress in nucleic research and molecular biology, vol. 62. Academic Press, San Diego, Calif. [DOI] [PubMed] [Google Scholar]
- 50.Smerdon, M. J., and M. W. Liberman. 1980. Distribution within chromatin of deoxyribonucleic acid repair synthesis occurring at different times after ultraviolet radiation. Biochemistry 19:2992-3000. [DOI] [PubMed] [Google Scholar]
- 51.Smerdon, M. J. 1986. Completion of excision repair in human cells: relationship between ligation and nucleosome formation. J. Biol. Chem. 261:244-252. [PubMed] [Google Scholar]
- 52.Smerdon, M. J., and F. Thoma. 1990. Site-specific DNA repair at the nucleosome level in a yeast minichromosome. Cell 61:675-684. [DOI] [PubMed] [Google Scholar]
- 53.Smith, J. S., E. Caputo, and J. D. Boeke. 1999. A genetic screen for ribosomal DNA silencing defects identifies multiple DNA replication and chromatin-modulating factors. Mol. Cell. Biol. 19:3184-3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sogo, J. M., and F. Thoma. 2003. The structure of rDNA chromatin, p. 1-15. In M. O. J. Olson (ed.), The nucleolus. Kluwer Academic/ Plenum, New York, N.Y.
- 55.Sollner-Webb, B., and E. B. Mougey. 1991. News from the nucleolus: rRNA gene expression. Trends Biochem. Sci. 16:58-62. [DOI] [PubMed] [Google Scholar]
- 56.Squires, S., J. A. Coates, M. Goldberg, L. H. Toji, S. P. Jackson, D. J. Clarke, and R. T. Johnson. 2004. p53 prevents the accumulation of double-strand DNA breaks at stalled-replication forks induced by UV in human cells. Cell Cycle 3:1543-1557. [DOI] [PubMed] [Google Scholar]
- 57.Tijsterman, M., R. De Pril, J. G. Tasseron-De Jong, and J. Brouwer. 1999. RNA polymerase II transcription suppresses nucleosomal modulation of UV-induced (6-4) photoproduct and cyclobutane pyrimidine dimer repair in yeast. Mol. Cell. Biol. 19:934-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tornaletti, S., and P. C. Hanawalt. 1999. Effect of DNA lesions on transcription elongation. Biochimie 81:139-146. [DOI] [PubMed] [Google Scholar]
- 59.Toussaint, M., G. Levasseur, M. Tremblay, M. Paquette, and A. Conconi. 2005. Psoralen photocrosslinking, a tool to study the chromatin structure of RNA polymerase I-transcribed ribosomal genes. Biochem. Cell Biol. 83:449-459. [DOI] [PubMed] [Google Scholar]
- 60.van Gool, A. J., R. Verhage, S. M. A. Swagemakers, P. van de Putte, J. Brouwer, C. Troelstra, D. Bootsma, and J. H. J. Hoeijmakers. 1994. RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6. EMBO J. 13:5361-5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Verhage, R. A., P. van de Putte, and J. Brouwer. 1996. Repair of rDNA in Saccharomyces cerevisiae: RAD4-independent strand-specific nucleotide excision repair of RNA polymerase I-transcribed genes. Nucleic Acids Res. 24:1020-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vrieling, H., A. A. van Zeeland, and L. H. Mullenders. 1998. Transcription-coupled repair and its impact on mutagenesis. Mutat. Res. 400:135-142. [DOI] [PubMed] [Google Scholar]
- 63.Wakasugi, M., and A. Sancar. Assembly, subunit composition, and footprinting of human DNA repair excision nuclease. Proc. Natl. Acad. Sci. USA 95:6669-6674. [DOI] [PMC free article] [PubMed]
- 64.Warner, J. R. 1989. Synthesis of ribosomes in Saccharomyces cerevisiae. Microbiol. Rev. 53:256-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weisenberger, D., and U. Scheer. 1995. A possible mechanism for the inhibition of ribosomal RNA gene transcription during mitosis. J. Cell Biol. 129:561-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wellinger, R. E., and F. Thoma. 1999. Nucleosome structure and positioning modulate excision repair in the non-transcribed strand of an active gene. EMBO J. 16:5046-5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu, Y., Y. Teng, H. Liu, S. H. Reed, and R. Waters. 2005. UV irradiation stimulates histone acetylation and chromatin remodeling at a repressed yeast locus. Proc. Natl. Acad. Sci. USA 102:8650-8655. [DOI] [PMC free article] [PubMed] [Google Scholar]