

Abstract
The Escherichia coli β sliding clamp protein is proposed to play an important role in effecting switches between different DNA polymerases during replication, repair, and translesion DNA synthesis. We recently described how strains bearing the dnaN159 allele, which encodes a mutant form of the β clamp (β159), display a UV-sensitive phenotype that is suppressed by inactivation of DNA polymerase IV (M. D. Sutton, J. Bacteriol. 186:6738-6748, 2004). As part of an ongoing effort to understand mechanisms of DNA polymerase management in E. coli, we have further characterized effects of the dnaN159 allele on polymerase usage. Three of the five E.coli DNA polymerases (II, IV, and V) are regulated as part of the global SOS response. Our results indicate that elevated expression of the dinB-encoded polymerase IV is sufficient to result in conditional lethality of the dnaN159 strain. In contrast, chronically activated RecA protein, expressed from the recA730 allele, is lethal to the dnaN159 strain, and this lethality is suppressed by mutations that either mitigate RecA730 activity (i.e., ΔrecR), or impair the activities of DNA polymerase II or DNA polymerase V (i.e., ΔpolB or ΔumuDC). Thus, we have identified distinct genetic requirements whereby each of the three different SOS-regulated DNA polymerases are able to confer lethality upon the dnaN159 strain, suggesting the presence of multiple mechanisms by which the actions of the cell's different DNA polymerases are managed in vivo.
The finding that Escherichia coli possesses five distinct DNA polymerases, while humans are currently known to possess at least three times this many (reviewed in references 15 and 61), suggests that organisms must employ a series of adaptable control systems in order to ensure that the correct DNA polymerase gains access to the replication fork at the appropriate time. To some extent, a cell can regulate the access to the replication fork of its different DNA polymerases by attenuating their respective expression levels, specific activities, and/or subcellular localizations (reviewed in reference 61). However, even with these control systems in place, situations will inevitably arise in which a cell must choose between multiple DNA polymerases. As selection of the incorrect polymerase could have catastrophic consequences, it seems likely that control systems in addition to those mentioned above must exist in order to ensure that in these situations, the correct DNA polymerase is recruited to the replication fork at the appropriate time.
When considering how E. coli might manage the actions of its five DNA polymerases to coordinately regulate DNA replication with DNA repair and translesion DNA synthesis, one cannot overstate the importance of the global SOS response: the steady-state levels of polymerase II (Pol II), Pol IV, and PolV vary as much as 10-fold or more as a function of SOS induction (23). Proper management of the E. coli SOS response requires the products of the lexA+ and recA+ genes. LexA protein acts as a transcriptional repressor. By binding to sequences termed SOS boxes located near promoters, LexA blocks access of RNA polymerase to the promoter, thereby effectively repressing transcription of more than 40 different genes (10, 16).
RecA protein, which is required for most homologous recombination (reviewed in reference 30), as well as for the repair of double-stranded DNA breaks (reviewed in references 24 and 30) and the restart of stalled replication forks (reviewed in reference 11), becomes activated for its role in SOS induction by binding to single-stranded DNA (ssDNA), which presumably results from the cell's failed attempts to copy over lesions in the DNA (reviewed in references 16 and 60). Upon binding to ssDNA, RecA forms a helical filament structure, referred to as “activated” RecA (30). Interaction of LexA with activated RecA mediates autodigestion of LexA, which largely inactivates its repressor function, leading to transcriptional derepression of the LexA-regulated genes (reviewed in reference 16).
Although many of the SOS-regulated genes, including polB (Pol II) and dinB (Pol IV), are expressed at modest levels in the absence of DNA damage, others, such as umuDC (Pol V), are expressed at easily detectable levels only following SOS induction (16, 60). Moreover, catalytic activity of Pol V requires that its umuD-encoded subunit undergo a RecA/ssDNA-facilitated autodigestion that is mechanistically similar to that of LexA (7, 41, 45, 53): intact UmuD, together with UmuC, acts in a DNA damage checkpoint control (38, 43), while autodigestion of UmuD results in the removal of its N-terminal 24 residues to generate UmuD′ (7, 41, 53), which leads to the release of the checkpoint and concomitant activation of Pol V-dependent translesion DNA synthesis. In addition to its role in the expression and posttranscriptional activation of Pol V, RecA also plays an as yet poorly understood role in stimulating the catalytic activity of Pol V (46, 47, 49, 52, 61).
In addition to SOS regulation, work from a variety of laboratories (2, 5, 8, 12, 27, 29, 67), including our own (56-59), indicates that the E. coli β sliding clamp and the γ clamp loader complex play important roles in coordinating access of the different DNA polymerases to the replication fork. The β clamp is a ring-shaped, homodimeric protein that topologically encircles double-stranded DNA. It is loaded onto DNA by the γ complex, which is comprised of six distinct polypeptides (τ2, γ, δ, δ′, ψ, and χ) (6, 34). Although the mechanism by which the β clamp is loaded onto DNA is not yet fully understood, it is clear that γ complex binds to the β clamp in an ATP-dependent fashion to catalyze the opening of the clamp. After positioning the opened clamp on the DNA so that it encircles double-stranded DNA, the γ complex undergoes a conformational change that is triggered by the ATPase activity of the γand τ subunits, which effects release of the clamp, resulting in its loading (1, 20). Once loaded, the β clamp moves freely along the DNA, acting as a mobile platform (55).
All five E. coli DNA polymerases interact with β, and this interaction serves to tether the polymerases to the DNA, endowing them with various degrees of processivity (5, 29, 31, 33, 63, 67). In the case of Pol IV, this interaction also serves to enhance its affinity for deoxyribonucleotides (3). Moreover, it has recently become clear that all five DNA polymerases bind to overlapping surfaces on the β clamp (28, 69), suggesting that competition of the different DNA polymerases for interaction with the β clamp may help to coordinate polymerase access to the replication fork (12, 28, 66, 67, 69).
Consistent with the idea that the β clamp and the γ clamp loader complex help to coordinate access to the replication fork of the different DNA polymerases, Viguera et al. (65) determined that a ΔholD::kan mutation, which affects the ψ subunit of the γclamp loader complex, conferred a temperature-sensitive phenotype that was suppressed by deletion of polB, and to a lesser extent by deletion of dinB. Furthermore, we previously reported that the dnaN159 allele, which encodes a mutant form of the β sliding clamp bearing G66E and G174A substitutions (19, 42, 56), was impaired for interaction with the α catalytic subunit of Pol III and conferred a UV-sensitive phenotype in a nucleotide excision repair-deficient background that was suppressed by (not epistatic with) inactivation of Pol IV (56). Taken together, these findings suggest that mutations that impair either the β clamp or the efficiency of clamp loading lead to altered polymerase usage, consistent with a role for the β clamp in helping to regulate Pol access to the replication fork.
Presumably, the ability of the SOS-regulated DNA polymerases to impair growth of a dnaN159 or ΔholD::kan strain is due in large part to a replication defect conferred by the mutations. Although it is possible that the various polymerases compete with each other for access to the replication fork in a stochastic manner, the fact that only particular SOS-regulated DNA polymerases impaired growth in specific genetic backgrounds suggested to us that additional factors, such as the steady-state levels of the different DNA polymerases, which are heavily influenced by the SOS response, as well as other SOS-regulated gene products, could help regulate access of the SOS-regulated polymerases to the replication fork.
To test this hypothesis, we used different lexA and recA alleles to determine the individual effects of SOS induction and RecA activation on polymerase usage in the dnaN159 mutant. Our results indicate that elevated levels of Pol IV impaired growth of the dnaN159 mutant. However, the recA730 allele, which expresses a mutant form of the RecA protein (RecA730) that displays an enhanced ability to form RecA/ssDNA nucleoprotein filaments, conferred a Pol II- and Pol V-dependent lethality in the dnaN159 mutant. Lethality conferred by Pol V did not correlate with its expression level, but rather was dependent upon recA730. Taken together, these results suggest that the LexA and RecA proteins play an important role in effecting polymerase selection and/or polymerase switching at the replication fork following DNA damage. These findings are discussed in terms of a model to describe how E. coli manages the actions of its different DNA polymerases.
MATERIALS AND METHODS
E. coli strains and bacteriological techniques.
E. coli strains are described in Table 1 and were routinely grown in Luria-Bertani (LB) medium (36). When necessary, the following antibiotics were used at the indicated concentrations: ampicillin, 150 μg/ml; chloramphenicol, 20 μg/ml; kanamycin, 60 μg/ml; spectinomycin, 50 μg/ml; streptomycin, 50 μg/ml; tetracycline, 10 μg/ml; and rifampin, 100 μg/ml. All strains are derivatives of E. coli K-12 and were constructed using P1vir-mediated generalized transduction (36). Transduction of the recA730 and recA441 alleles was achieved by selection for tetracycline resistance conferred by the closely linked srlC300::Tn10 allele, and the correct sequence for each allele was confirmed by automated nucleotide sequence analysis of the PCR-amplified recA gene. Transduction of the dnaN159 allele was achieved by selection for the closely linked tnaA300::Tn10 or zid-3162::Tn10kan alleles and was confirmed by verifying temperature sensitivity and by nucleotide sequence analysis of the PCR-amplified dnaN allele, as previously described (56).
TABLE 1.
E. coli strains and plasmids used in this studya
| Strain or plasmid | Relevant genotype | Source or construction |
|---|---|---|
| Strain | ||
| AM107 | ΔrecR252::Tn10∼9kan | J. Courcelle (9) |
| CAG18497 | fadR613::Tn10 | CGSCb |
| CAG18558 | zid-3162::Tn10kan | B. Michel (19) |
| DE298 | recA441 srlC300::Tn10 | R. Woodgate |
| FC1237 | Δ(dinB-yafN)::kan | P. Foster (54) |
| GW8028 | Δara-714 leu::Tn10 | G. Walker |
| JJC1209 | dnaN159 zid-3162::Tn10 | B. Michel (19) |
| RW120 | ΔumuDC595::cat | R. Woodgate (71) |
| STL1336 | Δ(araD-polB)::Ω | M. Goodman (4) |
| MS100 | tnaA300::Tn10 | This laboratory (56) |
| MS101b | dnaN159 tnaA300::Tn10 | This laboratory (56) |
| RW542 | lexA51(Def) | R. Woodgate (21) |
| MS104 | lexA51(Def) tnaA300::Tn10 | This laboratory (56) |
| MS105 | lexA51(Def) dnaN159 tnaA300::Tn10 | This laboratory (56) |
| RM100 | lexA51(Def) Δ(dinB-yafN)::kan | P1(FC1237) × RW542 |
| RM101 | lexA51(Def) ΔumuDC595::cat | P1(RW120) × RW542 |
| RM102 | lexA51(Def) Δara-714 leu::Tn10 | P1(GW8028) × RW542 |
| RM103 | lexA51(Def) Δ(araD-polB)::Ω | P1(STL1336) × RM102 |
| RM104 | lexA51(Def) Δ(araD-polB)::Ω tnaA300::Tn10 | P1(MS100) × RM103 |
| RM105 | lexA51(Def) Δ(araD-polB)::Ω dnaN159 tnaA300::Tn10 | P1(MS101) × RM103 |
| RM106 | lexA51(Def) Δ(dinB-yafN)::kan tnaA300::Tn10 | P1(MS100) × RM100 |
| RM107 | lexA51(Def) Δ(dinB-yafN)::kan dnaN159 tnaA300::Tn10 | P1(MS101) × RM100 |
| RM108 | lexA51(Def) ΔumuDC595::cat tnaA300::Tn10 | P1(MS100) × RM101 |
| RM109 | lexA51(Def) ΔumuDC595::cat dnaN159 tnaA300::Tn10 | P1(MS101) × RM101 |
| RW540 | lexA51(Def) uvrA6 | R. Woodgate |
| RM110 | lexA51(Def) uvrA6 tnaA300::Tn10 | P1(MS100) × RW540 |
| RM111 | lexA51(Def) uvrA6 dnaN159 tnaA300::Tn10 | P1(MS101) × RW540 |
| RM112 | lexA51(Def) uvrA6 recA441 srlC300::Tn10 | P1(DE298) × RW576 |
| RM113 | lexA51(Def) uvrA6 recA441 srlC300::Tn10 zid3162::Tn10kan | P1(CAG18558) × RM112 |
| RM114 | lexA51(Def) uvrA6 recA441 srlC300::Tn10 dnaN159 zid-3162::Tn10kan | P1(JJC1209) × RM112 |
| RW576 | lexA51(Def) uvrA6 recA730 | R. Woodgate |
| RM115 | lexA51(Def) uvrA6 recA730 tnaA300::Tn10 | P1(MS100) × RW576 |
| RM116 | lexA51(Def) uvrA6 recA730 Δ(dinB-yafN)::kan | P1(FC1237) × RW576 |
| RM117 | lexA51(Def) uvrA6 recA730 Δ(dinB-yafN)::kan tnaA300::Tn10 | P1(MS100) × RM116 |
| RM118 | lexA51(Def) uvrA6 recA730 ΔumuDC595::cat | P1(RW120) × RW576 |
| RM119 | lexA51(Def) uvrA6 recA730 ΔumuDC595::cat tnaA300::Tn10 | P1(MS100) × RM118 |
| RM120 | lexA51(Def) uvrA6 recA730 ΔumuDC595::cat dnaN159 tnaA300::Tn10 | P1(MS101) × RM118 |
| RM121 | lexA51(Def) uvrA6 recA730 ara-714 leu::Tn10 | P1(GW8028) × RW576 |
| RM122 | lexA51(Def) uvrA6 recA730 Δ(araD-polB)::Ω | P1(STL1336) × RM121 |
| RM123 | lexA51(Def) uvrA6 recA730 Δ(araD-polB)::Ω tnaA300::Tn10 | P1(MS100) × RM122 |
| RM124 | lexA51(Def) uvrA6 recA730 Δ(araD-polB)::Ω dnaN159 tnaA300::Tn10 | P1(MS101) × RM122 |
| RM125 | ΔumuDC595::cat fadR613::Tn10 | P1(CAG18497) × RW120 |
| RM126 | lexA51(Def) uvrA6 recA730 Δ(araD-polB)::Ω ΔumuDC595::cat fadR613::Tn10 | P1(RM125) × RM122 |
| RM127 | lexA51(Def) uvrA6 recA730 Δ(araD-polB)::Ω ΔumuDC595::cat fadR613::Tn10 zid-3162::Tn10kan | P1(CAG18558) × RM126 |
| RM128 | lexA51(Def) uvrA6 recA730 Δ(araD-polB)::Ω ΔumuDC595::cat fadR613::Tn10 dnaN159 zid3162::Tn10kan | P1(JJC1209) × RM126 |
| RM129c | lexA51(Def) uvrA6 recA730 dnaN159 tnaA300::Tn10 srd-1 | P1(MS101) × RW576 |
| RM130c | lexA51(Def) uvrA6 recA730 dnaN159 tnaA300::Tn10 srd-2 | P1(MS101) × RW576 |
| RM131a | lexA51(Def) uvrA6 recA730 dnaN159 tnaA300::Tn10 srd-1 ΔumuDC595::cat | P1(RW120) × RM129 |
| RM132 | lexA51(Def) uvrA6 recA730 ΔrecR252::Tn10∼9kan | P1(AM107) × RW576 |
| RM133 | lexA51(Def) uvrA6 recA730 ΔrecR252::Tn10∼9kan tnaA300::Tn10 | P1(MS100) × RM132 |
| RM134 | lexA51(Def) uvrA6 recA730 ΔrecR252::Tn10∼9kan dnaN159 tnaA300::Tn10 | P1(MS101) × RM132 |
| RM135c | lexA51(Def) uvrA6 recA730 zid-3162::Tn10kan srd-1 | P1(CAG18558) × RM129 |
| RM136c | lexA51(Def) uvrA6 recA730 zid-3162::Tn10kan srd-2 | P1(CAG18558) × RM130 |
| Plasmids | ||
| pWSK29 | Apr; pSC101 derivative | S. Kushner (68) |
| pRM100 | Apr; pWSK29 bearing polA (Pol I) | This work |
| pRM101 | Apr; pWSK29 bearing polB (Pol II) | This work |
| pRM102 | Apr; pWSK29 bearing dinB (Pol IV) | This work |
| pRM103 | Apr; pWSK29 bearing umuD′ and umuC (Pol V) | This work |
See Materials and Methods for a description of specific strain constructions. Strains AM107 to STL1336 (lines 1 to 9 in the table) were used as donors for P1vir-mediated transduction. Strains MS100 to RM109 (lines 10 to 24 in the table) and RM125 (line 42) are derivatives of strain RW118 [thr-1 araD139 Δ(gpt-proA)62 lacY1 tsx-33 glnV44(AS) galK2(Oc) hisG4(Oc) rpsL31 xylA5 mtl-1 argE3(Oc) thi-1 sulA211]. Strains RM110 to RM136 (lines 26 to 53 in the table), except for RM125, are derivatives of strain RW540 [line 25 in the table; thr-1 araD139 Δ(gpt-proA)62 lacY1 tsx-33 glnV44(AS) galK2(Oc) hisG4(Oc) rpsL31 xylA5 mtl-1 argE3(Oc) thi-1 sulA211 leuB6(Am) uvrA6 recA730].
The dnaN159 allele, which encodes two amino acid substitutions (G66E and G174A), was originally referred to as dnaN59 but has since been renamed by the E.coli Genetic Stock Center (CGSC).
These strains contain an unknown mutation that suppresses recA730 dnaN159 synthetic lethality. These suppressor loci are referred to as srd-1 and srd-2, for suppressor of recA730 dnaN159 synthetic lethality. See the footnotes to Table 3 and the text for further details.
Transduction of the Δ(araD-polB)::Ω, Δ(dinB-yafN)::kan, and ΔumuDC595::cat alleles was done by virtue of antibiotic resistance conferred by the indicated marker. Due to the high background of spectinomycin-resistant CFU occasionally observed when selecting for strains bearing the Ω fragment (regardless of the spectinomycin concentration used; data not shown), we transduced strains that we intended to make Δ(araD-polB)::Ω to tetracycline resistance with leu::Tn10, which is adjacent to polB, and then replaced leu::Tn10 with Δ(araD-polB)::Ω. The presence of each allele was subsequently confirmed by colony PCR. The Δ(araD-polB)::Ω allele was confirmed using primers 5′-CCG ACG GGA TCA ATC AGA AAG GTG-3′ and 5′-TCT GTC CTG GCT GGG AAC GA-3′, which amplify a ∼817-base-pair fragment from Δ(araD-polB)::Ω and yield no product using the polB+ allele.
The Δ(dinB-yafN)::kan allele was confirmed using primers 5′-cgc gaa ttc cat ATG CGT AAA ATC ATT CAT GTG GAT ATG G-3′ (the first 12 bases bear no sequence similarity to the dinB gene; rather, they introduce an NdeI restriction site that was used for cloning dinB into an overexpression vector) and 5′-CCG GTT GAT CAA TAA AGT ATT TAG CTG GG-3′, which amplify a ∼1,000-base-pair fragment from the dinB+-yafN+ region, and a ∼1,250-base-pair fragment from Δ(dinB-yafN)::kan. The ΔumuDC595::cat allele was confirmed using primers 5′-AGG CCA CGT GAG CAC AAG ATA AGA-3′ and 5′-ATA GGT ACA TTG AGC AAC TGA CTG-3′, which amplify a 530-base-pair fragment from ΔumuDC595::cat strains and yield no product using the umuD+C+ alleles.
Plasmid construction and transformation assay.
Plasmid DNAs are described in Table 1. Genes encoding Pol I (polA), Pol II (polB), and Pol IV (dinB) were PCR amplified from genomic DNA, and a synthetic operon encoding Pol V (umuD′C+) was PCR amplified from plasmid pGW3751 (41) using the following primer pairs: polA-promoter, 5′-CTT GCG TGA AAC GGG CGC CTT-3′ and polA-end, 5′-ggg aca cct agg TTA GTG CGC CTG ATC CCA G-3′ (the first 12 nucleotides are not complementary to polA and were included for cloning purposes); polB-promoter, 5′-CAC TAT CTG CGT AAG CAT GGC GCG AAG GC-3′ and polB-end, 5′-ggg aca cct agg TCA GAA TAG CCC AAG TTG C-3′ (the first 12 nucleotides are not complementary to polB and were included for cloning purposes); dinB-promoter, 5′-CAA TAA GAA TTC CGT CAA TCG CCA TCT GTT TGC CGG G-3′ and dinB-end, 5′-cgc aca aag ctt ggt acc TCA TAA TCC CAG CAC CAG TTG TC-3′ (the first 18 nucleotides are not complementary to dinB); and umuDC-promoter, 5′-CTG CTG GCA AGA ACA GAC-3′ and umuDC-end, 5′-CGT GAT CTG TTC GGT CGC TAA TCC-3′.
PCR fragments were blunt-end ligated into pCR-BluntII-Topo vector (Invitrogen, Carlsbad, CA) as per the manufacturer's recommendations. After verifying that each clone contained the correct nucleotide sequence (Roswell Park Cancer Institute Biopolymer Facility, Buffalo, NY), fragments were subcloned into pWSK29 by digestion with EcoRI. The correct orientation (downstream of the lac promoter) was confirmed by diagnostic PCR using the M13 reverse primer, which is homologous to the sequence upstream of the multiple cloning site in pWSK29, paired with second primer homologous to the 3′-end of the cloned insert.
Transformation assays were preformed with the indicated E. coli strains which were made chemically component using rubidium chloride (51). Fifty microliters of component cells (∼5.2 × 108 cells) was incubated with 200 ng of each purified plasmid DNA on ice for 30 min. Reactions were heat shocked at 42°C for 2 min, followed by incubation at 30°C for 1 h. Aliquots of each reaction were then plated onto LB plates supplemented with ampicillin, and colonies were counted after overnight incubation at 30°C.
Mutagenesis assays.
UV-induced mutagenesis was preformed using cultures grown overnight in M9 minimal medium supplemented with glucose (0.2%), thiamine (1 μg/ml), and Casamino Acids (0.5%) essentially as described previously (56, 57). Briefly, overnight cultures were subcultured in the same medium at 30°C with shaking until they reached mid-exponential growth (optical density at 595 nm [OD595] of ∼0.6). One milliliter of culture was then transferred to a sterile glass 15 mm petri dish and either irradiated with 3 J/m2 UV light (254 nm) using a 15-watt germicidal bulb (General Electric) or mock-irradiated. Cultures were then transferred to sterile 25 mm glass bubbler tubes containing 9 ml of supplemented M9 medium, followed by overnight incubation at 30°C with shaking. The following day, 100-μl aliquots of the irradiated and mock-irradiated overnight cultures were plated in duplicate onto LB plates containing rifampin in order to measure mutagenesis, while appropriate dilutions were plated onto LB without rifampin in order to determine cell titers. UV-induced mutation frequency was then calculated by determining the number of rifampin-resistant (Rifr) colonies following UV irradiation minus the number of Rifr colonies induced by mock UV irradiation divided by the cell titer. Spontaneous mutagenesis was measured in a similar manner except that cultures were not UV irradiated.
RESULTS
Access to the replication fork of the dinB-encoded Pol IV is regulated in part by its steady-state level.
We previously reported that the lexA51(Def) allele exacerbated the temperature-sensitive growth phenotype of the dnaN159 strain such that it was unable to grow at temperatures above 34°C (56). Since (i) the UV sensitivity of the dnaN159 strain was suppressed by inactivation of Pol IV and (ii) the dinB-encoded Pol IV is SOS regulated, we hypothesized that the enhanced temperature sensitivity of the dnaN159 lexA51(Def) strain was attributable to elevated levels of Pol IV. We tested this hypothesis in two different ways. In our first experiment, we asked whether SOS-induced levels of Pol IV or one of the other SOS-regulated DNA polymerases (i.e., Pol II or Pol V) led to the growth defect of the dnaN159 lexA51(Def) strain. For this, we compared the growth properties of various isogenic lexA51(Def) strains bearing either dnaN+ or dnaN159 together with deletions of polB (Pol II), dinB (Pol IV), or umuDC (Pol V). Our results indicated that inactivation of Pol IV [Δ(dinB-yafN)::kan] suppressed the temperature-sensitive growth phenotype of the dnaN159 lexA51(Def) strain at both 35 and 37°C (Table 2). In contrast, inactivation of Pol II [Δ(araD-polB)::Ω] or Pol V (ΔumuDC595::cat) had only a minimal effect on the growth phenotype of the dnaN159 lexA51(Def) strain.
TABLE 2.
Plating efficiencies of different isogenic lexA51(Def) strains
| Straina | Relevant genotypeb
|
Efficiency of growthc (relative to growth at 30°C):
|
||||||
|---|---|---|---|---|---|---|---|---|
| dnaN | lexA | polB | dinB | umuDC | 35°C | 37°C | 39°C | |
| MS100 | + | + | + | + | + | 1.1 | 1.1 | 1.0 |
| MS101 | 159 | + | + | + | + | 1.4 | 1.1 | 1.9 × 10−4 |
| MS104 | + | 51(Def) | + | + | + | 1.0 | 1.4 | 0.41 |
| MS105 | 159 | 51(Def) | + | + | + | 3.0 × 10−3 | 2.0 × 10−3 | <1.0 × 10−6 |
| RM104 | + | 51(Def) | Δ | + | + | 1.2 | 1.4 | 0.75 |
| RM105 | 159 | 51(Def) | Δ | + | + | 1.0 × 10−3 | 2.0 × 10−4 | <1.0 × 10−6 |
| RM106 | + | 51(Def) | + | Δ | + | 4.3 | 3.9 | 3.2 |
| RM107 | 159 | 51(Def) | + | Δ | + | 2.6 | 1.9 | <1.0 × 10−6 |
| RM108 | + | 51(Def) | + | + | Δ | 2.1 | 1.1 | 2.9 |
| RM109 | 159 | 51(Def) | + | + | Δ | 4.0 × 10−2 | 8.1 × 10−2 | <1.0 × 10−6 |
Strains are described in Table 1.
Abbreviations: +, wild type; 159, dnaN159; 51(Def), lexA51(Def); Δ, deletion; ΔpolB (Pol II), Δ(araD-polB)::Ω; ΔdinB (Pol IV), Δ(dinB-yafN)::kan; and ΔumuDC (Pol V), ΔumuDC595::cat.
Representative clones for each strain were grown overnight in LB medium at 30°C, and serial dilutions of each culture were plated at 30, 35, 37, and 39°C. Values shown are the ratio of CFU observed at the indicated temperature after overnight incubation divided by the number observed for the same strain at 30°C. The number of CFU/ml observed at 30°C for each strain was: MS100, 5.3 × 108; MS101, 3.8 × 108; MS104, 1.9 × 108; MS105, 2.1 × 108; RM104, 2.4 × 108; RM105, 3.4 × 108; RM106, 0.73 × 108; RM107, 0.54 × 108; RM108, 0.65 × 108; and RM109, 0.59 × 108.
In our second experiment, we asked whether plasmids expressing DNA polymerase I, II, IV, or V impaired growth of the dnaN+ lexA(Def) and/or the dnaN159 lexA51(Def) strains described above. As shown in Fig. 1A, each of these plasmids transformed strain MS104 [dnaN+ lexA51(Def)] with an efficiency that was comparable to that of the pWSK29 control plasmid. In contrast, we were unable to obtain more than a few transformants of the dnaN159 lexA51(Def) strain (MS105) using the Pol IV-expressing plasmid. With the exception of the Pol I-expressing plasmid, which displayed a 10-fold reduction in transformation efficiency, the other plasmids behaved like the pWSK29 control plasmid with respect to their transformation efficiency (Fig. 1A). Addition of isopropylthiogalactopyranoside (IPTG) to 50 μM, which leads to maximal induction of the lac promoter in pWSK29 (57) (data not shown), had no effect on these results (data not shown). Very similar results were observed using strains RM110 [dnaN+ lexA51(Def) uvrA6] and RM111 [dnaN159 lexA51(Def) uvrA6] (Fig. 1B), indicating that, unlike the UV sensitivity of the dnaN159 strain that was severely enhanced by the ΔuvrB::cat or ΔuvrC::cat allele (56), the Pol IV-dependent growth defect of the dnaN159 strain was independent of nucleotide excision repair. Taken together, these results suggest that LexA-dependent SOS regulation of the Pol IV steady-state level is important for helping to regulate its access to the replication fork.
FIG. 1.

Effects of plasmids directing expression of DNA polymerase I, II, IV, or V on growth of different dnaN+ and dnaN159 lexA51(Def) strains. Plasmids expressing Pol I, II, IV, or V were transformed into E. coli strains MS104 [dnaN+ lexA51(Def)] and MS105 [dnaN159 lexA51(Def)] (A) and RM110 [dnaN+ lexA51(Def) uvrA6] and RM111 [dnaN159 lexA51(Def) uvrA6] (B). Transformation efficiencies are expressed as the number of CFU obtained per microgram of supercoiled plasmid DNA (note that the y-axis is a log10 scale). Values represent the averages and ranges of two independent experiments. Plasmids: pWSK29, control; pRM100, polA (Pol I); pRM101, polB (Pol II); pRM102, dinB (Pol IV); pRM103, umuD′C (Pol V).
Synthetic lethality of the dnaN159 recA730 strain is suppressed by mutations that inactivate DNA Pol II or DNA Pol V.
Results discussed above suggest that access of Pol IV to the replication fork can be regulated, at least in part, by its steady-state level relative to the other polymerases. RecA protein is required for induction of the global SOS response, as well as most homologous recombination, the repair of double-stranded DNA breaks, the restart of stalled replication forks, and Pol V-dependent translesion DNA synthesis (reviewed in references 11, 16, 24, 30, and 61). In order to determine whether RecA affects polymerase selection following SOS induction, we combined dnaN159 with different recA alleles under the premise that certain combinations might confer synthetic phenotypes that could be modified by mutations affecting one or more of the various E. coli polymerases. Inactivation of recA (ΔrecA::cat) had only a minor effect on the dnaN159 strain, resulting in a slightly enhanced temperature-sensitive growth phenotype (data not shown), consistent with previous reports (19). This phenotype was similar to that of the dnaN159 lexA3(Ind−) strain (56), suggesting that it resulted from a lack of Pol V, and was not further characterized.
We next examined recA alleles that displayed enhanced DNA binding activity. The recA441 allele, which bears E38K and I298V substitutions, displays an enhanced rate of association with ssDNA in vitro (25, 26). The recA730 allele, which was isolated from recA441 by Witkin et al. (70), contains only the E38K substitution, and the mutant RecA730 protein displays an even more robust ssDNA binding activity in vitro than does RecA441 (25). Despite the fact that we could efficiently transduce the dnaN159 allele into the recA441 lexA51(Def) strain (RM112) by selection for kanamycin resistance conferred by the nearby zid-3162::Tn10kan allele (Table 3), transduction of dnaN159 into the isogenic recA730 lexA(Def) strain RW576 was remarkably inefficient (Table 3). Similar results were observed when trying to transduce dnaN159 into another recA730 strain [NR9350 (genotype: ara thi Δ(pro-lac) sulA211 srlC300::Tn10 recA730) (13)] using a P1vir lysate prepared on a zid-3162::Tn10kan dnaN159 strain (data not shown), indicating that the inability to do so was specific to combination of dnaN159 and recA730 and not to some other allele(s).
TABLE 3.
Genetic requirements for dnaN159 recA730 synthetic lethality
| Straina | Relevant genotypeb
|
Transduction efficiencyc
|
|||
|---|---|---|---|---|---|
| recA | lexA | Other | No. of transductants selected (Tcr or Kmr CFU) | No. ts/total no. screened (% of total) | |
| RW540 | + | 51(Def) | 94 | 40/48 (83) | |
| RM112 | 441 | 51(Def) | 89 | 41/50 (82) | |
| RW576 | 730 | 51(Def) | 552 | 2/94 (2)e | |
| RM122 | 730 | 51(Def) | ΔpolB | 1,227 | 59/98 (60) |
| RM116 | 730 | 51(Def) | ΔdinB | 37 | 0/35 (<3) |
| RM118 | 730 | 51(Def) | ΔumuDC | 65 | 5/11 (46) |
| RM126d | 730 | 51(Def) | ΔpolB, ΔumuDC | 616 | 42/52 (81) |
| RM135e | 730 | 51(Def) | srd-1 | 920 | 77/100 (77) |
| RM136e | 730 | 51(Def) | srd-2 | 2,008 | 64/100 (64) |
| RM132 | 730 | 51(Def) | ΔrecR | 58 | 8/20 (40) |
Strains are described in Table 1.
Abbreviations: +, wild type; 441, recA441; 730, recA730; 51(Def), lexA51(Def); ΔpolB (Pol II), Δ(araD-polB)::Ω; ΔdinB (Pol IV), Δ(dinB-yafN)::kan; ΔumuDC (Pol V), ΔumuDC595::cat; srd-1 and srd-2, suppressors of recA730 dnaN159 synthetic lethality; and ΔrecR, ΔrecR252::Tn10∼9kan.
Transductions were performed using a lysate propagated on a dnaN159 tnaA300::Tn10 strain except for strains RM112 and RM126, which used a lysate propagated on a dnaN159 zid-3162::Tn10kan strain (due to the fact that they contained the srlC300::Tn10 and fadR613::Tn10 alleles, respectively). The efficiency of cotransduction of dnaN159 and tnaA300::Tn10 (or zid-3162::Tn10kan) was measured by patching out transductants in duplicate, followed by incubation overnight at 30 and 42°C. Ratios represent the number of temperature-sensitive (ts) transductants divided by the number of clones patched out. Cotransduction frequencies for tnaA300::Tn10-dnaN159 (or zid-3162::Tn10kan-dnaN159) are indicated. Theoretical cotransduction frequencies were calculated to be 78% for tnaA300::Tn10 and dnaN, and 81% for zid-3162::Tn10kan and dnaN using the following formula: F = (1 − d/L)3, where F is the frequency of cotransduction, d is the distance between dnaN and zid or tnaA, and L is the size of the fragment that can be transduced by P1 vir (2.1 min) (40).
Transduction of the ΔumuDC595::cat allele into recA730 Δ(araD-polB)::Ω strain RM122 was inefficient (data not shown), suggesting that Pol II or Pol V was required by (or at least important to) the recA730 strain. Therefore, we constructed a ΔumuDC595::cat fadR613::Tn10 strain (RM125) so that we could measure the efficiency of cotransduction of ΔumuDC and fadR using the recA730 Δ(araD-polB)::Ω strain (RM122) and selection for Tcr. Cotransduction frequency for ΔumuDC595::cat and fadR613::Tn10 was calculated to be 85%. Our finding that 39 of 45 (86%) Tcr transductants tested (from a total of 119 Tcr transductants) were also Cmr was in excellent agreement with the theoretical value of 85%, indicating that neither Pol II nor Pol V is required by the recA730 strain for viability.
The two tetracycline-resistant and temperature-sensitive dnaN159 recA730 strains identified were colony purified and named RM129 and RM130. The dnaN159 allele in strains RM129 and RM130 was replaced by dnaN+ using a P1vir lysate grown on the dnaN+ zid-3162::Tn10kan strain. Three hundred eighty-four Kmr transductants were obtained with RM129, while 408 were obtained with RM130. Thirty-nine of 40 tested for RM129 were temperature resistant, and 35 of the 39 temperature-resistant clones were Tcs. Thirty-eight of 40 tested for RM130 were temperature resistant, and 31 of the 38 temperature-resistant clones were Tcs. Four temperature-resistant and Tcs clones for each strain were picked and colony purified. One of each colony-purified strain was selected (named RM135 and RM136; see Table 1) for transduction with a P1vir lysate grown on the dnaN159 tnaA300::Tn10 strain, the results of which are shown above.
In order to rule out the possibility that the recA730 mutation suppressed the temperature-sensitive growth phenotype of the dnaN159 strain, we determined the nucleotide sequence of the PCR-amplified dnaN allele from six separate strains; all six corresponded to the wild-type dnaN sequence (data not shown). This result indicates that recA730 does not suppress the temperature-sensitive growth phenotype of dnaN159 and suggests that the combination of the dnaN159 and recA730 alleles results in a synthetic lethal phenotype.
Given our finding that SOS-induced levels of Pol IV exacerbated the temperature-sensitive growth phenotype of the dnaN159 mutant (Table 2), we hypothesized that SOS-induced levels of one or more of the three SOS-regulated DNA polymerases, in combination with RecA730, might result in lethality in the dnaN159 strain. We therefore constructed a series of isogenic RW576 derivatives in which we combined the recA730 allele with mutations inactivating each of the SOS-regulated polymerases (Pol II, Pol IV, and Pol V) and tested these strains for their ability to be transduced to temperature sensitivity with dnaN159.
In striking contrast to our results discussed above indicating that inactivation of Pol IV suppressed the growth defect of the dnaN159 lexA51(Def) strain (see Table 2 and Fig. 1), inactivation of Pol IV [Δ(dinB-yafN)::kan] had no discernible effect onthe transduction efficiency of dnaN159 into the recA730 strain(Table 3). Conversely, inactivation of either Pol II [Δ(araD-polB)::Ω] or Pol V (ΔumuDC595::cat) allowed efficient transduction of dnaN159 into the recA730 strain (Table 3). Furthermore, inactivation of both polB and umuDC in the same strain (RM126) resulted in an even slightly higher efficiency of transduction for dnaN159 (Table 3). These findings indicate that lethality of the dnaN159 recA730 strain is the result of Pol II and/or Pol V. The fact that UmuD′ levels were similar in the recA441 and recA730 strains (Fig. 2) indicates that lethality is not due simply to the presence of elevated levels of Pol V or to the checkpoint function of umuDC (since intact UmuD was poorly detectable), but rather some property of the RecA730 protein in the dnaN159 strain which affects one or more functions of Pol II and Pol V.
FIG. 2.

Steady-state levels of the different umuD gene products in recA+, recA441, and recA730 strains. E. coli strains were grown in LB medium to an OD595 of 0.5. A 2-ml aliquot of each culture was collected by centrifugation. Cells were resuspended in 20 μl of sterile 0.8% NaCl and lysed by addition of 50 μl of 4X sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) loading dye (200 mM Tris-HCl [pH 6.8], 8% SDS, 0.4% bromophenyl blue, 40% glycerol) containing 10% β-mercaptoethanol prior to visualization by Western blot analysis using purified polyclonal antibodies specific to the UmuD and UmuD′ proteins. The salient features of each strain's genotype are indicated. Strain examined: MS104, dnaN+ recA+ recR+; MS105, dnaN159 recA+ recR+; RM112, dnaN+ recA441 recR+; RM113, dnaN159 recA441 recR+; RM133, dnaN+ recA730 ΔrecR252::Tn10∼9kan; RM134, dnaN159 recA730 ΔrecR252::Tn10∼9kan; RM115, dnaN+ recA730 recR+; RM129, dnaN159 srd-1 recA730 recR+; and RM130, dnaN159 srd-2 recA730 recR+. Levels of UmuD and UmuD′ in strains RM110 [dnaN+ lexA51(Def) uvrA6] and RM111 [dnaN159 lexA51(Def) uvrA6] were essentially identical to those observed in strains MS104 and MS105, respectively (data not shown).
Genetic characterization of viable dnaN159 recA730 strains provides further evidence for a role of DNA polymerase V-dependent replication in conferring synthetic lethality.
Despite the fact that the dnaN159 allele was synthetically lethal with recA730, we were able to identify two dnaN159 transductants of the recA730 strain that acquired temperature-sensitive growth (RM129 and RM130; see Tables 1 and 3). Based on nucleotide sequence analysis, these two strains did in fact contain the recA730 and dnaN159 alleles (data not shown). In addition, both strains contained the correct sequence for polB (Pol II) and umuDC (Pol V) (data not shown). In order to determine whether these strains contained a suppressor of the synthetic lethality, we first replaced the dnaN159 allele in RM129 and RM130 with dnaN+ by selection for the nearby zid-3162::Tn10kan allele (which is located on the opposite side of dnaN, as is tnaA300::Tn10), resulting in strains RM135 and RM136, respectively (see Table 3 footnotes). We next attempted to transduce strains RM135 and RM136 to temperature sensitivity with dnaN159 by selection for the nearby tnaA300::Tn10 allele. The results of this experiment indicated that both strains could be efficiently transduced with dnaN159, demonstrating that each of these strains contained at least one mutation that did not map within polB or umuDC and was unlinked to dnaN that acted to suppress the lethal effects of dnaN159 in the recA730 strain (Table 3).
The fact that strains RM129 and RM130 were viable but lacked mutations in polB and umuDC indicated that lethality could be suppressed by mutations affecting genes other than Pol II and Pol V. The RecF, RecO, and RecR proteins have been shown to play important roles in enhancing RecA function (37). We therefore asked whether the ΔrecR252::Tn10∼9kan allele would allow efficient construction of a recA730 dnaN159 strain. As shown in Table 3, dnaN159 was transduced efficiently into the recA730 ΔrecR252::Tn10∼9kan strain. Nonetheless, nucleotide sequence analysis of the recF, recO, and recR genes from strains RM129 and RM130 revealed that each contained the correct sequence, indicating that suppression in these two strains was conferred by a mutation affecting a different gene(s).
In order to better understand the basis for lethality of the recA730 dnaN159 strain, as well as the suppression of lethality by the ΔrecR252::Tn10∼9kan, srd-1, and srd-2 alleles, we investigated whether these alleles exerted an effect upon Pol V-dependent UV-induced and/or spontaneous mutagenesis.
It is known that RecA is required for catalytic activity of Pol V (48, 50, 62, 64). Thus, if ΔrecR252::Tn10∼9kan, srd-1, and/or srd-2 impairs RecA function, Pol V function may also be impaired. Consistent with this hypothesis, the dnaN+ and dnaN159 ΔrecR252::Tn10∼9kan strains displayed ∼5-fold and ∼2-fold lower UV-induced mutation rates, respectively, compared to the dnaN+ recR+ parent (Fig. 3A). Since these strains contained the lexA51(Def) allele, these results demonstrate an important role for the RecR protein in Pol V-dependent mutagenesis independent of SOS induction. Consistent with this finding, spontaneous mutation rates were modestly reduced in both the dnaN+ and dnaN159 ΔrecR252::Tn10∼9kan strains (Fig. 3B). Thus, the phenotype of the dnaN159 ΔrecR252::Tn10∼9kan strain is consistent with a model in which deletion of recR suppresses synthetic lethality by impairing Pol V function.
FIG. 3.

UV-induced and spontaneous mutation rates in different dnaN+ and dnaN159 lexA51(Def) recA730 strains. UV-induced (A) and spontaneous (B) mutagenesis frequencies were measured as described in Materials and Methods. Values presented are relative to the dnaN+ recA730 strain (RM115), which was set equal to 100%. Strain RM115 yielded 44.8 Rifr CFU/107 survivors for UV-induced mutagenesis and 2.2 Rifr CFU/107 survivors for spontaneous mutagenesis. Other strains examined: RM123, dnaN+ recA730 Δ(araD-polB)::Ω; RM124, dnaN159 recA730 Δ(araD-polB)::Ω; RM119, dnaN+ recA730 ΔumuDC595::cat; RM120, dnaN159 recA730 ΔumuDC595::cat; RM133, dnaN+ recA730 ΔrecR252::Tn10∼9kan; RM134, dnaN159 recA730 ΔrecR252::Tn10∼9kan; RM129, dnaN159 recA730 srd-1; RM130, dnaN159 recA730 srd-2; and RM131, dnaN159 recA730 srd-1 ΔumuDC595::cat. Results shown are averages of at least three experiments ± standard deviation.
In order to determine whether srd-1 or srd-2 mediated suppression via an effect on Pol V function, we measured UV-induced SOS mutagenesis of strains RM129 and RM130. Pol V-dependent UV-induced mutagenesis was reduced ∼5-fold in strain RM129 (srd-1) relative to the dnaN+ recA730 control strain RM115 (Fig. 3A). In contrast, proficiency in SOS mutagenesis of strain RM130 (srd-2) was similar to that observed for RM115 (Fig. 3A). We also measured spontaneous mutagenesis frequencies in both the dnaN159 recA730 srd-1 (RM129) and dnaN159 recA730 srd-2 (RM130) strains. While strains RM130 (srd-2) and RM115 displayed similar spontaneous mutation frequencies, spontaneous mutagenesis in strain RM129 (srd-1) was elevated more than sixfold relative to the dnaN+ control strain RM115 (Fig. 3B). Furthermore, this elevated mutation frequency was completely dependent upon Pol V, as the ΔumuDC595::cat allele eliminated it (Fig. 3B). These results indicate that suppression of the dnaN159 recA730 synthetic lethality can be achieved simply by attenuating access to the replication fork of Pol V rather than affecting the catalytic activity of the enzyme per se. Based on these results, we conclude that the srd-1 allele in strain RM129 affects the way in which Pol V is recruited to the replication fork.
Functionally distinct roles for Pol II and Pol V in conferring recA730 dnaN159 synthetic lethality.
In order to determine whether the dnaN159 recA730 synthetic lethality required the concerted actions of Pol II and Pol V or whether lethality resulted from the combined effects of their independent actions, we measured proficiency in Pol V-dependent SOS mutagenesis of the dnaN+ and dnaN159 Δ(araD-polB)::Ω strains. As shown in Fig. 3, the frequencies of UV-induced and spontaneous mutagenesis in the dnaN+ and dnaN159 Δ(araD-polB)::Ω strains were within twofold of each other. Thus, taken together, these results indicate that Pol II does not significantly affect Pol V function in translesion DNA synthesis, suggesting that synthetic lethality of the dnaN159 recA730 strain results from the combined effects of Pol II and Pol V and not from their concerted actions.
To rule out the possibility that Pol II and Pol V were working together in some facet of replication that did not result in an increased mutation frequency, but did confer lethality in the recA730 strain, we also examined the growth rates of the different recA730 strains. The rapid growth rate of the dnaN159 Δ(araD-polB)::Ω recA730 lexA51(Def) strain (RM124) suggests that inactivation of Pol II resulted in efficient suppression of synthetic lethality (Fig. 4). In contrast, the very slow growth rate of the dnaN159 ΔumuDC595::cat recA730 lexA51(Def) strain (RM120) suggests that either (i) suppression of lethality by inactivation of Pol V is inefficient or (ii) despite its ability to confer lethality in the dnaN159 recA730 genetic background, Pol V nonetheless plays one or more important roles in the dnaN159 strain. This latter possibility was suggested by our previous observation that the umuDC gene products play an important role in protecting the dnaN159 mutant from the lethal effects of UV irradiation (56).
FIG. 4.

Growth rates of different dnaN+ and dnaN159 lexA51(Def) recA730 strains. Overnight cultures of E. coli strains grown in LB medium were subcultured into the same medium to a starting OD595 of 0.04. Cultures were grown at 30°C with shaking, and the OD595 was measured and recorded every 20 min. Strains examined: RM115, dnaN+ recA730; RM123, dnaN+ recA730 Δ(araD-polB)::Ω; RM124, dnaN159 recA730 Δ(araD-polB)::Ω; RM119, dnaN+ recA730 ΔumuDC595::cat; RM120, dnaN159 recA730 ΔumuDC595::cat; RM133, dnaN+ recA730 ΔrecR252::Tn10∼9kan; RM134, dnaN159 recA730 ΔrecR252::Tn10∼9kan; RM129, dnaN159 recA730 srd-1; RM130, dnaN159 recA730 srd-2; and RM131, dnaN159 recA730 srd-1 ΔumuDC595::cat. Results shown represent the average of triplicates ± standard deviation.
In order to distinguish between these two possibilities, we examined the growth properties of the dnaN+ and dnaN159 recA730 lexA51(Def) strains bearing both the ΔpolB and the ΔumuDC alleles. The results of this analysis indicated that inactivation of Pol II largely suppressed the poor growth of the dnaN159 ΔumuDC595::cat recA730 lexA51(Def) strain (Fig. 4, inset). Thus, these findings are consistent with a model in which Pol V competes with Pol II for access to the replication fork and that inactivation of Pol V alone suppresses synthetic lethality less efficiently than does inactivation of Pol II.
We also examined the growth rates of the ΔrecR, srd-1, and srd-2 strains in order to compare their efficiency of suppression to those observed for the ΔpolB and ΔumuDC strains. Our finding that strain RM134 (dnaN159 recA730 ΔrecR252::Tn10∼9kan) grew almost as well as the dnaN+ recA730 strain (Fig. 4) suggests that, in addition to impairing Pol V function, the ΔrecR252::Tn10∼9kan allele also affects Pol II function, presumably by impairing RecA730 activity. Both the srd-1 (RM129) and srd-2 (RM130) strains grew as efficiently as the strain carrying the Δ(araD-polB)::Ω allele, suggesting that these mutations were effective suppressors of synthetic lethality (Fig. 4).
recA730 allele suppresses UV sensitivity of the dnaN159 strain.
The model that begins to emerge from our results discussed above is that although transcriptional derepression of the SOS regulon appears to play an important role in allowing Pol IV to gain access to the replication fork, RecA protein subsequently facilitates access of Pol II and Pol V. If, in the course of promoting access of Pol II and Pol V to the fork, RecA mitigates access of Pol IV, either actively or as a result of increased competition with Pol II and Pol V, then it follows that the recA730 allele would suppress the UV sensitivity of the dnaN159 strain, much the same as the Δ(dinB-yafN)::kan allele did (functional Pol IV, encoded by dinB, conferred UV sensitivity upon the dnaN159 strain [56]).
As a test of this hypothesis, we measured the UV sensitivity of the dnaN159 recA730 srd-1 (RM129) and srd-2 (RM130) strains as well as the dnaN+ recA730 srd° parent, RM115 (we were unable to examine the UV sensitivity of the dnaN159 recA730 srd° strain due to the fact that it was inviable). As hypothesized, RM129 and RM130 were each indistinguishable from the dnaN+ parent strain with respect to UV sensitivity (Fig. 5). Although we do not yet know the nature of the srd-1 and srd-2 alleles, and hence we do not know the effect(s) that these mutations may have on UV sensitivity, these results are nonetheless consistent with a model in which RecA, either directly or indirectly, plays a role in attenuating access of Pol IV to the replication fork as part of the global SOS response. Consistent with this conclusion, Pol IV is reportedly inefficient at replicating a RecA-coated DNA template in vitro (32, 63, 67).
FIG. 5.
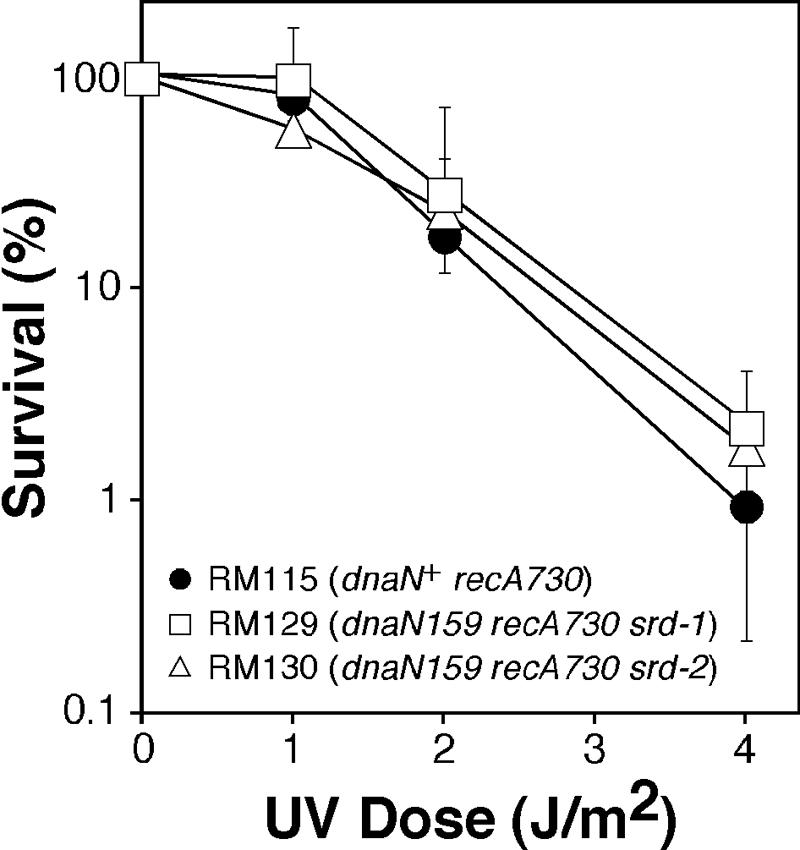
UV sensitivity of E. coli srd-1 and srd-2 strains RM129 and RM130. Overnight cultures of strains RM115 (dnaN+ recA730), RM129 (dnaN159 recA730 srd-1), and RM130 (dnaN159 recA730 srd-2) grown in M9 minimal medium supplemented with glucose (0.2%), thiamine (1 μg/ml), and Casamino Acids (0.5%) were subcultured into the same medium and grown at 30°C with shaking until reaching an OD595 of ∼0.6. A 5-ml aliquot of each culture was irradiated as indicated previously (57), and appropriate dilutions of each irradiated and unirradiated sample were plated onto LB plates and incubated for two days at 30°C. Results shown are the average of two independent experiments ± the range.
DISCUSSION
The steady-state levels and relative affinities of the various E. coli DNA polymerases for the β clamp, as well as possibly other proteins that help to guide them to the replication fork, have presumably been optimized through evolution to ensure an ideal balance between accurate replication and capacity for translesion DNA synthesis. The goal of the work discussed in this report was to better understand the roles of LexA and RecA in mediating this process. We hypothesized that since Pol II, Pol IV, and Pol V are all SOS regulated, the global SOS response might act to help manage the actions of these polymerases by regulating their access to the replication fork. As a test of this hypothesis, we used different lexA and recA alleles to determine the individual effects of SOS induction and RecA activation on Pol usage in the dnaN159 mutant.
Our results indicate that inactivation of LexA (Table 2) or transformation with a plasmid directing expression of Pol IV (Fig. 1) is sufficient for this polymerase to impair growth of the dnaN159 strain. In contrast, both SOS induction and activated RecA protein, provided by the recA730 allele, were required in order for Pol II or Pol V to impair growth of the dnaN159 strain (Table 3). The fact that RecA730 is chronically active due to a mutation and therefore cannot return to a resting state appears to be vital for its synthetic lethality with dnaN159. Thus, our inability to observe a similar phenotype in a recA+ dnaN159 strain is presumably due to the fact that RecA is transiently activated in response to DNA damage and activated RecA levels dissipate as the damage is repaired or tolerated. Nonetheless, we have recently determined that the UV sensitivity of the dnaN159 ΔumuDC strain is suppressed by inactivation of Pol II (M. D. Sutton and J. M. Duzen, submitted), indicating that Pol V and Pol II do confer a conditionally lethal phenotype in the recA+ dnaN159 strain following UV irradiation. Taken together, these results suggest that RecA protein, either directly or indirectly, influences the ability of the different SOS-regulated polymerases to gain access to the replication fork in vivo (Fig. 6).
FIG. 6.

Model to describe the role of the E. coli β sliding clamp, RecA protein and the global SOS response in managing polymerase usage at the replication fork. (A) Summary of the phenotypes observed for the dnaN159 mutant strain. Transcriptional de-repression of the SOS regulon by the lexA51(Def) allele leads to a conditional growth defect of the dnaN159 strain that is suppressed by inactivation of the dinB-encoded Pol IV. Subsequent introduction of the recA730 allele into the dnaN159 lexA51(Def) strain leads to a synthetic lethal phenotype that is suppressed by inactivation of the polB-encoded Pol II or the umuDC-encoded Pol V. (B) Model to describe the role of the SOS response in polymerase selection and switching in E. coli. Polymerases in bold are proposed to have the ability to gain access to the replication fork under the indicated conditions, and hence play prominent roles in DNA replication, while those in italics, although present, are proposed to play more minor roles. The lack of a polymerase indicates that it is not expressed to a significant level (or is not active) under the indicated conditions. UmuD2C participates in DNA damage checkpoint control and is an inactive precursor to Pol V (38, 43). RecA/ssDNA-facilitated autodigestion of UmuD to yield UmuD′ serves to release the checkpoint while simultaneously activating Pol V (UmuD′2C) for translesion DNA synthesis. See the text for details.
Our finding that recA730 suppressed the UV sensitivity of the dnaN159 strain (Fig. 5) is consistent with a model in which RecA can impair access to the fork of Pol IV. However, it should be stressed that Pol IV can still presumably compete with Pol II and Pol V for access to the replication fork in the presence of activated RecA protein: indeed, the Δ(dinB-yafN)::kan allele modestly enhanced Pol V-dependent UV-induced mutagenesis of a recA+ dnaN159 strain (data not shown), consistent with a model in which the RecA protein can act to attenuate competition between Pol IV and polymerases II and V. Thus, our findings suggesting that the duration and the degree of SOS induction influences polymerase selection (Fig. 6) could explain why the recA+ lexA+ ΔholD::kan strain displayed a conditional lethality that was dependent upon Pol II, and to a lesser extent, upon Pol IV (65), while a similar dnaN159 strain displayed a conditional lethality dependent upon Pol IV (56): the ΔholD::kan strain appears to be induced to a greater level for the SOS response than is the dnaN159 strain (56, 65), arguing that levels of activated RecA protein are higher. This, in turn, presumably results in the Pol II-dependent effect in the ΔholD::kan strain. The inability of Pol V to impair growth of the ΔholD::kan strain may be due to the fact that activated RecA levels were insufficient to allow the accumulation of a sufficient steady-state level of Pol V to impair growth. Our finding that Pol II was more effective than Pol V at impairing growth of the dnaN159 recA730 strain (Fig. 4) is consistent with this model.
Our finding that deletion of either polB (Pol II) or umuDC (Pol V) suppressed the lethality of the dnaN159 recA730 strain suggests that Pol II and Pol V are able to gain access to the replication fork in a RecA-mediated fashion. Our findings that deletion of polB suppressed the growth defect of the dnaN159 recA730 ΔumuDC595::cat strain (Fig. 4), and conferred a ∼2-fold effect on Pol V-dependent mutagenesis in the recA730 strain (Fig. 3), irrespective of the dnaN allele, suggest that Pol II and Pol V compete with each other for access to the replication fork. Consistent with this conclusion, deletion of polB in the dnaN159 lexA51(Def) recA+ strain (RM105) resulted in a ∼17-fold increase in spontaneous argE3(Oc)→Arg+ reversion (data not shown). Subsequent deletion of umuDC confirmed that spontaneous mutagenesis was Pol V dependent, suggesting that in the dnaN159 strain, Pol II competed effectively with Pol V for access to the replication fork, effectively suppressing mutagenesis. The fact that Pol V requires RecA for catalytic activity indicates that RecA was activated in the cells that displayed Pol V-dependent spontaneous mutagenesis, consistent with our genetic analyses suggesting that RecA effects Pol selection.
Given that β159 is impaired for interaction with the α catalytic subunit of Pol III (56) and that lagging-strand Pol III must cycle to a new primer every ∼1 second during lagging-strand synthesis (39), it is possible that the conditional lethality of Pol IV in the dnaN159 lexA51(Def) strain is due to competition of Pol IV with the lagging-strand polymerase, impairing Okazaki fragment synthesis. Alternatively, Pol IV might compete with Pol I to impair Okazaki fragment maturation and/or ssDNA gap repair. Our observation that Pol I is essential in the dnaN159 strain (56) is consistent with the idea that this strain displays an increased dependence upon ssDNA gap repair. Hence, competition between Pol IV and Pol I and/or Pol III for access to nascent Okazaki fragments might result in persistent ssDNA gaps in the lagging strand. These gaps, in turn, could allow the formation of RecA/ssDNA nucleoprotein filaments, leading to the chronic, low-level SOS induction observed in the dnaN159 strain (56). Pol II and Pol V might similarly compete with Pol III and Pol I for nascent Okazaki fragments, resulting in ssDNA gaps. However, our observation that the ΔumuDC595::cat allele did not suppress the lethality of the dnaN159 ΔpolA::kan strain (data not shown) indicates that the ΔpolA::kan and recA730 alleles affect the dnaN159 strain in different ways.
It was recently reported that dinB transcription is induced in E. coli by β-lactam-mediated inhibition of cell wall synthesis in a lexA- and recA-independent manner (35, 44). Importantly, β-lactam-mediated transcriptional induction of dinB correlates with an increase in the frequency of +1 lacZ frameshift mutagenesis in vivo (44). This finding is consistent with our results, suggesting that, in the dnaN159 strain, access to the replication fork of Pol IV is regulated largely by its expression level (although our results do not rule out the possibility that additional SOS-regulated gene products may be required for this process; see Table 1 and Fig. 1). The finding that Pol IV is expressed at 6 to 12 times higher steady-state levels than the other SOS-regulated polymerases is consistent with the idea that it is able to outcompete the other polymerases for binding to the β clamp and subsequent access to the replication fork (23). This conclusion is further supported by reports that modest overexpression of Pol IV from a multicopy plasmid significantly increases the frequency of untargeted mutagenesis, indicating Pol IV-dependent replication (22).
Thus, E. coli appears to utilize different control systems to manage the actions of each of its three SOS-regulated polymerases: the actions of Pol IV appear to be largely regulated by transcriptional controls, while the actions of Pol II and Pol V appear to be regulated in a far more complex manner: in the case of Pol V, these controls are incredibly complex and range from RecA/ssDNA-mediated autodigestion of UmuD to yield UmuD′ (7, 45, 53) to ClpXP- and Lon-mediated proteolysis of Pol V (14, 17, 18).
In conclusion, our results indicate that the global SOS response plays important roles in helping to manage the actions of Pol II, Pol IV, and Pol V. Furthermore, our findings suggest that RecA protein is able to attenuate the function of Pol II and Pol V as well as possibly Pol III and Pol IV in vivo. Further characterization of the roles of RecA protein in polymerase function as well as the identification of the srd-1 and srd-2 gene products will lead to a better understanding of polymerase management in E. coli.
Acknowledgments
We thank Mary Berlyn (E. coli Genetic Stock Center, Yale University) for advice regarding the nomenclature for the suppressor mutations in strains RM129 and RM130 and Roger Woodgate (NICHD, NIH) for his generous gift of antibodies specific to the UmuD/UmuD′ proteins. We also thank Mary Berlyn, Justin Courcelle (Portland State University), Bénédicte Michel (Institut National de la Recherche Agronomique), Graham Walker (Massachusetts Institute of Technology), and Roger Woodgate for generously providing E. coli strains, Laurie Sanders for her comments on the manuscript, and the members of our laboratory for helpful discussions.
This work was supported by Public Service Health grant GM066094 to M.D.S.
REFERENCES
- 1.Ason, B., J. G. Bertram, M. M. Hingorani, J. M. Beechem, M. O'Donnell, M. F. Goodman, and L. B. Bloom. 2000. A model for Escherichia coli DNA polymerase III holoenzyme assembly at primer/template ends. DNA triggers a change in binding specificity of the gamma complex clamp loader. J. Biol. Chem. 275:3006-3015. [DOI] [PubMed] [Google Scholar]
- 2.Becherel, O. J., R. P. Fuchs, and J. Wagner. 2002. Pivotal role of the β-clamp in translesion DNA synthesis and mutagenesis in E. coli cells. DNA Repair (Amsterdam) 1:703-708. [DOI] [PubMed] [Google Scholar]
- 3.Bertram, J. G., L. B. Bloom, M. O'Donnell, and M. F. Goodman. 2004. Increased dNTP binding affinity reveals a nonprocessive role for Escherichia coli beta clamp with DNA polymerase IV. J. Biol. Chem. 279:33047-33050. [DOI] [PubMed] [Google Scholar]
- 4.Bonner, C. A., S. Hays, K. McEntee, and M. F. Goodman. 1990. DNA polymerase II is encoded by the DNA damage-inducible dinA gene of Escherichia coli. Proc. Natl. Acad. Sci. USA 87:7663-7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonner, C. A., P. T. Stukenberg, M. Rajagopalan, R. Eritja, M. O'Donnell, K. McEntee, H. Echols, and M. F. Goodman. 1992. Processive DNA synthesis by DNA polymerase II mediated by DNA polymerase III accessory proteins. J. Biol. Chem. 267:11431-11438. [PubMed] [Google Scholar]
- 6.Bowman, G. D., E. R. Goedken, S. L. Kazmirski, M. O'Donnell, and J. Kuriyan. 2005. DNA polymerase clamp loaders and DNA recognition. FEBS Lett. 579:863-867. [DOI] [PubMed] [Google Scholar]
- 7.Burckhardt, S. E., R. Woodgate, R. H. Scheuermann, and H. Echols. 1988. UmuD mutagenesis protein of Escherichia coli: overproduction, purification, and cleavage by RecA. Proc. Natl. Acad. Sci. USA 85:1811-1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burgers, P. M., A. Kornberg, and Y. Sakakibara. 1981. The dnaN gene codes for the β subunit of DNA polymerase III holoenzyme of Escherichia coli. Proc. Natl. Acad. Sci. USA 78:5391-5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Courcelle, J., C. Carswell-Crumpton, and P. C. Hanawalt. 1997. recF and recR are required for the resumption of replication at DNA replication forks in Escherichia coli. Proc. Natl. Acad. Sci. USA 94:3714-3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Courcelle, J., A. Khodursky, B. Peter, P. O. Brown, and P. C. Hanawalt. 2001. Comparative gene expression profiles following UV exposure in wild-type and SOS-deficient Escherichia coli. Genetics 158:41-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cox, M. M., M. F. Goodman, K. N. Kreuzer, D. J. Sherratt, S. J. Sandler, and K. J. Marians. 2000. The importance of repairing stalled replication forks. Nature 404:37-41. [DOI] [PubMed] [Google Scholar]
- 12.Dalrymple, B. P., K. Kongsuwan, G. Wijffels, N. E. Dixon, and P. A. Jennings. 2001. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc. Natl. Acad. Sci. USA 98:11627-11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fijalkowska, I. J., R. L. Dunn, and R. M. Schaaper. 1997. Genetic requirements and mutational specificity of the Escherichia coli SOS mutator activity. J. Bacteriol. 179:7435-7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frank, E. G., D. G. Ennis, M. Gonzalez, A. S. Levine, and R. Woodgate. 1996. Regulation of SOS mutagenesis by proteolysis. Proc. Natl. Acad. Sci. USA 93:10291-10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedberg, E. C., R. Wagner, and M. Radman. 2002. Specialized DNA polymerases, cellular survival, and the genesis of mutations. Science 296:1627-1630. [DOI] [PubMed] [Google Scholar]
- 16.Friedberg, E. C., G. C. Walker, and W. Siede. 1995. DNA repair and mutagenesis. ASM Press, Washington, D.C.
- 17.Gonzalez, M., E. G. Frank, A. S. Levine, and R. Woodgate. 1998. Lon-mediated proteolysis of the Escherichia coli UmuD mutagenesis protein: in vitro degradation and identification of residues required for proteolysis. Genes Dev. 12:3889-3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez, M., F. Rasulova, M. R. Maurizi, and R. Woodgate. 2000. Subunit-specific degradation of the UmuD/D′ heterodimer by the ClpXP protease: the role of trans recognition in UmuD′ stability. EMBO J. 19:5251-5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grompone, G., M. Seigneur, S. D. Ehrlich, and B. Michel. 2002. Replication fork reversal in DNA polymerase III mutants of Escherichia coli: a role for the β clamp. Mol. Microbiol. 44:1331-1339. [DOI] [PubMed] [Google Scholar]
- 20.Hingorani, M. M., and M. O'Donnell. 1998. ATP binding to the Escherichia coli clamp loader powers opening of the ring-shaped clamp of DNA polymerase III holoenzyme. J. Biol. Chem. 273:24550-24563. [DOI] [PubMed] [Google Scholar]
- 21.Ho, C., O. I. Kulaeva, A. S. Levine, and R. Woodgate. 1993. A rapid method for cloning mutagenic DNA repair genes: isolation of umu-complementing genes from multidrug resistance plasmids R391, R446b, and R471a. J. Bacteriol. 175:5411-5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim, S. R., G. Maenhaut-Michel, M. Yamada, Y. Yamamoto, K. Matsui, T. Sofuni, T. Nohmi, and H. Ohmori. 1997. Multiple pathways for SOS-induced mutagenesis in Escherichia coli: an overexpression of dinB/dinP results in strongly enhancing mutagenesis in the absence of any exogenous treatment to damage DNA. Proc. Natl. Acad. Sci. USA 94:13792-13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim, S. R., K. Matsui, M. Yamada, P. Gruz, and T. Nohmi. 2001. Roles of chromosomal and episomal dinB genes encoding DNA pol IV in targeted and untargeted mutagenesis in Escherichia coli. Mol. Genet. Genomics 266:207-215. [DOI] [PubMed] [Google Scholar]
- 24.Kowalczykowski, S. C., D. A. Dixon, A. K. Eggleston, S. D. Lauder, and W. M. Rehrauer. 1994. Biochemistry of homologous recombination in Escherichia coli. Microbiol. Rev. 58:401-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavery, P. E., and S. C. Kowalczykowski. 1992. Biochemical basis of the constitutive repressor cleavage activity of recA730 protein. A comparison to recA441 and recA803 proteins. J. Biol. Chem. 267:20648-20658. [PubMed] [Google Scholar]
- 26.Lavery, P. E., and S. C. Kowalczykowski. 1990. Properties of recA441 protein-catalyzed DNA strand exchange can be attributed to an enhanced ability to compete with SSB protein. J. Biol. Chem. 265:4004-4010. [PubMed] [Google Scholar]
- 27.Lenne-Samuel, N., J. Wagner, H. Etienne, and R. P. Fuchs. 2002. The processivity factor β controls DNA polymerase IV traffic during spontaneous mutagenesis and translesion synthesis in vivo. EMBO Rep. 3:45-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez de Saro, F. J., R. E. Georgescu, M. F. Goodman, and M. O'Donnell. 2003. Competitive processivity-clamp usage by DNA polymerases during DNA replication and repair. EMBO J. 22:6408-6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopez de Saro, F. J., and M. O'Donnell. 2001. Interaction of the beta sliding clamp with MutS, ligase, and DNA polymerase I. Proc. Natl. Acad. Sci. USA 98:8376-8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lusetti, S. L., and M. M. Cox. 2002. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu. Rev. Biochem. 71:71-100. [DOI] [PubMed] [Google Scholar]
- 31.Maki, S., and A. Kornberg. 1988. DNA polymerase III holoenzyme of Escherichia coli. III. Distinctive processive polymerases reconstituted from purified subunits. J. Biol. Chem. 263:6561-6569. [PubMed] [Google Scholar]
- 32.Maor-Shoshani, A., K. Hayashi, H. Ohmori, and Z. Livneh. 2003. Analysis of translesion replication across an abasic site by DNA polymerase IV of Escherichia coli. DNA Repair (Amsterdam) 2:1227-1238. [DOI] [PubMed] [Google Scholar]
- 33.Maor-Shoshani, A., and Z. Livneh. 2002. Analysis of the stimulation of DNA polymerase V of Escherichia coli by processivity proteins. Biochemistry 41:14438-14446. [DOI] [PubMed] [Google Scholar]
- 34.McHenry, C. S. 2003. Chromosomal replicases as asymmetric dimers: studies of subunit arrangement and functional consequences. Mol. Microbiol. 49:1157-1165. [DOI] [PubMed] [Google Scholar]
- 35.Miller, C., L. E. Thomsen, C. Gaggero, R. Mosseri, H. Ingmer, and S. N. Cohen. 2004. SOS response induction by beta-lactams and bacterial defense against antibiotic lethality. Science 305:1629-1631. [DOI] [PubMed] [Google Scholar]
- 36.Miller, J. H. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Plainview, N.Y.
- 37.Morimatsu, K., and S. C. Kowalczykowski. 2003. RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair. Mol. Cell 11:1337-1347. [DOI] [PubMed] [Google Scholar]
- 38.Murli, S., T. Opperman, B. T. Smith, and G. C. Walker. 2000. A role for the umuDC gene products of Escherichia coli in increasing resistance to DNA damage in stationary phase by inhibiting the transition to exponential growth. J. Bacteriol. 182:1127-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naktinis, V., J. Turner, and M. O'Donnell. 1996. A molecular switch in a replication machine defined by an internal competition for protein rings. Cell 84:137-145. [DOI] [PubMed] [Google Scholar]
- 40.Neidhardt, F. C., and R. Curtiss. 1996. Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed. ASM Press, Washington, D.C.
- 41.Nohmi, T., J. R. Battista, L. A. Dodson, and G. C. Walker. 1988. RecA-mediated cleavage activates UmuD for mutagenesis: mechanistic relationship between transcriptional derepression and posttranslational activation. Proc. Natl. Acad. Sci. USA 85:1816-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohmori, H., M. Kimura, T. Nagata, and Y. Sakakibara. 1984. Structural analysis of the dnaA and dnaN genes of Escherichia coli. Gene 28:159-170. [DOI] [PubMed] [Google Scholar]
- 43.Opperman, T., S. Murli, B. T. Smith, and G. C. Walker. 1999. A model for a umuDC-dependent prokaryotic DNA damage checkpoint. Proc. Natl. Acad. Sci. USA 96:9218-9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perez-Capilla, T., M. R. Baquero, J. M. Gomez-Gomez, A. Ionel, S. Martin, and J. Blazquez. 2005. SOS-independent induction of dinB transcription by beta-lactam-mediated inhibition of cell wall synthesis in Escherichia coli. J. Bacteriol. 187:1515-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perry, K. L., S. J. Elledge, B. B. Mitchell, L. Marsh, and G. C. Walker. 1985. umuDC and mucAB operons whose products are required for UV light- and chemical-induced mutagenesis: UmuD, MucA, and LexA proteins share homology. Proc. Natl. Acad. Sci. USA 82:4331-4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pham, P., J. G. Bertram, M. O'Donnell, R. Woodgate, and M. F. Goodman. 2001. A model for SOS-lesion-targeted mutations in Escherichia coli. Nature 409:366-370. [DOI] [PubMed] [Google Scholar]
- 47.Pham, P., E. M. Seitz, S. Saveliev, X. Shen, R. Woodgate, M. M. Cox, and M. F. Goodman. 2002. Two distinct modes of RecA action are required for DNA polymerase V-catalyzed translesion synthesis. Proc. Natl. Acad. Sci. USA 99:11061-11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reuven, N. B., G. Arad, A. Maor-Shoshani, and Z. Livneh. 1999. The mutagenesis protein UmuC is a DNA polymerase activated by UmuD′, RecA, and SSB and is specialized for translesion replication. J. Biol. Chem. 274:31763-31766. [DOI] [PubMed] [Google Scholar]
- 49.Reuven, N. B., G. Arad, A. Z. Stasiak, A. Stasiak, and Z. Livneh. 2001. Lesion bypass by the Escherichia coli DNA polymerase V requires assembly of a RecA nucleoprotein filament. J. Biol. Chem. 276:5511-5517. [DOI] [PubMed] [Google Scholar]
- 50.Reuven, N. B., G. Tomer, and Z. Livneh. 1998. The mutagenesis proteins UmuD′ and UmuC prevent lethal frameshifts while increasing base substitution mutations. Mol. Cell 2:191-199. [DOI] [PubMed] [Google Scholar]
- 51.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 52.Schlacher, K., K. Leslie, C. Wyman, R. Woodgate, M. M. Cox, and M. F. Goodman. 2005. DNA polymerase V and RecA protein, a minimal mutasome. Mol. Cell 17:561-572. [DOI] [PubMed] [Google Scholar]
- 53.Shinagawa, H., H. Iwasaki, T. Kato, and A. Nakata. 1988. RecA protein-dependent cleavage of UmuD protein and SOS mutagenesis. Proc. Natl. Acad. Sci. USA 85:1806-1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strauss, B. S., R. Roberts, L. Francis, and P. Pouryazdanparast. 2000. Role of the dinB gene product in spontaneous mutation in Escherichia coli with an impaired replicative polymerase. J. Bacteriol. 182:6742-6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stukenberg, P. T., P. S. Studwell-Vaughan, and M. O'Donnell. 1991. Mechanism of the sliding beta-clamp of DNA polymerase III holoenzyme. J. Biol. Chem. 266:11328-11334. [PubMed] [Google Scholar]
- 56.Sutton, M. D. 2004. The Escherichia coli dnaN159 mutant displays altered DNA polymerase usage and chronic SOS induction. J. Bacteriol. 186:6738-6748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sutton, M. D., J. M. Duzen, and R. W. Maul. 2005. Mutant forms of the Escherichia coli β sliding clamp that distinguish between its roles in replication and DNA polymerase V-dependent translesion DNA synthesis. Mol. Microbiol. 55:1751-1766. [DOI] [PubMed] [Google Scholar]
- 58.Sutton, M. D., M. F. Farrow, B. M. Burton, and G. C. Walker. 2001. Genetic interactions between the Escherichia coli umuDC gene products and the β processivity clamp of the replicative DNA polymerase. J. Bacteriol. 183:2897-2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sutton, M. D., T. Opperman, and G. C. Walker. 1999. The Escherichia coli SOS mutagenesis proteins UmuD and UmuD′ interact physically with the replicative DNA polymerase. Proc. Natl. Acad. Sci. USA 96:12373-12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sutton, M. D., B. T. Smith, V. G. Godoy, and G. C. Walker. 2000. The SOS response: recent insights into umuDC-dependent mutagenesis and DNA damage tolerance. Annu. Rev. Genet. 34:479-497. [DOI] [PubMed] [Google Scholar]
- 61.Sutton, M. D., and G. C. Walker. 2001. Managing DNA polymerases: coordinating DNA replication, DNA repair, and DNA recombination. Proc. Natl. Acad. Sci. USA 98:8342-8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang, M., I. Bruck, R. Eritja, J. Turner, E. G. Frank, R. Woodgate, M. O'Donnell, and M. F. Goodman. 1998. Biochemical basis of SOS-induced mutagenesis in Escherichia coli: reconstitution of in vitro lesion bypass dependent on the UmuD′2C mutagenic complex and RecA protein. Proc. Natl. Acad. Sci. USA 95:9755-9760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tang, M., P. Pham, X. Shen, J. S. Taylor, M. O'Donnell, R. Woodgate, and M. F. Goodman. 2000. Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature 404:1014-1018. [DOI] [PubMed] [Google Scholar]
- 64.Tang, M., X. Shen, E. G. Frank, M. O'Donnell, R. Woodgate, and M. F. Goodman. 1999. UmuD′2C is an error-prone DNA polymerase, Escherichia coli pol V. Proc. Natl. Acad. Sci. USA 96:8919-8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Viguera, E., M. Petranovic, D. Zahradka, K. Germain, D. S. Ehrlich, and B. Michel. 2003. Lethality of bypass polymerases in Escherichia coli cells with a defective clamp loader complex of DNA polymerase III. Mol. Microbiol. 50:193-204. [DOI] [PubMed] [Google Scholar]
- 66.Wagner, J., H. Etienne, R. Janel-Bintz, and R. P. Fuchs. 2002. Genetics of mutagenesis in E. coli: various combinations of translesion polymerases (Pol II, IV and V) deal with lesion/sequence context diversity. DNA Repair (Amsterdam) 1:159-167. [DOI] [PubMed] [Google Scholar]
- 67.Wagner, J., S. Fujii, P. Gruz, T. Nohmi, and R. P. Fuchs. 2000. The β clamp targets DNA polymerase IV to DNA and strongly increases its processivity. EMBO Rep. 1:484-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang, R. F., and S. R. Kushner. 1991. Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100:195-199. [PubMed] [Google Scholar]
- 69.Wijffels, G., B. P. Dalrymple, P. Prosselkov, K. Kongsuwan, V. C. Epa, P. E. Lilley, S. Jergic, J. Buchardt, S. E. Brown, P. F. Alewood, P. A. Jennings, and N. E. Dixon. 2004. Inhibition of protein interactions with the β2 sliding clamp of Escherichia coli DNA polymerase III by peptides from β2-binding proteins. Biochemistry 43:5661-5671. [DOI] [PubMed] [Google Scholar]
- 70.Witkin, E. M., J. O. McCall, M. R. Volkert, and I. E. Wermundsen. 1982. Constitutive expression of SOS functions and modulation of mutagenesis resulting from resolution of genetic instability at or near the recA locus of Escherichia coli. Mol. Gen. Genet. 185:43-50. [DOI] [PubMed] [Google Scholar]
- 71.Woodgate, R. 1992. Construction of a umuDC operon substitution mutation in Escherichia coli. Mutat. Res. 281:221-225. [DOI] [PubMed] [Google Scholar]