

Abstract
Transforming growth factor β (TGF-β) has been implicated in the maintenance of homeostasis in various organs, including the gastric epithelium. In particular, TGF-β-induced signaling was shown to be required for the differentiation-associated physiological apoptosis of gastric epithelial cells, but its mechanism has not been well understood. In this study, the molecular mechanism of TGF-β-induced apoptosis was analyzed in a human gastric epithelial cell line, SNU16, as an in vitro model. Expression of Smad7 and Bcl-XL, but not viral FLIP, was shown to prevent TGF-β-induced apoptosis, indicating an exclusive requirement of the activation of Smad signaling pathway and mitochondrial dysfunction followed by activation of caspase-9. In addition, treatment with TGF-β induced binding of Bim, a proapoptotic Bcl-2 homology domain 3 (BH3)-only protein, to Bcl-XL, which is dependent on the activation of Smad, and reduction in the expression of Bim by RNA interference decreased the sensitivity to TGF-β-induced apoptosis. Moreover, we found abnormalities in the gastric epithelium of both Bim and caspase-9 knockout mice; these abnormalities were associated with a defect of physiological apoptosis in gastric epithelial cells. These results indicate for the first time that TGF-β is involved in the physiological loss of gastric epithelial cells by activating apoptosis mediated by Smad, Bim, and caspase-9.
Gastric epithelial cells exhibit a rapid rate of turnover, which requires an appropriate balance between the proliferation of progenitor cells and the loss of mature cells (12). Loss of mature epithelial cells at the gastric surface is thought to be mediated mainly by physiological cell death, namely, apoptosis, which always occurs at the surface of the gastrointestinal tract. In the gastric mucosa, 1 to 3% of the epithelial cells were reported to show morphological features of apoptosis at any given time under physiological conditions (47). The physiological loss of gastric epithelial cells is necessary for the maintenance of tissue homeostasis associated with the exchange of mature epithelial cells to fresh proliferating cells, and any defects in this epithelial cell death pathway may be a contributing factor to disease development. However, little direct evidence linking the gastric epithelial cell death and apoptosis-related molecules has been obtained and the molecular regulation of apoptosis of gastric epithelial cells largely remains unclear. In this study, we focused on transforming growth factor β (TGF-β) as one of the key regulators in this physiological cell death.
TGF-β is a multifunctional cytokine, which has essential roles in a variety of physiological or pathological processes (9, 42). While TGF-β functions as a potent suppressor of cell proliferation, it can also induce or suppress apoptosis in certain types of cells (39, 42). Previous studies have established that the acquisition of resistance to TGF-β is a critical step for carcinogenesis in many organs (8). Abnormal expression of TGF-β receptors and inactive mutations of Smad family genes are frequently detected in human gastric carcinomas (17, 18, 32). In addition, genetic approaches with mutant mice in which TGF-β signaling was abolished indicated that some of these mutants exhibited not only unique epithelial hyperplasia in the gastric mucosa but also an increased incidence of gastric cancer (3, 6, 11, 26, 41, 45). Thus, TGF-β has been shown to play an important role in the morphogenesis and maintenance of structure and function of the gastric epithelium. It is noteworthy that levels of physiological apoptosis are abnormally low in the epithelium of glandular stomachs and forestomachs in Runx3-deficient mice (26) and Smad7-transgenic mice (13), respectively, both of which exhibit defects in TGF-β-induced signaling. Hyperplasia in the gastric epithelium of these mutant mice might be caused not only by loss of cell proliferation-inhibitory activity but also by reduced apoptotic loss of epithelial cells from the surface of the stomach. These previous studies prompted us to speculate that TGF-β is involved in the regulation of turnover of gastric epithelial cells through their proapoptotic activity. Although a number of previous studies proposed that TGF-β could control apoptosis through the up-regulation or posttranslational modification of some proapoptotic effectors, including mitogen-activated protein (MAP) kinases such as JNK and p38 MAP kinase, death-associated protein (DAP) kinase, DAXX, SHIP, and ARTS (for a review, see reference 42), these reports were often contradictory and the ability of TGF-β to induce or suppress apoptosis seems to vary markedly depending on the cellular lineage or experimental condition. The exact molecular regulation of TGF-β-dependent apoptosis is therefore still unclear.
In this study, we examined the signaling pathway of TGF-β1-induced apoptosis in a human gastric epithelial cell line, SNU16. SNU16 cells are highly sensitive to TGF-β1 (21, 26), while most gastric carcinoma-derived cell lines are resistant to TGF-β1 due to defects in the TGF-β signaling machinery. We speculated that the response of SNU16 cells to TGF-β1 might mimic the response of normal gastric epithelial cells in vivo. We show here that TGF-β1-induced apoptosis is mediated by the sequential activation of Smad; Bim, a member of Bcl-2 homology domain 3 (BH3)-only protein family; and caspase-9. Moreover, analysis of the stomachs of Bim−/− and caspase-9−/− mice revealed a profound role for TGF-β-mediated activation of Bim and caspase-9 in the physiological turnover of the gastric epithelium.
MATERIALS AND METHODS
Cells and plasmids.
Human gastric carcinoma-derived SNU16 cells were kindly provided by Y. Ito and K. Ito (Institute for Molecular and Cell Biology, Singapore). SNU16 cells were cultured in RPMI 1640 medium supplemented with 15% (vol/vol) fetal bovine serum, 20 mM HEPES (pH 7.4), 50 μM β-mercaptoethanol, 50 units/ml penicillin, and 50 μg/ml streptomycin. Expression constructs for FLAG-tagged viral FLICE/caspase-8 inhibitory protein (v-FLIP) E8, FLAG-tagged Bcl-XL, and green fluorescence protein (GFP) were previously prepared in our laboratories using pME18S vector (15). Expression constructs for FLAG-tagged Smad7 and Myc-tagged dominant-negative MKK7 were kindly provided by K. Miyazono (University of Tokyo) and E. Nishida (Kyoto University), respectively. SNU16 cells were transfected with expression vectors by use of LipofectAMINE Plus (Invitrogen) according to the manufacturer's instructions.
Detection of apoptotic cells.
The DNA ladder assay was performed as previously described (28). Apoptotic cells with subdiploid DNA content were detected as previously reported (15). In brief, cells were harvested and stained with 50 μg/ml propidium iodide (PI) in phosphate-buffered saline containing 0.1% sodium citrate, 0.1% Triton X-100, and 50 μg/ml RNase A. The DNA content of cells was analyzed with an EPICS XL flow cytometer (Beckman Coulter). For the analysis of GFP-expressing cells, cells were transfected with a GFP expression vector together with various other expression vectors. Transfected cells were fixed first in 3.7% formaldehyde and then in cold 70% ethanol. After treatment with 50 μg/ml RNase A solution, cells were stained with 50 μg/ml PI. The DNA content of the GFP-expressing cells was determined by two-color analysis with a flow cytometer. Cells with activated caspase-3 were detected using a phycoerythrin (PE)-fluorescein isothiocyanate (FITC)-conjugated monoclonal active caspase-3 antibody apoptosis kit (BD PharMingen) according to the manufacturer's instructions. FITC- or PE-positive cells were analyzed with a flow cytometer.
Western blotting and immunoprecipitation.
Cells were suspended in cold lysis buffer (20 mM HEPES-KOH [pH 7.4] containing 150 mM NaCl, 1 nM EDTA, 1% Triton X-100, and protease inhibitor mixture [Roche]). For immunoprecipitation, cell lysate was incubated first with appropriate antibodies for 1 h and subsequently with protein A-Sepharose (Amersham) overnight. Cell lysates and immunoprecipitates were analyzed by Western blotting as described previously (25). To fractionate whole cell lysates into cytosolic and membrane-rich fractions, cells were suspended in 20 mM HEPES-KOH (pH 7.4) containing 10 mM KCl, 1.5 mM MgCl2, 1 nM EDTA, 1 mM EGTA, 1 mM dithiothreitol, and 1 mM phenymethylsulfonyl fluoride and incubated for 15 min on ice. Cells were disrupted using a Dounce homogenizer, and the homogenate was centrifuged at 600 × g for 10 min. The supernatant was further centrifuged at 10,000 × g for 15 min to obtain a cytosolic fraction and heavy membrane-rich pellet.
To detect conformational change of Bax, cells were suspended in 10 mM HEPES-KOH (pH 7.4) containing 150 mM NaCl, 1% 3-[(3-cholamidopropyl)dimethyl-ammonio] propanesulfonic acid (CHAPS), and the protease inhibitor mixture. Cell lysates were immunoprecipitated with anti-Bax monoclonal antibody (6A7 BD; PharMingen), and immunoprecipitates were then subjected to Western blotting with anti-Bax polyclonal antibody (Cell Signaling). To examine Bax oligomerization, the heavy membrane-rich fraction was incubated for 30 min in 20 mM HEPES-NaOH (pH 7.0) containing 150 mM NaCl and 1 mM BMH cross-linker (Pierce). The cross-linking reaction was quenched by adding dithiothreitol, and Western blotting was then performed with anti-Bax antibody (Cell Signaling).
Also used in this study were antibodies to caspase-8 (5F7; MBL), caspase-9 (Cell Signaling), JNK1 (C-17; Santa Cruz), p38 (5F11; Cell Signaling), Smad2 (34G6; Cell Signaling), phospho-p38 (Cell Signaling), phospho-JNK (G9; Cell Signaling), phospho-Smad2 (Cell Signaling), cytochrome c (2C12; BD PharMingen), COX-4 (20E8; Molecular Probes), Bcl-X (BD PharMingen), Bcl-XL (for immunoprecipitation; Cell Signaling), Bcl-2 (BD PharMingen), Mcl-1 (BD PharMingen), Bad (C-7; Santa Cruz), Bid (Cell Signaling), Bim (14A8; Chemicon), Bmf (9G10; Alexis), actin (Chemicon), and FLAG epitope tag (M2; Sigma).
RNA interference.
Expression vectors for short hairpin RNA (shRNA) were based on the pSUPER RNAi system (OligoEngine). Each expression vector was constructed to generate short interference RNA corresponding to the following sequences: for Bimα1 cDNA (GenBank accession number AB071195), nucleotide (nt) 6 to 28 (2C2), nt 122 to 144 (2F6), and nt 505 to 527 (2H4); and for Bmf cDNA (GenBank accession number NM_033503), nt 553 to 575 (2A12) and nt 492 to 514 (2H7).
Mice and histological analyses of stomachs.
C57BL/6 mice were purchased from CLEA Japan Inc. The caspase-9−/− mice (22) and Bim−/− mice (37) on a C57BL/6 background were generated by backcrossing more than 10 times with C57BL/6 mice. The genotype of the mutant mice was identified by PCR assay with DNA isolated from tails. All mice were housed under specific-pathogen-free conditions with autoclaved water and controlled feeding until use.
Gastric tissues from newborn mice were fixed with 4% paraformaldehyde, embedded in paraffin, and serially sectioned at 8 μm. For the detection of apoptotic cells, sections were subjected to terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining, which was performed with an in situ cell death detection kit (Roche) according to the manufacturer's instructions. Sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). For histological analyses, paraffin-embedded tissue sections were stained with the periodic acid-Schiff (PAS) reaction solution and hematoxylin.
RESULTS
SNU16 cells undergo caspase-dependent apoptotic cell death in response to TGF-β1.
We analyzed the effects of TGF-β1 treatment on SNU16 cells. After 24 h of culture with 10 ng/ml TGF-β1, these cells showed decreased viability as well as severe growth arrest, and after 48 h of treatment with TGF-β1, more than 90% of SNU16 cells had died (Fig. 1A). TGF-β1-treated SNU16 cells exhibited morphological changes of the nucleus (chromosomal condensation) (data not shown), which is a classical feature of apoptotic cells. We also detected DNA fragmentation (Fig. 1B) and exposure of phosphatidylserine on the outer cell surface (see Fig. S1 in the supplemental material), both hallmarks of apoptosis, confirming that TGF-β1 induced typical apoptotic cell death in SNU16 cells.
FIG. 1.

TGF-β1 induces caspase-dependent apoptosis in a gastric epithelial cell line, SNU16. (A) Cells were treated with 10 ng/ml TGF-β1 for the period indicated, and cell viability was then determined by quantifying the amount of cellular ATP. The data shown represent the means ± standard deviations (SD) (n = 3). (B) Cleaved genomic DNA was extracted from SNU16 cells treated with TGF-β1 for the period indicated and resolved by 2.0% agarose gel electrophoresis. (C) After incubation with (bold line) or without (dotted line) 10 ng/ml TGF-β1 for 24 h, cells were stained with FITC-conjugated anti-active caspase-3 (casp-3) antibody and activation of caspase-3 was investigated by flow cytometry. Percentages of caspase-3-activated cells are indicated in the figure. (D) After incubation with 10 ng/ml TGF-β1 for the period indicated, cell lysate was analyzed by Western blotting with antibodies to casapse-8 (casp-8) or caspase-9 (casp-9). (E) Cells were pretreated with or pancaspase inhibitor zVAD-fmk (25 μM) for 1 h or left untreated and were subsequently treated with 10 ng/ml TGF-β1 for 48 h. Apoptotic subdiploid cells were analyzed by flow cytometry after staining with PI. Percentages of subdiploid cells are indicated in the figure. (A to E) Data are representative of at least three independent experiments. DMSO, dimethyl sulfoxide.
To assess the involvement of caspases in TGF-β1-induced apoptosis, SNU16 cells were stained with FITC-conjugated anti-active caspase-3 antibody and analyzed by flow cytometry. Activation of caspase-3 was observed in 58% of cells treated with TGF-β1 for 24 h (Fig. 1C). In addition, processed forms of the initiator caspases, caspase-8 and caspase-9, were detected in apoptotic cells (Fig. 1D). These results indicated that the caspase cascade was activated in SNU16 cells in response to TGF-β1. Addition of zVAD-fmk, a broad-range caspase inhibitor, prevented both DNA fragmentation (data not shown) and the appearance of cells with a subdiploid DNA content (Fig. 1E) in TGF-β1-treated cells, but it did not affect TGF-β1-induced growth arrest (Fig. 1E). This indicates that activation of the caspase is critical for TGF-β1-induced apoptosis but not growth arrest in SNU16 cells.
Activation of Smad pathway and subsequent de novo protein synthesis are required for TGF-β1-induced apoptosis in SNU16 cells.
TGF-β-induced signaling was reported to modulate the transcription of a large number of genes in various tissues and cells (40). To analyze whether de novo protein synthesis is required for TGF-β1-induced apoptosis, SNU16 cells were stimulated with TGF-β1 in the presence of cycloheximide (CHX), an inhibitor of protein synthesis. Addition of CHX inhibited DNA fragmentation (data not shown), the appearance of subdiploid cells (Fig. 2A), and processing of caspase-8 and caspase-9 (Fig. 2B). Thus, CHX as well as zVAD-fmk can inhibit TGF-β1-induced apoptosis. However, the cell cycle was clearly blocked at G1 in TGF-β1-treated cells in the presence of zVAD-fmk but not in the presence of CHX (Fig. 1C and 2A). These results indicate that CHX inhibits both TGF-β1-induced growth arrest and apoptosis, while zVAD-fmk can inhibit only the latter. Thus, TGF-β1-induced apoptosis requires de novo protein synthesis and subsequent activation of the caspase cascade.
FIG. 2.

TGF-β1-induced apoptosis requires activation of Smad and de novo protein synthesis. (A) Cells were pretreated with 1 μg/ml CHX for 1 h and subsequently incubated with 10 ng/ml TGF-β1 for 24 h in the presence or absence of CHX. Apoptotic subdiploid cells were measured by flow cytometry after staining with PI. Percentages of subdiploid cells are indicated in the figure. Data are representative of three independent experiments. (B) Western blot analysis was performed with antibodies to caspase-8 (pro-casp-8) or caspase-9 (pro-casp-9) on cells as described for panel A. (C) Western blot analysis was performed with antibodies to phosphorylated JNK (p-JNK), phosphorylated p38 (p-p38), or phosphorylated Smad2 (p-Smad2) on cells incubated with 10 ng/ml TGF-β1 for the periods indicated. As a positive control, UV-irradiated cells (10 kJ/cm2) were similarly analyzed. (D) After transfection with an expression vector for Smad7 (inhibitory Smad) or an empty vector together with a GFP expression vector, cells were treated with 10 ng/ml TGF-β1 for 24 h and then stained with PE-conjugated anti-active caspase-3 (active-casp-3) antibody. Expression of GFP and activation of caspase-3 were simultaneously analyzed by two-color flow cytometry. Percentages of caspase-3-activated cells among GFP+ cells are indicated in the figure. Data are representative of three independent experiments. (E) Experiments described for panel D were repeated three times; means ± SD of caspase-3-activated cells (percent) in both GFP− and GFP+ populations are indicated. The effect of MKK7-KL was examined using the same method that was used to investigate the effect of Smad7.
TGF-β has been shown to stimulate several intercellular signaling pathways, including the Smad and MAP kinase pathways (9). In particular, previous studies of TGF-β-induced apoptosis have focused on the Smad, p38, or JNK signaling pathways (for a review, see reference 42). We therefore examined whether TGF-β1 activates Smad, p38, and JNK in our system by Western blotting using antibodies that detect exclusively the phosphorylated (activated) form of these proteins. In SNU16 cells, phosphorylated p38 and JNK could be readily detected after UV irradiation but not in response to TGF-β1 treatment (Fig. 2C). In contrast, phosphorylation of Smad2, one of the receptor-Smads in TGF-β1-induced signaling, was observed within 30 min of stimulation with TGF-β1 (Fig. 2C).
To further investigate the role of Smad-mediated signaling in TGF-β1-induced apoptosis, we evaluated the effect of overexpressed Smad7, an inhibitory Smad, on TGF-β1-induced apoptosis in SNU16 cells. SNU16 cells were cotransfected with a vector encoding Smad7 together with a vector for GFP and then cultured for 24 h in the presence or absence of TGF-β1. Expression of exogenous Smad7 was confirmed by Western blotting of total lysate of the transfected cells (see Fig. S2A in the supplemental material). We then examined TGF-β1-induced caspase activation in GFP-positive (GFP+) and GFP-negative (GFP−) cells by flow cytometry by staining with an anti-active caspase-3 antibody. TGF-β1-induced activation of caspase-3 was absent in GFP+ cells but was readily observed in GFP− cells (Fig. 2D and E). In addition, TGF-β1-induced accumulation of subdiploid cells was repressed in Smad7-expressing cells (see Fig. S2B in the supplemental material). Expression of Smad7 was also shown to prevent TGF-β1-induced G1 cell cycle arrest, ensuring that the exogenous Smad7 was sufficient to perturb the TGF-β1-induced transcriptional response (see Fig. S2B in the supplemental material). These results indicate that Smad7 inhibits TGF-β1-induced apoptosis in SNU16 cells. In contrast, when either empty vector or a vector encoding MKK7-KL, which is a dominant-negative form of MKK7, JNK kinase, was transfected together with an expression vector for GFP, GFP+ cells underwent apoptosis similar to that seen with GFP− cells in response to TGF-β1 treatment (Fig. 2D and E; also see Fig. S2B in the supplemental material). Moreover, we observed that treatment of SNU16 cells with a specific chemical inhibitor of p38 (SB103580) or JNK (SP600125) had no effect on TGF-β1-induced apoptosis (data not shown). Collectively, these results demonstrated that the Smad pathway, but not the p38 or JNK pathways, plays an essential role in TGF-β1-induced apoptosis in SNU16 cells.
TGF-β1-induced apoptosis is initiated by activation of caspase-9 downstream of mitochondrial dysfunction.
The intercellular signaling pathways that activate the caspase cascade can be separated into at least two categories: the caspase-8-initiated cascade downstream of death receptors (extrinsic pathway) and the caspase-9-initiated cascade downstream of the release of cytochrome c from mitochondria (intrinsic pathway) (7, 43). TGF-β was originally reported to induce apoptosis in a caspase-8-dependent manner, while other studies proposed that TGF-β-induced apoptosis is mediated by a caspase-9-dependent mechanism (38). To clarify which initiator caspase is dominant in the activation of the caspase cascade in TGF-β1-treated SNU16 cells, we examined the effect of v-FLIP E8 and Bcl-XL, which inhibit activation of the extrinsic and intrinsic pathways of the caspase cascade, respectively (2, 5).
SNU16 cells were cotransfected with a vector for v-FLIP E8 or Bcl-XL together with a vector for GFP, and we examined the sensitivity of the GFP+ and GFP− cells for various apoptosis-inducing stimuli. The expression of the (FLAG epitope-tagged) transfected apoptosis inhibitors was confirmed by Western blotting with anti-FLAG antibody (see Fig. S3A in the supplemental material). By monitoring the activation of caspase-3, we first confirmed that v-FLIP E8 inhibited Fas-mediated but not UV-initiated apoptosis in SNU16 cells, whereas Bcl-XL did the opposite, showing the inhibitory effects of v-FLIP E8 and Bcl-XL on the activation of caspase-8 and caspase-9, respectively (Fig. 3A; also see Fig. S3B in the supplemental material). We then examined the effects of v-FLIP E8 and Bcl-XL on caspase activation induced by the treatment with TGF-β1. Bcl-XL was found to suppress the activation of caspase-3, whereas v-FLIP E8 had no effect (Fig. 3B; see Fig. S3C in the supplemental material), indicating a critical role of activation of caspase-9 in TGF-β1-induced apoptosis in SNU16 cells. These results were also confirmed by quantifying the number of apoptotic cells with a subdiploid DNA content (see Fig. S3C in the supplemental material).
FIG. 3.
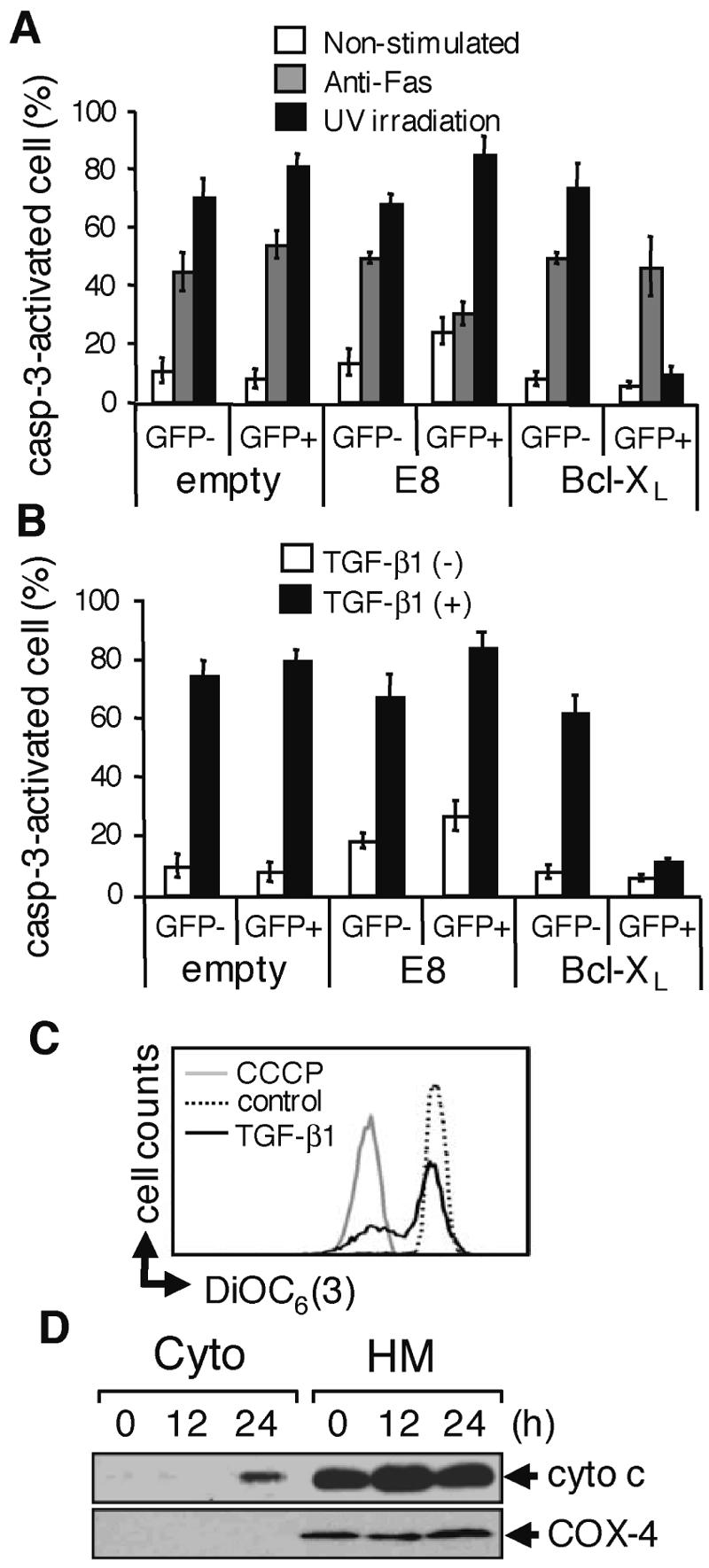
TGF-β1 induces apoptosis in SNU16 cells through the mitochondrial apoptosis pathway. (A) Cells were transfected with the indicated expression vectors (empty or viral FLIP E8 or Bcl-XL; both FLAG tagged) together with a vector for GFP. Transfected cells were stimulated with 1 μg/ml anti-Fas antibody (CH-11) for 12 h or irradiated with 20 kJ/cm2 UV and cultured for 12 h. Caspase-3 (casp-3)-activated cells in the GFP− or GFP+ population were examined as described for Fig. 2D (see Fig. S3B in the supplemental material). Experiments were performed three times, and the means ± SD of caspase-3-activated cells are indicated for the GFP− and GFP+ populations as described for Fig. 2E. (B) Cells transfected as described for panel A were treated with 10 ng/ml TGF-β1 for 24 h and examined as described for panel A (see Fig. S3C in the supplemental material). (C) Cells were treated with 10 ng/ml TGF-β1 for 24 h or left untreated (control) and were stained with DiOC6 (3). As a positive control, cells were similarly analyzed in the presence of carbonyl cyanide m-chlorophenylhydrazone (CCCP). (D) After incubation of SNU16 with TGF-β1 for the period indicated, cytosolic (Cyto) and heavy membrane (HM) fractions were prepared and subjected to Western blotting with anti-cytochrome c (cyto c) antibody. As a control for purity of the subcellular fractionation, the blot was also probed with an antibody to cytochrome oxidase subunit 4 (COX-4).
Activation of caspase-9 is generally thought to be induced downstream of outer mitochondrial membrane disruption and the release of cytochrome c into the cytosol (7). To investigate the effects of TGF-β1 treatment on mitochondria, we stained SNU16 cells with DiOC6(3) (3,3′-dihexyloxacarbocyanine iodide), which monitors the loss of mitochondrial electric potential. Flow cytometric analysis indicated that treatment with TGF-β1 induced loss of the mitochondrial electric potential in SNU16 cells (Fig. 3C). In addition, we observed the release of cytochrome c from mitochondria in TGF-β1-treated cells (Fig. 3D). These results indicate that TGF-β1 induces apoptosis in SNU16 cells by the intrinsic pathway of caspase cascade.
TGF-β1-induced outer mitochondrial membrane disruption is mediated by proapoptotic members of the Bcl-2 family.
Numerous studies have indicated that Bcl-2 family proteins are essential regulators of the release of cytochrome c from mitochondria (5). We therefore analyzed the expression of Bcl-2 family members in SNU16 cells treated with TGF-β1. We first examined the expression of the anti-apoptotic Bcl-2, Bcl-XL, and Mcl-1 in a mitochondrion-rich fraction, but no clear change in their expression was observed after the treatment with TGF-β1 (Fig. 4A). We next examined Bax, one of the multi-BH domain members of the Bcl-2 family, and found that its expression was not altered in SNU16 cells after treatment with TGF-β1 (Fig. 4B, upper panel, arrow). While Bax is generally inactive in healthy cells, apoptosis-inducing stimuli were reported to promote a conformational change and oligomerization of Bax (27). We therefore performed an immunoprecipitation experiment using an antibody specific for an N-terminal exposed form of Bax (available for binding only in active Bax). When SNU16 cells were incubated with TGF-β1 for 24 h, active Bax could be immunoprecipitated with the antibody, indicating that Bax was in its apoptosis-promoting form (Fig. 4B, middle panel, arrowhead). We confirmed the activation of Bax by detecting its oligomerized form by use of a cross-linking reagent, BMH (Fig. 4B, bottom panel). These observations suggested that TGF-β1 induced mitochondrial dysfunction by inducing activation of Bax.
FIG. 4.

TGF-β1 induces a conformational change of Bax and expression of Bim and Bmf in SNU16 cells. (A) The heavy-membrane fraction from SNU16 cells treated with 10 ng/ml TGF-β1 for the period indicated was subjected to Western blotting to examine the expression of antiapoptotic molecules, Bcl-2, Bcl-XL, and Mcl-1 (Mcl-1 was below the level of detection). (B) SNU16 cells were treated with 10 ng/ml TGF-β1 for the period indicated and then subjected to Western blotting with anti-Bax antibody (top panel). To analyze the activation of Bax, immunoprecipitation was carried out with a specific antibody for the active form of Bax. The immunoprecipitated Bax was detected by Western blotting using a polyclonal antibody that binds all forms of Bax (middle panel). Oligomerization of Bax was examined by Western blotting with an anti-Bax antibody after incubation of cell lysates with the chemical cross-linker BMH (bottom panel). Stars indicate nonspecific bands. (C) SNU16 cells were treated with 10 ng/ml TGF-β1 for the period indicated and then subjected to Western blotting with antibodies to the BH3-only proteins indicated and actin. (D) Total RNA was extracted from SNU16 cell treated with 10 ng/ml TGF- β1 for the indicated times and analyzed by real-time PCR using specific primers for human BimEL sequence (forward primer, 5′-GCCCCTACCTCCCTACAGAC-3′; reverse primer, 5′-AAGATGAAAAGCGGGGATCT-3′). The expression level of Bim mRNA was normalized to that of GAPDH and is expressed as the ratio to the expression level of TGF-β1-untreated cells. The data shown represent the means ± SD (n = 3). (E) The indicated 3.9-kb nucleotide fragment of human bim gene (including the 5′ flanking region, exon 1, intron 1, and exon 2) was isolated by PCR, and the nucleotide fragment was cloned into a pBV-B2 vector upstream of the firefly luciferase gene. The transcription start site is numbered +1. (F) SNU16 cells transfected with the human bim reporter construct and the renilla luciferase expression construct pRT-TK were treated with 10 ng/ml TGF-β1 for 12 h, after which a dual-luciferase assay was performed. The relative luciferase activity of firefly was determined after normalization to the activity of renilla. The data shown represent the means ± SD (n = 3). cont, control. (G) SNU16 cells were transfected with an expression vector for Smad7 or an empty vector, and the dual-luciferase activity was analyzed as described in for panel F. The data shown represent the means ± SD (n = 3).
The well-recognized molecular model for the release of cytochrome c from mitochondria by Bcl-2 family proteins has indicated that the multi-BH domain members of this family, Bax and Bak, function as death effectors after being directly or indirectly activated by proapoptotic BH3-only proteins (37). We therefore speculated that TGF-β1 signaling might activate Bax through the proapoptotic function of BH3-only proteins. We assessed here the expression of the BH3-only proteins Bad, Bid, Bik, Bim, Bmf, Hrk, Puma, and hNoxa at the mRNA and/or protein level. Reverse transcription-PCR analysis detected the mRNAs for Bad, Bid, Bim (BimEL and BimL), and Bmf but not for Bik, Hrk, BimS, Puma, or hNoxa in SNU16 cells (data not shown). We then investigated the expression of these BH3-only proteins before and after the treatment with TGF-β1 by Western blotting. Bad and Bid were readily detected in SNU16 cells, but their expression was not affected by treatment with TGF-β1 (Fig. 4C, top and second panels). We could not detect the cleavage-associated activation of Bid within 24 h of treatment with TGF-β1, indicating that caspase-mediated activation of Bid was not a major player in this pathway to apoptosis (Fig. 4C, second panel). While Bim was detected as two isoforms, BimEL and BimL, BimEL was dominantly expressed in SNU16 cells. Bim was detected even in unstimulated cells, but the expression of Bim was increased about twofold after treatment with TGF-β1 at both the protein (Fig. 4C, third panel) and mRNA (Fig. 4D) levels. Bmf was also increased after the treatment with TGF-β1 (Fig. 4C, fourth panel).
In SNU16 cells, TGF-β1-induced transcriptional activation of Bim was indicated by a dual-luciferase analysis, and the up-regulation of Bim mRNA was inhibited by the expression of an inhibitory Smad, Smad7 (Fig. 4E to G). The induction level and the time course of the induction of Bim mRNA were similar to those of Bim protein. All the results suggest that TGF-β1-mediated up-regulation of Bim was induced at the transcription level, while a significant amount of Bim was already present in untreated SNU16 cells, and the level of the up-regulation was not so high (about twofold at both the RNA and protein levels) (Fig. 4C and D).
Bim is a key initiator of TGF-β1-induced apoptosis.
BH3-only proteins trigger apoptosis in large part by binding and thereby inactivating antiapoptotic Bcl-2 family members, such as Bcl-XL (14). We therefore assessed activation of Bad, Bim, and Bmf by detecting their interaction with Bcl-XL. To detect interaction of Bcl-XL and BH3-only proteins that occur upstream of caspase activation, we first performed coimmunoprecipitation with SNU16 cells overexpressing Bcl-XL, in which activation of the caspase cascade was prevented as indicated in Fig. 3B. As a consequence, endogenous BimEL and Bmf, but not Bad, were coimmunoprecipitated with overexpressed Bcl-XL in lysates from cells treated for 24 h with TGF-β1 (Fig. 5A). Furthermore, we performed an immunoprecipitation assay with anti-Bcl-XL antibody on parental SNU16 cells and detected interaction of endogenous Bcl-XL with endogenous BimEL, but not with Bmf, in cells treated with TGF-β1 but not in untreated cells (Fig. 5B). While the addition of caspase inhibitor zVAD-fmk prevented the appearance of TGF-β1-induced subdiploid apoptotic cells, this had no effect on coimmunoprecipitation of BimEL with Bcl-XL (Fig. 5C), confirming that the interaction between endogenous Bim and Bcl-XL was induced prior to and independent of caspase activation. In addition, TGF-β1-induced interaction between endogenous Bim and Bcl-XL was shown to be repressed in Smad7-expressing cells (Fig. 5D), indicating that the interaction was dependent on TGF-β1-induced activation of Smad. These results indicated that Bim was induced to interact with Bcl-XL in TGF-β1-treated SNU16 cells, resulting in oligomerization of Bax and subsequent mitochondrial dysfunction.
FIG. 5.

TGF-β1 induces activation of Bim in SNU16 cells. (A) SNU16 cells transfected with an expression vectors for FLAG-tagged Bcl-XL were treated with 10 ng/ml TGF-β1 for the period indicated; immunoprecipitation (IP) was then performed with anti-FLAG antibody M2. The immunoprecipitate was subjected to Western blotting with antibodies to the BH3-only proteins indicated. (B) Cells were treated with 10 ng/ml TGF-β1 for 24 h and divided in two pools. One half was subjected to immunoprecipitation with anti-Bcl-XL antibody. The other half was stained with PI, and the percentage of apoptotic cells was quantified by flow cytometry as described for Fig. 1E. (C) SNU16 cells were treated with 10 ng/ml TGF-β1 in the presence or absence of 25 μM zVAD-fmk for the indicated periods. Immunoprecipitation and detection of apoptotic cells were performed as described for panel B. DMSO, dimethyl sulfoxide. (D) Top and middle panels: after transfection with an expression vector for Myc-tagged Smad7 or an empty vector together with an expression vectors for FLAG-tagged Bcl-XL, cells were treated with 10 ng/ml TGF-β1 for 24 h and then immunoprecipitation and Western blotting were performed as described for panel A. Bottom panel: the expression of Smad7 was examined in the total lysate.
To directly examine the role of Bim in TGF-β1-induced apoptosis, we utilized the RNA interference (RNAi) technique to reduce the expression of endogenous Bim protein. We prepared three distinct shRNA expression vectors termed 2C2, 2F6, and 2H4. Because the expression vector 2H4 targeted an mRNA sequence of Bimα1 (Bimα1 is one of the isoforms of Bim not detected in SNU16 cells), this vector was assessed to be used as a control. Expression vector 2C2 targeted mRNA sequences of both BimEL and BimL, and 2F6 targeted a specific mRNA sequence of BimEL alone. These shRNA expression vectors had no effect on the level of actin or another BH3-only protein, Bad, but SNU16 cells transfected with the vectors 2C2 and 2F6, but not those expressing the control vector 2H4, showed a marked reduction in BimEL expression (Fig. 6A). This RNAi-mediated inhibition of Bim expression was found to afford SNU16 with significant protection against TGF-β1-induced cell death (Fig. 6B). Flow cytometric analysis confirmed that activation of caspase-3 was also prevented in TGF-β1-treated cells in which the expression of endogenous Bim was reduced (Fig. 6C). In contrast, RNAi-mediated inhibition of Bmf expression (Fig. 6D) showed no significant protection against TGF-β1-induced cell death even when the expression of Bmf was suppressed in SNU16 cells transfected with the Bim-specific vector 2C2 (Fig. 6E). These results demonstrate that TGF-β1-induced apoptosis in SNU16 cells requires the apoptosis-promoting function of Bim but not that of Bmf.
FIG. 6.

Reduction of Bim expression by the RNAi method renders SNU16 cells resistant to TGF-β1-induced apoptosis, but that of Bmf-expression does not. (A) SNU16 cells were transfected with an expression vector encoding Bim-specific shRNA (2H4, 2C2, or 2F6) together with the puromycin-resistant gene and then cultured with 1 μg/ml puromycin for 3 days. Viable cells were incubated with 10 ng/ml TGF-β1 for 24 h and then subjected to Western blotting with antibodies to Bim, Bad, or actin. (B) SNU16 cells were transfected with an expression vector for the indicated shRNA or with a Bcl-XL expression vector together with GFP and then incubated with 10 ng/ml TGF-β1 for the period indicated. Transfected cells were lysed, and the fluorescence intensity of GFP was quantified. Cell viability was determined from the ratio of fluorescence before and after the treatment with TGF-β1. Means ± SD of three independent experiments are shown. (C) Activation of caspase-3 (casp3) was analyzed as described for Fig. 2D and Fig. 3 for TGF-β1-treated or UV-irradiated SNU16 cells transfected with expression vectors for the indicated shRNA or Bcl-XL as in described for panel B. Experiments were performed three times, and the means ± SD for caspase-3-activated cells in the GFP-positive population are indicated. (D) SNU16 cells were transfected with an empty vector or vectors for Bim-specific shRNA (2C2) or Bmf-specific shRNA (2A12 and 2H7) together with FLAG-tagged BimEL or Bmf expression vectors. Total cell lysate was extracted and subjected to Western blotting using anti-FLAG M2 antibody. (E) SNU16 cells were transfected with expression vectors for Bcl-XL, Bim-specific shRNA (2C2), and/or Bmf-specific shRNA (2A12 and 2H7) together with that of GFP, and then cell viability was quantified after the treatment with TGF-β1 as described for panel B. Means ± SD of three independent experiments are shown.
Gastric abnormality of newborn mice with defects in the TGF-β-dependent apoptosis pathway.
We speculated that the physiological apoptosis in the gastric epithelium might depend on TGF-β signaling, because mutant mice lacking TGF-β1 or Runx3 exhibited a characteristic hyperplasia of the gastric mucosa (6, 26, 41). These reports led us to predict that similar phenotypes might be observed in the stomachs of mice with defects in the TGF-β-dependent apoptosis pathway. In addition, Bim was shown to express in the epithelium of glandular stomachs of C57BL/6 mice by an immunohistochemical analysis (see Fig. S4 in the supplemental material). Therefore, we analyzed the stomachs of caspase-9 and Bim knockout mice, both of which were previously generated (4, 22). We carefully compared glandular stomachs of caspase-9 knockout (caspase-9−/−) and Bim knockout (Bim−/−) mice with those of control mice on a C57BL/6 background by TUNEL and histochemical (PAS-HE) staining (Fig. 7).
FIG. 7.

Histological features of gastric tissues from caspase-9−/− and Bim−/− mice. (A to F) TUNEL (B, D, and F; green) and DAPI (A, C, and E; blue) staining results for paraffin-embedded sections from newborn caspase-9−/− mice (C and D) and Bim−/− mice (E and F) were compared with those from control littermates (WT) (A and B). (G to L) Gastric epithelial abnormalities of caspase-9−/− mice (I and J) and Bim−/− mice (K and L) were analyzed by PAS-HE staining. Gastric tissues from control littermates were also analyzed (G and H). Bars: 100 μm (A to F, G, I, and K) and 10 μm (H, J, and L).
When the spontaneously occurring apoptosis in the gastric epithelium was evaluated by TUNEL staining, a significant number of apoptotic cells were observed in the gastric epithelium of wild-type mice (Fig. 7B), consistent with previous experiments. In contrast, the numbers of apoptotic cells in the epithelium were significantly decreased in both caspase-9−/− and Bim−/− mice (Fig. 7D and F), suggesting that caspase-9 and Bim are involved in the physiological apoptosis observed in gastric epithelial cells. PAS-HE staining indicated that the surface epithelial cells in both mutant mice consisted of a larger number of more-elongated cells than in normal littermates, while the phenotype of gastric epithelium of Bim−/− mice was less prominent than that of caspase-9−/− mice (Fig. 7G, H, I, J, K, and L). It should be noted that nuclei were located near the cell surface area in the surface epithelial cells of caspase-9−/− stomachs whereas they were at basal area in control stomach (Fig. 7I and J). These disordered observations might imply an excessive accumulation of cells with longer life in the gastric epithelial layer of caspase-9−/− and Bim−/− mice. While the accumulation of epithelial cells could be caused either by overproliferation of progenitor cells or by reduction of loss of epithelial cells by apoptosis, the latter appears more likely in this case, since TUNEL staining of epithelial cells of these mutant mice revealed much lower numbers of apoptotic cells than in wild-type mice.
DISCUSSION
In this study, TGF-β1-induced apoptosis was demonstrated to be mediated by sequential activation of Smad, Bim, and caspase-9 in human metastatic gastric carcinoma-derived SNU16 cells (see Fig. S5 in the supplemental material). Although previous reports proposed that TGF-β induced apoptosis in several cell lines through TGF-β-dependent activation of MAP kinases, especially p38 and JNK (31, 33, 50), we could not detect any evidence for the involvement of these MAP kinase pathways in TGF-β1-induced apoptosis in SNU16 cells. In contrast, we observed that TGF-β1-induced apoptosis could be suppressed by expression of the inhibitory Smad, Smad7 (Fig. 2), and expression of Smad7 was shown to inhibit TGF-β1-induced activation of Bim (Fig. 5D). It should be noted that transgenic mice expressing a Smad7 transgene exclusively in epithelial tissues have abnormally low numbers of apoptotic cells in their forestomach epithelium (13). Thus, TGF-β1-induced apoptosis is mediated by a Smad-dependent pathway, at least in the stomach.
TGF-β1-induced apoptosis was also demonstrated to require the caspase cascade in SNU16 cells. Although activation of both initiator caspases, caspase-8 and caspase-9, was detected by Western blotting (Fig. 1C), experiments using the inhibitory proteins, v-FLIP E8 and Bcl-XL for caspase-8 and caspase-9, respectively, indicated that TGF-β1 triggered the activation of caspase-9. The cleavage and activation of caspase-8 might be a secondary event caused by activated executioner caspases (i.e., caspase-3, caspase-6, and caspase-7). Moreover, we observed not only the activation of the multi-BH domain Bcl-2 family member Bax (Fig. 4B) but also the loss of mitochondrial electric potential (Fig. 3C) and release of cytochrome c from mitochondria (Fig. 3D) in TGF-β1-treated SNU 16 cells, which could induce activation of caspase-9. From these results, we conclude that TGF-β1-induced apoptosis in SNU16 cells is dependent on a caspase-9-mediated mechanism downstream of mitochondrial dysfunction induced by the activation of Bax.
TGF-β1-induced apoptosis in SNU16 cells required the up-regulation of a TGF-β1-responsive gene(s) and de nova protein synthesis prior to activation of the caspase cascade downstream of caspase-9 (Fig. 2A and B). TGF-β has been reported to regulate apoptosis through the expression of inositol phosphatase SHIP (46), Smad7 (23), DAP kinase (16), GADD45b (49), or a mitochondrial septin-like protein called ARTS (24). However, the precise molecular mechanism by which these molecules participate in activation of the apoptosis-promoting machinery, such as the caspase cascade, death receptors, or Bcl-2 family proteins, remains to be clarified. In this study, we found a principal role for the BH3-only Bcl-2 family member Bim in TGF-β1-induced apoptosis in SNU16 cells (Fig. 6). Some studies proposed that other BH3-only proteins, including Bad and Bid, might participate in TGF-β-induced apoptosis (19, 20), but we did not observe the cleavage-associated activation of Bid (Fig. 5A) or a detectable interaction of Bad with anti-apoptotic Bcl-XL in TGF-β1-treated SNU16 cells even when caspase activation was blocked (Fig. 5B). However, we cannot exclude the possible involvement of other proapoptotic mediators in this process, because overexpression of Bcl-XL inhibited TGF-β1-induced apoptosis of SNU16 cells more effectively than reduced expression of Bim by the RNAi method (Fig. 6B and C). We speculated that this result might be caused by the residual expression of Bim or partial involvement of Bmf, another BH3-only protein found to increase by treatment with TGF-β1. However, RNAi-mediated inhibition of Bmf expression was shown to be unable to protect TGF-β1-induced cell death (Fig. 5E). In conclusion, there is no doubt that Bim is activated and functions as a main initiator in TGF-β1-induced apoptosis in SNU16 cells.
Bim was identified as a Bcl-2-interacting protein, and various isoforms exist, including BimEL, BimL, and BimS (29). Bim was reported to be expressed in hematopoietic, epithelial, neuronal, and germ cells (30). The proapoptotic function of Bim was reported to be regulated by a couple of mechanisms as follows (for a review, see reference 44): FOXO3a, a member of the Forkhead transcriptional factor family, has been implicated in the transcriptional activation of Bim (10); Bim was reported to be sequestered in the cytosol of healthy cells by its association with dynein light chain 1 (36); transcriptional and/or posttranslational regulation by JNK has been reported to be involved in Bim activation in neurons (34, 35); and Bim was shown to be inactivated by ERK-mediated phosphorylation, which causes its ubiquitination and proteasomal degradation in osteoclasts (1). While Bim was shown to be expressed in gastric epithelial cells (see Fig. S4 in the supplemental material), the role of Bim in gastric epithelium has not yet been examined. Interestingly, TGF-β2, another member of the TGF-β superfamily, was reported to induce expression of Bim in a B-cell line, WEHI-231, but only when transfected with Smad3 (48). In SNU16 cells, we also observed increased expression of Bim protein (Fig. 4C and 6A) as well as mRNA (Fig. 6D) in response to TGF-β1 treatment. All the data in Fig. 4 and 6 suggest that TGF-β1-mediated up-regulation of Bim was induced at the transcription level. However, a significant amount of Bim was already present in untreated SNU16 cells, and the level of the up-regulation was not so high (about twofold at both the RNA and protein levels) (Fig. 4C and D). Furthermore, increased expression of Bim was detected within 6 h of treatment with TGF-β1 both in RNA and protein levels, whereas apoptotic cells began to appear only after 18 h of treatment with TGF-β1 (Fig. 1 and Fig. 4C and D). These results indicate that the induction of Bim expression alone is insufficient to activate the caspase cascade, implying an additional, possibly posttranscriptional, mechanism for Bim activation in TGF-β1-induced apoptosis in SNU16 cells. An Smad-dependently TGF-β1-induced molecule(s) may play a role in the activation of Bim, since expression of Smad7 inhibited TGF-β1-induced activation of Bim (Fig. 5D). At the present time, however, the molecular mechanism by which TGF-β1 promotes binding of Bim to Bcl-XL has not been clarified. There is no evidence that previously described molecules, such as DAXX, DAP kinase, SHIP, GADD45b, and ARTS, are involved in the activation of Bim. We postulate that Bim is posttranslationally activated by an uncharacterized molecule(s) which is transcriptionally up-regulated downstream of the TGF-β1-dependent Smad pathway. This assumption seems to coincide with our in vivo observation that apoptosis is induced in a part of the gastric epithelial cells (Fig. 7B), although all the gastric epithelial cells express similar amounts of Bim (see Fig. S4 in the supplemental material).
On the basis of our studies with the gastric epithelial cell line SNU16 together with previously reported abnormalities in the stomachs of TGF-β1−/−, Runx3−/−, and Smad7 transgenic mice (6, 13, 26), we hypothesized that mice with defects in TGF-β-mediated apoptosis signaling might exhibit phenotypic abnormalities in their stomachs. To test our hypothesis, we investigated whether the stomachs of mutant mice lacking Bim or caspase-9 exhibited any abnormality. As shown in Fig. 7, severe and moderate abnormalities were observed in the gastric epithelium of caspase-9−/− and Bim−/− mice, respectively. Gastric epithelial cells on the surface from mutant mice were found to be more elongated with abnormal nuclear localization, suggesting that epithelial cells are excessively accumulated and more crowded there in both mutant mouse lines. It is possible that this phenotype is caused by the reduction in the TGF-β-dependent physiological apoptosis of epithelial cells demonstrated in the present study. The reduced apoptosis in gastric epithelium in both caspase-9−/− and Bim−/− mice was similarly observed in a previous study of Runx3−/− mice (26). The observed gastric phenotypes of Bim−/− or caspase-9−/− mice, however, seem to be somewhat less serious than those of TGF-β1−/− mice (6) or Runx3−/− mice (26). We speculated that the reason for this might be that the Bim- or caspase-9-deficient cells remain responsive to the growth-inhibitory activity of TGF-β, whereas Runx3 deficiency probably impairs both activities of TGF-β.
In conclusion, we identified here the key role of Bim and caspase-9 in the physiological death of gastric epithelial cells. Our results are the first direct evidence that proapoptotic molecules, Bim and caspase-9, control the apoptosis of gastric epithelial cells not only in vitro but also in vivo. On the grounds of our finding that TGF-β1 stimulates apoptosis through activation of Bim and caspase-9, together with previous genetic studies of TGF-β signaling molecules, we propose here that through the sequential activation of Smad, Bim, and caspase-9, the TGF-β-regulated apoptosis-promoting machinery contributes to the physiological turnover of gastric epithelial cells. While no clear incidence of gastric cancer has been shown in the caspase-9-deficient and Bim-deficient mice, probably because of their early mortality, abnormalities in components of this machinery may contribute to some types of pathological disorders, including gastric cancer, which must be intimately examined in the future.
Supplementary Material
Acknowledgments
We thank K. Miyazono, E. Nishida, P. Bouillet, Y. Ito, and K. Ito for providing materials. We also thank K. Sakamaki, A. Murakami, K. K. Lee, and K. Okamoto for useful comments.
This work was supported in part by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of the Japanese government. M.O. was supported by the 21st century program of MEXT of the Japanese government for the Graduate School of Biostudies and the Institute for Virus Research, Kyoto University.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Akiyama, T., P. Bouillet, T. Miyazaki, Y. Kadono, H. Chikuda, U. I. Chung, A. Fukuda, A. Hikita, H. Seto, T. Okada, T. Inaba, A. Sanjay, R. Baron, H. Kawaguchi, H. Oda, K. Nakamura, A. Strasser, and S. Tanaka. 2003. Regulation of osteoclast apoptosis by ubiquitylation of proapoptotic BH3-only Bcl-2 family member Bim. EMBO J. 22:6653-6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertin, J., R. C. Armstrong, S. Ottilie, D. A. Martin, Y. Wang, S. Banks, G. H. Wang, T. G. Senkevich, E. S. Alnemri, B. Moss, M. J. Lenardo, K. J. Tomaselli, and J. I. Cohen. 1997. Death effector domain-containing herpesvirus and poxvirus proteins inhibit both Fas- and TNFR1-induced apoptosis. Proc. Natl. Acad. Sci. USA 94:1172-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boivin, G. P., J. R. Molina, I. Ormsby, G. Stemmermann, and T. Doetschman. 1996. Gastric lesions in transforming growth factor β-1 heterozygous mice. Lab. Investig. 74:513-518. [PubMed] [Google Scholar]
- 4.Bouillet, P., D. Metcalf, D. C. Huang, D. M. Tarlinton, T. W. Kay, F. Kontgen, J. M. Adams, and A. Strasser. 1999. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286:1735-1738. [DOI] [PubMed] [Google Scholar]
- 5.Cory, S., and J. M. Adams. 2002. The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2:647-656. [DOI] [PubMed] [Google Scholar]
- 6.Crawford, S. E., V. Stellmach, J. E. Murphy-Ullrich, S. M. Ribeiro, J. Lawler, R. O. Hynes, G. P. Boivin, and N. Bouck. 1998. Thrombospondin-1 is a major activator of TGF-β1 in vivo. Cell 93:1159-1170. [DOI] [PubMed] [Google Scholar]
- 7.Danial, N. N., and S. J. Korsmeyer. 2004. Cell death: critical control points. Cell 116:205-219. [DOI] [PubMed] [Google Scholar]
- 8.de Caestecker, M. P., E. Piek, and A. B. Roberts. 2000. Role of transforming growth factor-β signaling in cancer. J. Natl. Cancer Inst. 92:1388-1402. [DOI] [PubMed] [Google Scholar]
- 9.Derynck, R., and Y. E. Zhang. 2003. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425:577-584. [DOI] [PubMed] [Google Scholar]
- 10.Gilley, J., P. J. Coffer, and J. Ham. 2003. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 162:613-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hahm, K. B., K. M. Lee, Y. B. Kim, W. S. Hong, W. H. Lee, S. U. Han, M. W. Kim, B. O. Ahn, T. Y. Oh, M. H. Lee, J. Green, and S. J. Kim. 2002. Conditional loss of TGF-β signalling leads to increased susceptibility to gastrointestinal carcinogenesis in mice. Aliment. Pharmacol. Ther. 16(Suppl. 2):115-127. [DOI] [PubMed] [Google Scholar]
- 12.Hall, P. A., P. J. Coates, B. Ansari, and D. Hopwood. 1994. Regulation of cell number in the mammalian gastrointestinal tract: the importance of apoptosis. J. Cell Sci. 107(Pt. 12):3569-3577. [DOI] [PubMed] [Google Scholar]
- 13.He, W., A. G. Li, D. Wang, S. Han, B. Zheng, M. J. Goumans, P. Ten Dijke, and X. J. Wang. 2002. Overexpression of Smad7 results in severe pathological alterations in multiple epithelial tissues. EMBO J. 21:2580-2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang, D. C., and A. Strasser. 2000. BH3-Only proteins—essential initiators of apoptotic cell death. Cell 103:839-842. [DOI] [PubMed] [Google Scholar]
- 15.Imai, T., S. Adachi, K. Nishijo, M. Ohgushi, M. Okada, T. Yasumi, K. Watanabe, R. Nishikomori, T. Nakayama, S. Yonehara, J. Toguchida, and T. Nakahata. 2003. FR901228 induces tumor regression associated with induction of Fas ligand and activation of Fas signaling in human osteosarcoma cells. Oncogene 22:9231-9242. [DOI] [PubMed] [Google Scholar]
- 16.Jang, C. W., C. H. Chen, C. C. Chen, J. Y. Chen, Y. H. Su, and R. H. Chen. 2002. TGF-β induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell Biol. 4:51-58. [DOI] [PubMed] [Google Scholar]
- 17.Ju, H. R., U. Jung, C. H. Sonn, S. R. Yoon, J. H. Jeon, Y. Yang, K. N. Lee, and I. Choi. 2003. Aberrant signaling of TGF-β1 by the mutant Smad4 in gastric cancer cells. Cancer Lett. 196:197-206. [DOI] [PubMed] [Google Scholar]
- 18.Kang, S. H., Y. J. Bang, Y. H. Im, H. K. Yang, D. A. Lee, H. Y. Lee, H. S. Lee, N. K. Kim, and S. J. Kim. 1999. Transcriptional repression of the transforming growth factor-beta type I receptor gene by DNA methylation results in the development of TGF-β resistance in human gastric cancer. Oncogene 18:7280-7286. [DOI] [PubMed] [Google Scholar]
- 19.Kim, B. C., M. Mamura, K. S. Choi, B. Calabretta, and S. J. Kim. 2002. Transforming growth factor β1 induces apoptosis through cleavage of BAD in a Smad3-dependent mechanism in FaO hepatoma cells. Mol. Cell. Biol. 22:1369-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim, S. G., H. S. Jong, T. Y. Kim, J. W. Lee, N. K. Kim, S. H. Hong, and Y. J. Bang. 2003. Transforming growth factor-β1 induces apoptosis through Fas ligand-independent activation of the Fas death pathway in human gastric SNU-620 carcinoma cells. Mol. Biol. Cell 15:420-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim, S. G., S. N. Kim, H. S. Jong, N. K. Kim, S. H. Hong, S. J. Kim, and Y. J. Bang. 2001. Caspase-mediated Cdk2 activation is a critical step to execute transforming growth factor-β1-induced apoptosis in human gastric cancer cells. Oncogene 20:1254-1265. [DOI] [PubMed] [Google Scholar]
- 22.Kuida, K., T. F. Haydar, C. Y. Kuan, Y. Gu, C. Taya, H. Karasuyama, M. S. Su, P. Rakic, and R. A. Flavell. 1998. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase-9. Cell 94:325-337. [DOI] [PubMed] [Google Scholar]
- 23.Landstrom, M., N. E. Heldin, S. Bu, A. Hermansson, S. Itoh, P. ten Dijke, and C. H. Heldin. 2000. Smad7 mediates apoptosis induced by transforming growth factor β in prostatic carcinoma cells. Curr. Biol. 10:535-538. [DOI] [PubMed] [Google Scholar]
- 24.Larisch, S., Y. Yi, R. Lotan, H. Kerner, S. Eimerl, W. Tony Parks, Y. Gottfried, S. Birkey Reffey, M. P. de Caestecker, D. Danielpour, N. Book-Melamed, R. Timberg, C. S. Duckett, R. J. Lechleider, H. Steller, J. Orly, S. J. Kim, and A. B. Roberts. 2000. A novel mitochondrial septin-like protein, ARTS, mediates apoptosis dependent on its P-loop motif. Nat. Cell Biol. 2:915-921. [DOI] [PubMed] [Google Scholar]
- 25.Lee, K. K., T. Ohyama, N. Yajima, S. Tsubuki, and S. Yonehara. 2001. MST, a physiological caspase substrate, highly sensitizes apoptosis both upstream and downstream of caspase activation. J. Biol. Chem. 276:19276-19285. [DOI] [PubMed] [Google Scholar]
- 26.Li, Q. L., K. Ito, C. Sakakura, H. Fukamachi, K. Inoue, X. Z. Chi, K. Y. Lee, S. Nomura, C. W. Lee, S. B. Han, H. M. Kim, W. J. Kim, H. Yamamoto, N. Yamashita, T. Yano, T. Ikeda, S. Itohara, J. Inazawa, T. Abe, A. Hagiwara, H. Yamagishi, A. Ooe, A. Kaneda, T. Sugimura, T. Ushijima, S. C. Bae, and Y. Ito. 2002. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 109:113-124. [DOI] [PubMed] [Google Scholar]
- 27.Makin, G. W., B. M. Corfe, G. J. Griffiths, A. Thistlethwaite, J. A. Hickman, and C. Dive. 2001. Damage-induced Bax N-terminal change, translocation to mitochondria and formation of Bax dimers/complexes occur regardless of cell fate. EMBO J. 20:6306-6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murakawa, M., S. K. Jung, K. Iijima, and S. Yonehara. 2001. Apoptosis-inducing protein, AIP, from parasite-infected fish induces apoptosis in mammalian cells by two different molecular mechanisms. Cell Death Differ. 8:298-307. [DOI] [PubMed] [Google Scholar]
- 29.O'Connor, L., A. Strasser, L. A. O'Reilly, G. Hausmann, J. M. Adams, S. Cory, and D. C. Huang. 1998. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 17:384-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Reilly, L. A., L. Cullen, J. Visvader, G. J. Lindeman, C. Print, M. L. Bath, D. C. Huang, and A. Strasser. 2000. The proapoptotic BH3-only protein bim is expressed in hematopoietic, epithelial, neuronal, and germ cells. Am. J. Pathol. 157:449-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park, H. J., B. C. Kim, S. J. Kim, and K. S. Choi. 2002. Role of MAP kinases and their cross-talk in TGF-β1-induced apoptosis in FaO rat hepatoma cell line. Hepatology 35:1360-1371. [DOI] [PubMed] [Google Scholar]
- 32.Park, K., S. J. Kim, Y. J. Bang, J. G. Park, N. K. Kim, A. B. Roberts, and M. B. Sporn. 1994. Genetic changes in the transforming growth factor beta (TGF-β) type II receptor gene in human gastric cancer cells: correlation with sensitivity to growth inhibition by TGF-β. Proc. Natl. Acad. Sci. USA 91:8772-8776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perlman, R., W. P. Schiemann, M. W. Brooks, H. F. Lodish, and R. A. Weinberg. 2001. TGF-β-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat. Cell Biol. 3:708-714. [DOI] [PubMed] [Google Scholar]
- 34.Putcha, G. V., S. Le, S. Frank, C. G. Besirli, K. Clark, B. Chu, S. Alix, R. J. Youle, A. LaMarche, A. C. Maroney, and E. M. Johnson, Jr. 2003. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron 38:899-914. [DOI] [PubMed] [Google Scholar]
- 35.Putcha, G. V., K. L. Moulder, J. P. Golden, P. Bouillet, J. A. Adams, A. Strasser, and E. M. Johnson. 2001. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron 29:615-628. [DOI] [PubMed] [Google Scholar]
- 36.Puthalakath, H., D. C. Huang, L. A. O'Reilly, S. M. King, and A. Strasser. 1999. The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell 3:287-296. [DOI] [PubMed] [Google Scholar]
- 37.Puthalakath, H., and A. Strasser. 2002. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 9:505-512. [DOI] [PubMed] [Google Scholar]
- 38.Sanchez-Capelo, A. 2005. Dual role for TGF-β1 in apoptosis. Cytokine Growth Factor Rev. 16:15-34. [DOI] [PubMed] [Google Scholar]
- 39.Schuster, N., and K. Krieglstein. 2002. Mechanisms of TGF-β-mediated apoptosis. Cell Tissue Res. 307:1-14. [DOI] [PubMed] [Google Scholar]
- 40.Shi, Y., and J. Massague. 2003. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113:685-700. [DOI] [PubMed] [Google Scholar]
- 41.Shull, M. M., I. Ormsby, A. B. Kier, S. Pawlowski, R. J. Diebold, M. Yin, R. Allen, C. Sidman, G. Proetzel, D. Calvin, et al. 1992. Targeted disruption of the mouse transforming growth factor-β 1 gene results in multifocal inflammatory disease. Nature 359:693-699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siegel, P. M., and J. Massague. 2003. Cytostatic and apoptotic actions of TGF-β in homeostasis and cancer. Nat. Rev. Cancer 3:807-821. [DOI] [PubMed] [Google Scholar]
- 43.Strasser, A., A. W. Harris, D. C. Huang, P. H. Krammer, and S. Cory. 1995. Bcl-2 and Fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J. 14:6136-6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Strasser, A., and M. Pellegrini. 2004. T-lymphocyte death during shutdown of an immune response. Trends Immunol. 25:610-615. [DOI] [PubMed] [Google Scholar]
- 45.Takaku, K., H. Miyoshi, A. Matsunaga, M. Oshima, N. Sasaki, and M. M. Taketo. 1999. Gastric and duodenal polyps in Smad4 (Dpc4) knockout mice. Cancer Res. 59:6113-6117. [PubMed] [Google Scholar]
- 46.Valderrama-Carvajal, H., E. Cocolakis, A. Lacerte, E. H. Lee, G. Krystal, S. Ali, and J. J. Lebrun. 2002. Activin/TGF-β induce apoptosis through Smad-dependent expression of the lipid phosphatase SHIP. Nat. Cell Biol. 4:963-969. [DOI] [PubMed] [Google Scholar]
- 47.von Herbay, A., and J. Rudi. 2000. Role of apoptosis in gastric epithelial turnover. Microsc. Res. Tech. 48:303-311. [DOI] [PubMed] [Google Scholar]
- 48.Wildey, G. M., S. Patil, and P. H. Howe. 2003. Smad3 potentiates transforming growth factor β (TGFβ)-induced apoptosis and expression of the BH3-only protein Bim in WEHI 231 B lymphocytes. J. Biol. Chem. 278:18069-18077. [DOI] [PubMed] [Google Scholar]
- 49.Yoo, J., M. Ghiassi, L. Jirmanova, A. G. Balliet, B. Hoffman, A. J. Fornace, Jr., D. A. Liebermann, E. P. Bottinger, and A. B. Roberts. 2003. Transforming growth factor-β-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. J. Biol. Chem. 278:43001-43007. [DOI] [PubMed] [Google Scholar]
- 50.Yu, L., M. C. Hebert, and Y. E. Zhang. 2002. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 21:3749-3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.