

Abstract
Our laboratory has shown that arsenite markedly increased the cancer rate caused by solar-simulation ultraviolet radiation (UVR) in the hairless mouse skin model. In the present study, we investigated how arsenite affected DNA photodamage repair and apoptosis after solar-simulation UVR in the mouse keratinocyte cell line 291.03C. The keratinocytes were treated with different concentrations of sodium arsenite (0.0, 2.5, 5.0 μM) for 24 hr and then were immediately irradiated with a single dose of 0.30 kJ/m2 UVR. At 24 hr after UVR, DNA photoproducts [cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts (6-4PPs)] and apoptosis were measured using the enzyme-linked immunosorbent assay and the two-color TUNEL (terminal deoxynucleotide transferase dUTP nick end labeling) assay, respectively. The results showed that arsenite reduced the repair rate of 6-4PPs by about a factor of 2 at 5.0 μM and had no effect at 2.5 μM. UVR-induced apoptosis at 24 hr was decreased by 22.64% at 2.5 μM arsenite and by 61.90% at 5.0 μM arsenite. Arsenite decreased the UVR-induced caspase-3/7 activity in parallel with the inhibition of apoptosis. Colony survival assays of the 291.03C cells demonstrate a median lethal concentration (LC50) of arsenite of 0.9 μM and a median lethal dose (LD50) of UVR of 0.05 kJ/m2. If the present results are applicable in vivo, inhibition of UVR-induced apoptosis may contribute to arsenite’s enhancement of UVR-induced skin carcinogenesis.
Keywords: apoptosis, arsenite, mouse keratinocyte, photodamage repair, skin cancer, solar-simulation UVR
Previous studies (Burns et al. 2004; Rossman et al. 2001) showed that arsenite markedly increased the cancer incidence of solar-simulation ultraviolet radiation (UVR) in hairless mice. The relationship between arsenite concentration in drinking water and the yield of squamous cell carcinomas in UVR-exposed mouse skin was linear up to 5 mg/L. UVR is a complete carcinogen that readily induces skin cancer (Zhuang et al. 2000). Although UVR induces a wide range of DNA damage, such as protein–DNA crosslinks, oxidative base damage, single-stand breaks, and double-strand breaks (de Gruijl et al. 2001), the major DNA damage is cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts (6-4PPs). Both of the latter lesions are repaired by nucleotide excision repair (NER) (Mitchell et al. 1985). Failure to repair CPDs and 6-4PPs leads to the signature mutations containing CC to TT and C to T transitions (Miller 1985; Ziegler et al. 1993) that are common in UVR-induced human skin cancers but not in other epithelial cancers (Brash et al. 1996). UVR can stimulate many signal transduction pathways (Bender et al. 1997) that may contribute to skin cancer progression.
Exposure to arsenite is associated with skin, bladder, lung, and probably kidney and liver cancers in humans (Rossman 2003). Studies of human cell lines have demonstrated that arsenite can reduce NER capacity by inhibiting the DNA incision step (Hartwig et al. 1997). Inhibition of the ligation step in base excision repair (Li and Rossman 1989) by arsenite has also been reported. Arsenite is known to alter the methylation status of the cell, which affects expression of a variety of genes (Zhao et al. 1997). Arsenite also shows high affinity toward vicinyl sulfhydryl groups, including zinc finger proteins identified as transcription factors and several DNA repair enzymes (Hartwig 2001).
The aim of the present study was to examine how arsenite affected the solar-simulation UVR-induced apoptosis and photodamage repair in mouse keratinocyte line 291.03C in vitro. The results showed that arsenite reduced the repair rate of 6-4PPs by about 50% at 5.0 μM but not at 2.5 μM and inhibited apoptosis in mouse keratinocyte cell line 291.03C.
Materials and Methods
Cell culture.
Mouse keratinocyte cell line 291.03C, generously provided by M. Kulesz-Martin, was grown in Eagle’s minimum essential medium (EMEM; Mediatech Cellgro, Herndon, VA) with nonessential amino acids (Cellgro) containing 5% fetal bovine serum, 1% antibiotic-antimycotic, and 10 ng/mL epidermal growth factor at 37°C in 5% CO2. The 291.03C line is a 7,12-dimethylbenz[a]anthracene–initiated clone derived from nontransformed 291 cells (Kulesz-Martin et al. 1985). It is a precursor of squamous cell carcinoma, which was selected on the basis of its ability to resist extracellular Ca2+-induced terminal differentiation, and has distinctive keratinocyte morphology. The gene expression patterns are substantially similar between the nontransformed cell line 291 and 291.03C. The p53 protein in 291.03C is wild type, even if its function is somewhat compromised (Wang et al. 2002).
Ultraviolet irradiation.
The UVR source was a bank of four FS20 sunlamps (Westinghouse, Bloomfield, NJ) mounted in parallel 15 cm apart. The UVR intensity was measured (30 cm below the source) by a calibrated radiometer/photometer (model IL1400A; International Light Inc., Newburyport, MA). According to manufacturer specifications, 85% of the lamp output was in the UVB (290–320 nm) range, < 1% was in the UVC (200–290 nm) range, 4% was in the UVA (320–400 nm) range, and the remainder was in the visible (>400 nm) range.
For assays of photodamage repair, apoptosis, and caspase activity, we seeded 2 × 106 291.03C keratinocytes in 100 mm culture dishes and incubated them for 24 hr with different concentrations (0.0, 2.5, and 5.0 μM) of sodium arsenite (Sigma, St. Louis, MO) beginning at about 70% of confluence. At 24 hr the cells were washed with Dulbecco’s phosphate-buffered saline (DPBS; Sigma) twice and exposed to UVR in the presence of 5 mL DPBS.
Colony survival assay.
Mouse 291.03C keratinocytes were seeded at a density of 300 cells/60 mm dish in EMEM. After 24 hr, sodium arsenite was added to the medium from a freshly prepared stock solution to final concentrations of 0.0, 0.05, 0.1, 0.5, 1.0, and 5.0 μM, and the cells were incubated for 7 days, followed by fixation in methanol and then staining with 0.5% crystal violet in 50% methanol. We counted colonies and determined the percentage of survival as the ratio of treated to control × 100. The survival after exposure to solar-simulation UVR was determined similarly at 7 days after single doses of 0.0, 0.05, 0.10, 0.20, and 0.30 kJ/m2.
Measurement of CPDs and 6-4PPs in genomic DNA by ELISA.
We isolated genomic DNA using the QIAamp Blood Kit (QIAGEN Inc., Valencia, CA). DNA concentrations were calculated from the absorbance at 260 nm measured by a Beckman DU 650 spectrophotometer (Beckman Instruments, Fullerton, CA). We determined the quantities of CPDs and 6-4PPs by enzyme-linked immunosorbent assay (ELISA) as described by Mori et al. (1991). In brief, Falcon polyvinyl-chloride flat-bottom 96-well assay plates (Becton Dickinson Labware, Franklin Lakes, NJ) precoated with 1% protamine sulfate (Sigma) were incubated with purified genomic DNA (15 ng for CPD detection and 150 ng for 6-4PP detection) in PBS at 37°C for 20 hr. For CPD detection, we used the TDM-2 antibody, and for 6-4PP detection we used the 64M-2 antibody (both antibodies were generously provided by T. Mori, Nara, Japan). After adding biotinylated F(ab′)2 goat anti-mouse IgG fragments and streptavidinperoxidase (Zymed, San Francisco, CA), we measured the optical density from o-phenylene diamine at 492 nm using a Bio Assay Reader HTS7000 (Perkin-Elmer Corp., Norwalk, CT). The percentage of the initial number of photoproducts was calculated at various times after UVR exposure by using standard curves obtained from DNA samples irradiated with UVR doses of 0.0, 0.06, 0.12, 0.24, and 0.36 kJ/m2.
Measurement of apoptosis by flow cytometry.
Apoptosis was detected using the APO-BRDU kit (Phoenix Flow Systems, Inc., San Diego, CA) following the protocol provided by the manufacturer. Briefly, attached cells were harvested by trypsinization and combined with free-floating cells that were harvested by centrifugation. After washing with PBS, the cells were fixed with 1% paraformaldehyde in PBS, followed by fixation with 70% ice-cold ethanol overnight. The fixed cells were washed twice with wash buffer included in the kit, and freshly prepared DNA labeling solution [containing terminal deoxynucleotidyl transferase and 5-bromo-2′-deoxyuridine (BrdU) 5′-triphosphate] was added to the cell pellet and incubated overnight at room temperature. Cells were labeled by fluorescein isothiocyanate–conjugated monoclonal antibody to BrdU (FITC~PBR-1 mAb), washed again, resuspended in staining solution containing propidium iodide and RNase, and incubated for 30 min at room temperature. After this, cells were immediately analyzed using a Coulter EPICS XL-MCL flow cytometer (Beckman Coulter, Miami, FL). We calculated the percentage of R1 (normal), R2 (apoptotic without loss of DNA), and R3 (hypodiploid) cells using EXPO32 Multifile software (Beckman Coulter).
Measurement of caspase-3/7 activity.
We measured Caspase-3/7 activity using the Apo-one Homogenous Caspase-3/7 Assay (Promega, Madison, WI) following the protocol provided by the manufacturer. In brief, cells were trypsinized, and 20,000 cells/sample were mixed with the same volume of the Apo-one Homogenous Caspase-3/7 reagent. After incubation at room temperature for 2 hr, cas-pase-3/7 activities were estimated from the fluorescence of each sample at the excitation wavelength of 485 nm and the emission wavelength of 535 nm using the Bio Assay Reader HTS7000. EMEM mixed with the same volume of Apo-one Homogenous Caspase-3/7 reagent served as a negative control.
Results
Toxicity of arsenite and UVR to 291.03C mouse keratinocytes.
Figure 1 shows the effects of arsenite (Figure 1A) and solar-simulation UVR (Figure 1B) on clonal survival of 291.03C mouse keratinocytes. The median lethal dose (LD50) of UVR was 0.05 kJ/m2. There was no measurable colony survival at UV doses > 0.30 kJ/m2. The median lethal concentration (LC50) of sodium arsenite was 0.9 μM. Arsenite did not show significant lethality < 0.5 μM and showed nearly 100% lethality > 5.0 μM.
Figure 1.

Effect of sodium arsenite (A) and solar-simulation UVR (B) on colony survival and cell cycle progression of mouse keratinocyte line 291.03C. Each point represents the mean ± SD (n = 3).
In a previous study (Burns et al. 2004), hairless mice were fed sodium arsenite in drinking water at concentrations ranging from 1.25 mg/L (9.6 μM) to approximately 10 mg/L (77.0 μM), and solar spectrum UVR exposure was applied to the dorsal skin at 1.0 kJ/m2 three times weekly. The arsenite concentrations and solar UVR dose used in the present in vitro study were 2.5 and 5 μM and 0.3 kJ/m2. These two arsenite concentrations were estimated to be equivalent to 26 and 52% of the lowest arsenite concentration (1.25 mg/L) used in the in vivo carcinogenesis study (Burns et al. 2004).
Arsenite effects on DNA photodamage repair.
The two photolesions, CPDs and 6-4PPs, were detected by ELISA. The 6-4PPs were 80% removed by 12 hr, whereas CPDs were not removed > 10% by 24 hr (Figure 2). According to the regression analysis of the data, arsenite showed no significant effect on CPDs repair. The 6-4PP repair rate after UVR was 11.95%/hr; when combined with 2.5 μM or 5.0 μM arsenite, the 6-4PP repair rates were 11.3%/hr and 6.19%/hr, respectively. Arsenite slowed the 6-4PP repair rate by 48% at 5.0 μM, but no difference was detected at 2.5 μM.
Figure 2.

The effect of sodium arsenite on the repair of CPDs (A) and 6-4PPs (B) from genomic DNA in 291.03C cells treated with arsenite (2.5 or 5.0 μM) for 24 hr and then exposed to solar-simulation UVR (0.3 kJ/m2). At different time points postexposure, genomic DNA was isolated, and the photodamage was detected by ELISA. Each point represents mean ± SD (n = 3).
Arsenite inhibits UVR-induced apoptosis.
Figure 3 shows that at 24 hr after UVR alone (0.30 kJ/m2) the percentage of apoptotic cells was 27.6% (Figure 3D). When UVR-treated cells were incubated in 2.5 μM or 5.0 μM arsenite, the percentage of apoptotic cells decreased to 21.4% (77.36% of UVR only; Figure 3E) and 10.5% (38.1% of UVR only; Figure 3F), respectively. Untreated control cells showed few apoptotic cells (Figure 3A), whereas 5.0 μM arsenite only showed 4.9% apoptotic cells at 48 hr after treatment (Figure 3B) and 8.1% at 60 hr (Figure 3C) after treatment. Apoptosis was not detected at 0 hr and 14 hr after UVR and was 51.56, 39.42, and 36.47% at 36 hr after UVR, UVR + 2.5 μM arsenite, and UVR + 5 μM arsenite, respectively (data not shown), indicating that apoptosis is progressing with the time. As shown in Figure 3, the R3 population was 5.28% (UVR alone), 3.71% (UVR + 2.5 μM arsenite), and 1.32% (UVR + 5.0 μM arsenite), indicating that apoptosis is more extensive after treatment with UVR alone compared with UVR plus arsenite.
Figure 3.

The effect of sodium arsenite on apoptosis caused by solar-simulation UVR in 291.03C cells treated with 2.5 or 5.0 μM arsenite for 24 hr and then exposed to 0.3 kJ/m2 solar-simulation UVR. (A) Negative control; (B) arsenite (5.0 μM) alone for 48 hr; (C) arsenite (5.0 μM) alone for 60 hr; (D) 24 hr post-UVR; (E) 24 hr post-UVR with arsenite (2.5 μM); (F) 24 hr post-UVR with arsenite (5.0 μM). Apoptosis was analyzed by flow cytometry as described in “Materials and Methods.” Data shown are mean ± SD (n = 3). The apoptotic cell population was calculated as R2 + R3.
The caspase-3/7 activities 24 hr after UVR are shown in Figure 4. Arsenite decreased the UVR-induced caspase-3/7 activity to 88.48% at 2.5 μM and to 58.83% at 5 μM. Arsenite alone did not affect the caspase level significantly. These results are consistent with the results in Figure 3 indicating arsenite inhibited UVR-induced apoptosis (Figure 4).
Figure 4.
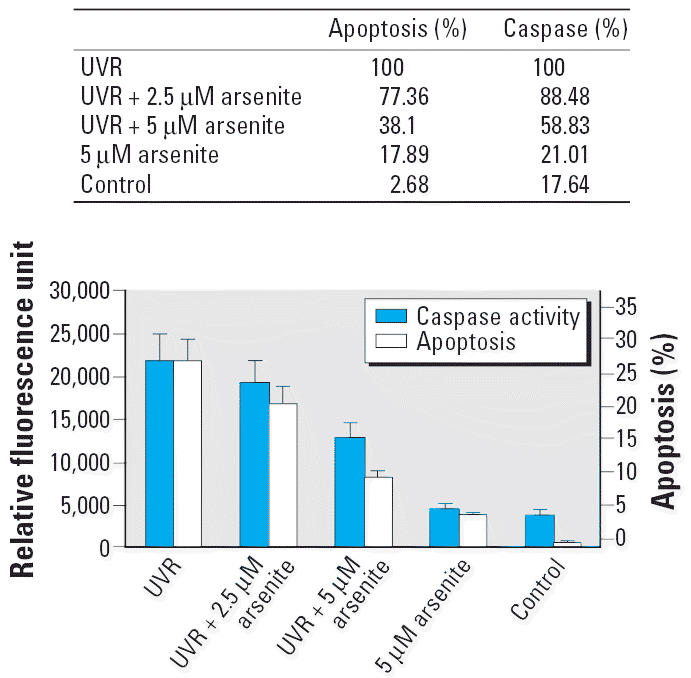
The effect of sodium arsenite on caspase-3/7 activity in 291.03C cells treated with 2.5 and 5.0 μM arsenite for 24 hr and then exposed to 0.3 kJ/m2 solar-simulation UVR. Twenty-four hours after UVR, caspase-3/7 activity was measured as described in “Materials and Methods,” The data show a close parallel with the apoptosis data from Figure 3. Data shown are mean ± SD (n = 3).
Discussion
The photoproducts (CPDs and 6-4PPs) produced by UVR may lead to mutations and cancer development if the damage is not removed from the DNA. There are two mechanisms for a cell to remove DNA damage: repairing the DNA damage or inducing apoptosis. Arsenite indeed increased the mutagenicity of UVB in Chinese hamster V79 cells (Li and Rossman 1991). As reported here, mouse keratinocytes did not repair UVR-induced CPDs efficiently, and arsenite did not affect the DNA photodamage repair rates significantly. The apoptosis inhibiting activity of arsenite may have converted a greater amount of DNA damage to mutations without substantially affecting DNA repair. If so, these findings might help to explain why skin cancer in mice is markedly increased by prolonged exposure to the combination of UVR and dietary arsenite.
Although it has been reported that arsenite inhibits DNA repair in a variety of cell types (Hartwig et al. 1997; Li and Rossman 1989; Yager and Wiencke 1997), the present study in a mouse keratinocyte cell line (Figure 2) shows little effect of arsenite on the removal of UVR-induced photoproducts from the genomic DNA except the reduced 6-4PP repair rate at 5 μM. In normal human epidermal keratinocytes (NHEK, Cambrex BIO Science, Walkersville, MA), 6-4PPs were removed at a rate of 30%/hr, whereas CPDs were removed at a rate of 2%/hr after 0.3 kJ/m2 solar-simulation UVR (data not shown). The mouse 291.03C keratinocyte line exhibited a 6-4PP removal rate of 13%/hr and a CPD removal rate of < 0.4%/hr (Figure 2). The repair rates of mouse keratinocytes were not > 20% for CPDs and > 40% for 6-4PPs compared with those of human keratinocytes.
There are two subpathways of NER: transcription-coupled repair (TCR) and global genomic repair (GGR). TCR refers to the preferential repair of transcribed strands of active genes, and GGR refers to repair anywhere else in the DNA. Many rodent cells have normal TCR, which is very important for clonal survival, but are deficient in GGR of CPDs, which is more important for suppressing muta-genesis (Hanawalt 2001). Because the ELISA method used here detects GGR, these results confirm that mouse keratinocyte line 291.03C performs GGR of DNA photodamage less efficiently than do human keratinocytes.
UVR can trigger apoptosis by damaging the DNA and activating the death receptors on the cell surface (Kulms and Schwarz 2000). Arsenite can induce Fas/FasL-dependent apoptosis at higher concentrations (≥5.0 μM) in primary human keratinocytes (Liao et al. 2004). The results reported here show that 5.0 μM arsenite produced a small increase (8%) in apoptosis after 60 hr of treatment, whereas a single 0.30 kJ/m2 dose of solar-simulation UVR produced 27% apoptosis by 24 hr and 51% by 36 hr, indicating that apoptosis increased gradually after exposure to UVR. Paradoxically, arsenite at the 5.0 μM concentration produced a small incidence of apoptosis by itself, but it was inhibitory when combined with a strongly apoptotic dose of UVR (Figure 3). The caspase results generally confirmed the apoptosis results obtained by flow cytometry. At 36 hr after UVR + 5.0 μM arsenite, apoptosis increased to 37% (30% less than UVR alone), indicating that arsenite delayed the onset of the apoptosis but did not prevent it completely.
In a previous study of Chinese hamster V79 cells (Li and Rossman 1991), the combination of UVB (0.2 kJ/m2) and sodium arsenite (10 μM and 15 μM) increased the mutation rates by 1.65-fold and 2.06-fold respectively, whereas survival was decreased to 43 and 11.8%, respectively. The inhibition of apoptosis may help to explain the higher mutation rates in the presence of arsenite. In conclusion, arsenite lessened the rate of DNA repair and inhibited apoptosis at 24 hr after a single exposure of mouse keratinocyte line 291.03C to 0.3 kJ/m2 of solar-simulation UVR. The consequences of this delay on mutation rates will be investigated in future studies.
Footnotes
This work was supported by National Institute of Environmental Health Sciences (NIEHS) grant ES09252 and National Aeronautics and Space Administration grant NAG9-1528, and is part of the Nelson Institute of Environmental Medicine and the NYU Cancer Institute programs supported by grant CA16087 from the National Cancer Institute and a Center Grant (ES00260) from the NIEHS.
References
- Bender K, Blattner C, Knebel A, Iordanov M, Herrlich P, Rahmsdorf HJ. UV-induced signal transduction. J Photochem Photobiol B. 1997;37:1–17. doi: 10.1016/s1011-1344(96)07459-3. [DOI] [PubMed] [Google Scholar]
- Brash DE, Ziegler A, Jonason AS, Simon JA, Kunala S, Leffell DJ. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion. J Investig Dermatol Symp Proc. 1996;1:136–142. [PubMed] [Google Scholar]
- Burns FJ, Uddin AN, Wu F, Nádas A, Rossman TG. Arsenic-induced enhancement of ultraviolet radiation carcinogenesis in mouse skin: a dose-response study. Environ Health Perspect. 2004;112:599–603. doi: 10.1289/ehp.6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gruijl FR, van Kranen HJ, Mullenders LH. UV-Induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J Photochem Photobiol B. 2001;63:19–27. doi: 10.1016/s1011-1344(01)00199-3. [DOI] [PubMed] [Google Scholar]
- Hanawalt PC. Revisiting the rodent repairadox. Environ Mol Mutagen. 2001;38:89–96. doi: 10.1002/em.1057. [DOI] [PubMed] [Google Scholar]
- Hartwig A. Zinc finger proteins as potential targets for toxic metal ions: differential effects on structure and function. Antioxid Redox Signal. 2001;3:625–634. doi: 10.1089/15230860152542970. [DOI] [PubMed] [Google Scholar]
- Hartwig A, Groblinghoff UD, Beyersmann D, Natarajan AT, Filon R, Mullenders LH. Interaction of arsenic (III) with nucleotide excision repair in UV-irradiated human fibroblasts. Carcinogenesis. 1997;18:399–405. doi: 10.1093/carcin/18.2.399. [DOI] [PubMed] [Google Scholar]
- Kulesz-Martin M, Yoshida MA, Prestine L, Yuspa SH, Bertram JS. Mouse cell clones for improved quantitation of carcinogen-induced altered differentiation. Carcinogenesis. 1985;6:1245–1254. doi: 10.1093/carcin/6.9.1245. [DOI] [PubMed] [Google Scholar]
- Kulms D, Schwarz T. Molecular mechanisms of UV-induced apoptosis. Photodermatol Photoimmunol Photomed. 2000;16:195–201. doi: 10.1034/j.1600-0781.2000.160501.x. [DOI] [PubMed] [Google Scholar]
- Li JH, Rossman TG. Inhibition of DNA ligase activity by arsenite: a possible mechanism of its comutagenesis. Mol Toxicol. 1989;2:1–9. [PubMed] [Google Scholar]
- Li JH, Rossman TG. Comutagenesis of sodium arsenite with ultraviolet radiation in Chinese hamster V79 cells. Biol Met. 1991;4:197–200. doi: 10.1007/BF01141180. [DOI] [PubMed] [Google Scholar]
- Liao WT, Chang KL, Yu CL, Chen GS, Chang LW, Yu HS. Arsenic induces human keratinocyte apoptosis by the FAS/FAS ligand pathway, which correlates with alterations in nuclear factor-kappa B and activator protein-1 activity. J Invest Dermatol. 2004;122:125–129. doi: 10.1046/j.0022-202X.2003.22109.x. [DOI] [PubMed] [Google Scholar]
- Miller JH. Mutagenic specificity of ultraviolet light. J Mol Biol. 1985;182:45–65. doi: 10.1016/0022-2836(85)90026-9. [DOI] [PubMed] [Google Scholar]
- Mitchell DL, Haipek CA, Clarkson JM. (6–4) Photoproducts are removed from the DNA of UV-irradiated mammalian cells more efficiently than cyclobutane pyrimidine dimers. Mutat Res. 1985;143:109–112. doi: 10.1016/s0165-7992(85)80018-x. [DOI] [PubMed] [Google Scholar]
- Mori T, Nakane M, Hattori T, Matsunaga T, Ihara M, Nikaido O. Simultaneous establishment of monoclonal antibodies specific for either cyclobutane pyrimidine dimer or (6–4) photoproduct from the same mouse immunized with ultraviolet-irradiated DNA. Photochem Photobiol. 1991;54:225–232. doi: 10.1111/j.1751-1097.1991.tb02010.x. [DOI] [PubMed] [Google Scholar]
- Rossman TG. Mechanism of arsenic carcinogenesis: an integrated approach. Mutat Res. 2003;533:37–65. doi: 10.1016/j.mrfmmm.2003.07.009. [DOI] [PubMed] [Google Scholar]
- Rossman TG, Uddin AN, Burns FJ, Bosland MC. Arsenite is a cocarcinogen with solar ultraviolet radiation for mouse skin: an animal model for arsenic carcinogenesis. Toxicol Appl Pharmacol. 2001;176:64–71. doi: 10.1006/taap.2001.9277. [DOI] [PubMed] [Google Scholar]
- Wang Z, Liu Y, Mori M, Kulesz-Martin M. Gene expression profiling of initiated epidermal cells with benign or malignant tumor fates. Carcinogenesis. 2002;23:635–643. doi: 10.1093/carcin/23.4.635. [DOI] [PubMed] [Google Scholar]
- Yager JW, Wiencke JK. Inhibition of poly (ADP-ribose) polymerase by arsenite. Mutat Res. 1997;386:345–351. doi: 10.1016/s1383-5742(97)00011-2. [DOI] [PubMed] [Google Scholar]
- Zhao CQ, Young MR, Diwan BA, Coogan TP, Waalkes MP. Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc Natl Acad Sci USA. 1997;94:10907–10912. doi: 10.1073/pnas.94.20.10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang L, Wang B, Sauder DN. Molecular mechanism of ultraviolet-induced keratinocyte apoptosis. J Interferon Cytokine Res. 2000;20:445–454. doi: 10.1089/10799900050023852. [DOI] [PubMed] [Google Scholar]
- Ziegler A, Leffell DJ, Kunala S, Sharma HW, Gailani M, Simon JA, et al. Mutation hotspots due to sunlight in the p53 gene of nonmelanoma skin cancers. Proc Natl Acad Sci USA. 1993;90:4216–4220. doi: 10.1073/pnas.90.9.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]