

Abstract
Chlamydia trachomatis is an obligate intracellular bacterium that develops within a parasitophorous vacuole termed an inclusion. The inclusion is nonfusogenic with lysosomes but intercepts lipids from a host cell exocytic pathway. Initiation of chlamydial development is concurrent with modification of the inclusion membrane by a set of C. trachomatis-encoded proteins collectively designated Incs. One of these Incs, IncA, is functionally associated with the homotypic fusion of inclusions. Inclusions also do not fuse when cultures are multiply infected with C. trachomatis and cultivated at 32°C. We obtained evidence linking these experimental observations by characterizing IncA localization in 32°C cultures. Analysis of inclusions by light and transmission electron microscopy confirmed that HeLa cells infected with multiple C. trachomatis elementary bodies and cultivated at 32°C for 24 h contained multiple, independent inclusions. Reverse transcriptase PCR and immunoblot analyses of C. trachomatis-infected HeLa cells demonstrated the presence of IncA at 24 h in 32°C cultures. When parallel cultures were probed with IncA-specific antibodies in indirect immunofluorescence assays, IncA was detectable in intracellular chlamydiae but not within the inclusion membrane. In addition, analysis of purified reticulate bodies from 37 and 32°C cultures showed that bacterium-associated pools of IncA are enriched in cultures grown at 32°C. Microscopic observation of infected cells revealed that some vacuoles had fused by 48 h postinfection, and this finding was correlated with the detection of IncA in inclusion membranes by immunofluorescence microscopy. The data are consistent with a requirement for IncA in fusions of C. trachomatis inclusions and suggest that the effect of incubation at 32°C is manifested by restricted export of IncA to the inclusion membrane.
Chlamydiae are gram-negative, obligate, intracellular parasites. The clinically significant species include Chlamydia trachomatis, the etiologic agent of sexually transmitted disease (serovars D to K), blinding trachoma (serovars A to C), and lymphogranuloma venereum (LGV 1, 2, and 3), the respiratory pathogen Chlamydia pneumoniae, and the animal pathogen Chlamydia psittaci. Chlamydiae exhibit a biphasic developmental cycle in which infectious, metabolically inert elementary bodies (EBs) differentiate into vegetative reticulate bodies (RBs) within parasitophorous vacuoles termed inclusions. EBs initially occupy individual tightly membrane-bound inclusions that are not fusogenic with lysosomes (12). The inclusions are detached from the host cell endocytic pathway, yet chlamydial development is accompanied by acquisition of sphingomyelin from an exocytic pathway(s) (8, 20) and host-derived phospholipids potentially by other routes (11, 27). C. trachomatis inclusions eventually expand into membrane-limited, spacious vacuoles that accommodate the proliferating RBs. When host cells are infected by multiple C. trachomatis EBs, individual Chlamydia-containing vacuoles eventually fuse to form a single inclusion (4, 15, 16). Infections with C. psittaci strain GPIC (guinea pig inclusion conjunctivitis), however, result in multilobed, independent inclusions, even when the infection is initiated by a single organism (17). Although the factors involved in establishing and maintaining this intracellular niche are poorly understood, chlamydiae express and secrete a set of proteins termed Incs which are hypothesized to be important in chlamydial pathogenesis. Incs lack overall sequence similarity and are poorly conserved among chlamydial species, but they are currently defined by common characteristics, including (i) localization to the inclusion membrane, (ii) the lack of an N-terminal, cleavable secretion signal, and (iii) bilobed hydrophobic domains composed of 40 to 60 amino acid residues.
First identified in C. psittaci (19), IncA has been the focus of numerous studies. IncA homologs have subsequently been identified in all C. trachomatis biovars (3) and in C. pneumoniae (2). C. trachomatis L2 incA encodes a ca. 30-kDa protein and is expressed by 10 h postinfection (10, 23). IncA is exported to the inclusion membrane, where its C terminus is exposed to the host cell cytoplasm (10, 18), and C. psittaci IncA is phosphorylated by unknown host cell kinases (18). Although functional data for most Incs is lacking, multiple studies have correlated C. trachomatis L2 IncA with homotypic fusion of inclusions. C. trachomatis L2 inclusions begin to fuse at ca.10 h postinfection (10, 15, 26), a time corresponding to the presence of IncA. Microinjection of IncA-specific antibodies into C. trachomatis L2-infected HeLa cells blocks homotypic fusion of inclusions (10). Importantly, clinical isolates of C. trachomatis which do not display homotypic fusion lack detectable IncA (25). Although C. psittaci GPIC expresses IncA, the similarity to C. trachomatis IncA is minimal, with only 22% identity over the length of the protein. C. psittaci GPIC IncA is also approximately 30% larger than C. trachomatis IncA. The multilobed inclusion structure of GPIC inclusions (17) suggests that IncA of C. psittaci GPIC does not promote fusion of inclusions as C. trachomatis IncA does. The failure of C. psittaci IncA to induce fusogenicity is supported by results obtained by using IncA in yeast two-hybrid systems. C. trachomatis IncA interacts with itself but not with C. psittaci IncA, while C. psittaci IncA does not react even with complementary C. psittaci IncA. BLAST searches have failed to identify an IncA homolog in C. pneumoniae.
Van Ooij et al. (26) have demonstrated that incubation of C. trachomatis cultures at 32°C impairs homotypic fusion of C. trachomatis inclusions. Since IncA has been implicated in homotypic fusion, we investigated whether the decrease in fusion of inclusions detected at 32°C may be manifested by a reduced presence of IncA. We demonstrate here that although expression of IncA in 32°C cultures is comparable to expression in control cultures incubated at 37°C, IncA export to the inclusion membrane is impaired. The appearance of IncA in the inclusion membrane in 32°C cultures correlates with detectable fusion of inclusions. Collectively, the data provide a mechanism by which 32°C incubation affects inclusion fusogenicity and support a working model in which IncA is essential for homotypic fusion of inclusions.
MATERIALS AND METHODS
Cell culture and organisms.
Human epithelial HeLa 229 cell cultures were maintained at 37°C in the presence of 5% CO2-95% humidified air in RPMI 1640 medium (Life Technologies, Baltimore, Md.) supplemented with 10% (vol/vol) fetal bovine serum and 10 μg of gentamicin (Life Technologies) per ml. HeLa cells were infected with C. trachomatis LGV-434, serovar L2 in Hanks' balanced salt solution (HBSS) (Life Technologies) at 37°C as previously described (5, 9) or at 4°C for RNA isolation experiments as described by Fields and Hackstadt (6). Infected cells were then incubated for the appropriate times at either 37 or 32°C in the presence of 5% CO2-95% humidified air.
Transmission electron microscopy (TEM).
HeLa cells were grown on 13-mm thermanox coverslips (Nunc, Naperville, Ill.) and infected with C. trachomatis L2 in HBSS at a multiplicity of infection (MOI) of approximately 5 as previously described (5, 9). Infected monolayers were incubated at either 37 or 32°C for 24 h. Cultures were fixed with 2.5% (wt/vol) glutaraldehyde-4% (wt/vol) paraformaldehyde in 0.1 M sodium cacodylate-0.05 M sucrose buffer. The samples were postfixed with 0.5% (wt/vol) osmium tetroxide-0.8% (wt/vol) potassium ferricyanide, followed by 1% (vol/vol) tannic acid, and stained overnight at 4°C en bloc in 1% (wt/vol) uranyl acetate. Samples were dehydrated with a graded ethanol series and embedded in Spurr's resin. Thin sections were cut with an RMC MT-7000 ultramicrotome (Ventana, Tucson, Ariz.) and stained with 1% uranyl acetate and Reynold's lead citrate before they were observed at 80 kV with a Philips CM-10 electron microscope (FEI, Hillsboro, Oreg.). Images were acquired with an AMT digital camera system (Advanced Microscopy Techniques, Chazy, N.Y.) and were processed by using Adobe Photoshop, version 5.5 (Adobe Systems, Inc., San Jose, Calif.).
Immunodetction analyses.
The presence of chlamydial proteins in RBs and in 37 and 32°C cultures was examined by performing immunoblot analyses of purified C. trachomatis serovar L2 RBs and whole-culture extracts of C. trachomatis serovar L2-infected HeLa monolayers, respectively. C. trachomatis serovar L2 RBs were harvested from 17-h cultures incubated at 37 or 32°C by density gradient purification through RenoCal-76 (Bracco Diagnostics, Princeton, N.J.) as described previously (5). Whole-culture extracts were generated by lysis after 24 h of infection by treatment with ice-cold water containing Complete protease inhibitor cocktail (Roche Diagnostic, Indianapolis, Ind.). For immunoblot analysis, proteins were precipitated by addition of trichloroacetic acid (Fischer Scientific, Suwanne, Ga.) to a concentration of 10% (vol/vol), solubilized in electrophoresis sample buffer (2.3% [wt/vol] sodium dodecyl sulfate, 5% [vol/vol] β-mercaptoethanol, 25% [vol/vol] glycerol, 60 mM Tris [pH 6.8]), and resolved in 12% (vol/vol) polyacrylamide gels by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (13). Resolved material was then transferred to Immobilon-P (Millipore Corp., Bedford, Mass.) in carbonate buffer (10 mM NaHCO3, 3 mM Na2CO3, 10% methanol; pH 9.9), and Chlamydia-specific proteins were detected by probing with anti-major outer membrane protein (anti-MOMP) (1), anti-IncA (22), or anti-IncG (22). Visualization was achieved by probing with alkaline phosphatase-conjugated anti-rabbit immunoglobulin G (IgG) (Sigma-Aldrich Inc., St. Louis, Mo.) and developing with nitroblue tetrazolium-5-bromo-4-chloro-3-indolylphosphate (NBT-BCIP) (Life Technologies).
Immunofluorescence localization of Inc proteins in C. trachomatis-infected HeLa monolayers was tested by indirect immunofluorescence. HeLa monolayers were cultivated on 12-mm coverslips and infected at an MOI of ca. 5. Cultures were fixed and permeablized at appropriate time points with methanol, blocked with phosphate-buffered saline (135 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; pH 8.0) supplemented with 5% (wt/vol) bovine serum albumin and 0.05% (vol/vol) Tween 20 (Sigma-Aldrich), and probed with specific antibodies. Samples were probed first with either anti-IncA or anti-IncG and then with Texas Red-conjugated anti-rabbit IgG antibodies (Jackson Immunoresearch Laboratories, West Grove, Pa.). Chlamydiae were then stained by probing with anti-GroEL (28), followed by fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG antibodies (Molecular Probes, Inc., Eugene, Oreg.). Fluorescence images were acquired with a ×60 apochromatic objective on an FXA photomicroscope (Nikon) and were digitized by using a Dage-MTI CCD 72 camera with a DSP2000 image processor. Digital images were prepared by using Adobe Photoshop, version 5.0 (Adobe Systems, Inc.). For experiments involving the quantitation of inclusions in cells, cultures were cultivated and stained with anti-IncG and anti-HSP60 as described above. The inclusion content was determined for 50 randomly selected cells in triplicate samples for each treatment.
Analysis of gene expression.
Expression of incA and incG in 32 and 37°C cultures was tested by reverse transcriptase (RT) PCR analysis of whole-culture RNA. HeLa monolayers were infected at an MOI of ca. 5 with C. trachomatis serovar L2 in HBSS for 1 h at 4°C to allow attachment but not internalization of chlamydiae. Whole-culture RNA was harvested at 24 h after a temperature shift by treatment with Trizol reagent (Life Technologies) according to the manufacturer's instructions. RNAs were treated with amplification grade DNase I (Life Technologies) to remove residual DNA, and Chlamydia-specific messages were amplified from total RNA by using an Access RT-PCR kit (Promega, Madison, Wis.). We used previously reported sequence-specific primer sets (23) for incA (ED12 [sense] and ED13 [antisense]) and incG (MC7 [sense] and MC9 [antisense]) or primers for 16S rRNA (ED91 [5′-AACACTGGGACTGAGACAC-3′] [sense] and ED92 [5′-CCTTTTCTCCATCTACCC-3′] [antisense]). RNAs were reverse transcribed for 45 min at 48°C by using avian myeloblastosis virus RT, and the resulting cDNAs were denatured for 2 min at 94°C prior to amplification for 40 cycles consisting of 30 s at 94°C, 30 s at 55°C, and 30 s at 72°C. Samples lacking RT were included as DNA contamination controls, and C. trachomatis L2 genomic DNA was amplified as a positive control. Amplified products from 32 and 37°C cultures were normalized to 16S rRNA content, resolved in 1.5% (wt/vol) agarose gels, and visualized by staining with ethidium bromide.
RESULTS
Analysis of 32°C cultures by electron microscopy.
Van Ooij et al. (26) reported that cultivation of C. trachomatis-infected epithelial cells at 32°C resulted in formation of clusters of nonfused inclusions. We first sought to confirm the existence of multiple, distinct inclusions in HeLa cells infected with C. trachomatis L2 at a high MOI and cultivated at 32°C. The inclusion morphology in 32°C cultures was compared to that in 37°C cultures by TEM analysis of equivalently infected HeLa monolayers that were cultivated for 24 h and differed only in the postinfection incubation temperature (Fig. 1). Consistent with previously reported immunofluorescence data (26), this comparative study revealed that 32°C incubation resulted in the presence of several apparently separate inclusions in each cell, whereas single inclusions were present in 37°C cultures. Overall, the inclusions in 32°C cultures were smaller than the inclusions in 37°C cultures, but incubation at 32°C did not prevent chlamydial development since vegetative RBs were readily detectable in 32°C inclusions. The developmental cycle, however, was obviously delayed compared to that in 37°C cultures since EBs were readily detected within inclusions in 37°C cultures while very few chlamydiae with condensed nuclei were seen when cultures were incubated at 32°C. Progeny EBs were present, however, at later time points (48 h) in 32°C cultures since significant numbers of inclusion-forming units were detectable in secondary passages (data not shown).
FIG. 1.

Analysis of 32°C cultures by TEM demonstrates the presence of multiple C. trachomatis inclusions. HeLa cell monolayers were infected at an MOI of 5 with C. trachomatis L2 and incubated at either 32 or 37°C for 24 h. Both the 32 and 37°C images were taken at a magnification of ×4,600. Bar = 2 μm for both panels.
incA expression in 32°C cultures.
Since the C. trachomatis protein IncA has been functionally implicated in homotypic inclusion fusion (10, 25), we asked whether the impairment of fusion detected in cultures incubated at 32°C was manifested by an alteration in incA expression. We first tested for the presence of the incA message in 32°C cultures by RT-PCR (Fig. 2) to verify incA expression. Whole-culture RNA was extracted at 24 h postinfection from parallel C. trachomatis cultures incubated at either 32 or 37°C, and 16S rRNA and messages for incA and incG were amplified by using specific primers and single-step RT-PCR. Loading of amplified products on resolving agarose gels was normalized to 16S rRNA content since the quantity of this molecule reflects the number of chlamydiae (7, 14). The differences in the 16S rRNA amounts in three separate experiments ranged from ca. 4- to 40-fold excess in 37°C cultures compared to 32°C cultures (data not shown). Since IncG has not been functionally implicated in homotypic fusion, the incG-specific message was amplified as a positive control. As expected, an incG-specific product was detected in both 32 and 37°C cultures. We also detected significant amounts of incA-specific signal in 32°C RNA samples, indicating that incA was indeed expressed at the transcriptional level at 32°C. RT-PCR detection of the hctA-specific message at 20 h postinfection in 32°C cultures (data not shown) suggests that late gene expression (23) is not significantly delayed in such cultures compared to 37°C cultures.
FIG. 2.

Transcription of incA is comparable in 32 and 37°C cultures. HeLa cell monolayers were infected at an MOI of 5 with C. trachomatis L2 and cultivated at either 32 or 37°C for 24 h. Whole-culture RNA was harvested, and incA and incG messages were amplified by RT-PCR. 16S rRNA was amplified as a control for RNA quality and to normalize analyzed samples. Products were amplified from C. trachomatis L2 genomic DNA and from RNA harvested from uninfected HeLa cells as positive and negative controls, respectively. Reactions were performed in the presence (+) or in the absence (−) of RT, and products were resolved in 1.5% agarose gels and stained with ethidium bromide.
We next extended our analysis of IncA production by employing immunoblot analysis of whole-culture extracts to test for the presence of IncA (Fig. 3). Equivalent volumes of whole-culture protein samples from 32 and 37°C cultures were serially diluted and analyzed by immunoblotting by using MOMP-specific antibodies to establish the relative chlamydial content in each sample. These experiments indicated that there was a ca. fourfold excess of MOMP at 37°C compared to the amount at 32°C. Culture samples were normalized for equal MOMP contents and probed with anti-MOMP to verify loading and with anti-IncA or anti-IncG. Essentially equal amounts of both IncA and IncG were detected in 32 and 37°C cultures. Hence, expression of incA by individual chlamydiae is not significantly affected by incubation of cultures at 32°C, suggesting that the effect of low temperature on homotypic fusion is not manifested by a lack of IncA expression.
FIG. 3.

Comparison of IncA in 32 and 37°C cultures by immunoblot analysis. (A) Proteins were resolved in sodium dodecyl sulfate-12% (wt/vol) polyacrylamide gel electrophoresis gels, and anti-MOMP was used to detect MOMP in serially diluted samples from whole-culture lysates to establish chlamydial contents. mw, molecular weight. (B) Material corresponding to 8% of a 32°C culture or 2% of a 37°C culture was probed with anti-MOMP, anti-IncA, or anti-IncG. Multiple bands in the IncG panel likely represent different phosphorylation states (21). Proteins were visualized by probing with secondary antibodies conjugated to alkaline phosphatase and development with NBT-BCIP.
Localization of IncA in 32°C cultures.
According to current working models, IncA exposure at the cytoplasmic face of the inclusion membrane is required to successfully promote homotypic fusion. Since individual chlamydiae in cultures incubated at 32°C expressed levels of IncA comparable to the levels in cultures incubated at 37°C, we next examined whether the nonfused inclusions detected at 32°C contained membrane-localized IncA by indirect immunofluorescence microscopy (Fig. 4). C. trachomatis-infected cultures were incubated at either 32 or 37°C and probed with GroEL-specific antibodies to show the locations of chlamydiae (green) and either IncA- or IncG-specific antibodies (red). Both IncA- and IncG-specific signals were detectable in 37°C cultures and exhibited the characteristic rim-like staining indicative of inclusion membrane localization (Fig. 4A). In addition, Inc- and GroEL-specific signals did not colocalize in merged images, indicating that the majority of each Inc was localized to the inclusion membrane. Although the inclusion size was significantly smaller in 32°C cultures, the inclusion membrane-specific staining was maintained for IncG (Fig. 4B). Conversely, an IncA-specific signal was not detectable in inclusion membranes when cultures were maintained at 32°C. Instead, the IncA-specific signals overlapped with those of GroEL, indicating that IncA colocalized with chlamydiae. These data indicated that IncA was being produced in 32°C cultures but not exported to the inclusion membrane in detectable amounts.
FIG. 4.

IncA is not detectable in inclusion membranes in cultures incubated at 32°C. IncA localization was detected by indirect immunofluorescence in C. trachomatis L2-infected HeLa cell cultures infected at an MOI of 5 and cultivated at 37°C (A) or 32°C (B) for 24 h. GroEL staining (green) with anti-GroEL (α-GroEL) was used to localize chlamydiae, and cultures were probed with anti-IncG (α-IncG) (as a control) or anti-IncA (α-IncA) (red). Individual channel and merged images are shown. Proteins were visualized by probing with FITC-conjugated (for GroEL) or Texas Red-conjugated (for IncG and IncA) secondary antibodies. Bars = 5 μm.
Although IncA and IncG are secreted proteins, RBs harvested from 37°C cultures contain amounts of both of these proteins which are detectable in immunoblots (22). We reasoned that if IncA export was perturbed, RBs isolated from cultures incubated at 32°C should contain comparatively more IncA. We tested this hypothesis by performing an immunoblot analysis of RB lysates derived from RBs purified from 17-h C. trachomatis L2-infected HeLa cultures (Fig. 5). Equal amounts (based on MOMP reactivity in immunoblots) of 32°C-derived RBs and 37°C-derived RBs were probed with MOMP-, IncA- and IncG-specific antibodies. Lysates from uninfected HeLa cells and 20-h C. trachomatis L2-infected cultures were used as antibody specificity controls. Essentially equal amounts of MOMP were detected in samples obtained from 32 and 37°C cultures. The IncA-specific signal was more pronounced in 32°C-derived RBs than in 37°C-derived RBs. Interestingly, IncG was also enriched in the 32°C samples. These data indicate that both IncA export and IncG export may be impaired at 32°C and suggest that there is a more global effect on protein secretion.
FIG. 5.

IncA and IncG are enriched in RBs isolated from 32°C cultures compared to RBs isolated from 37°C cultures. Proteins from whole-culture lysates of 20-h infected (L2+HeLa) or uninfected (HeLa) HeLa cell monolayers, as well as whole-cell lysates of Renografin-purified RBs were resolved in 12% (wt/vol) acrylamide gels. IncA and IncG were detected with anti-IncA and anti-IncG antibodies, respectively. Immunoblot analysis with MOMP-specific antibodies was used to normalize samples for chlamydial content of RB samples. Proteins were visualized by probing with secondary antibodies conjugated to alkaline phosphatase and development with NBT-BCIP.
The observation that IncG secretion may occur at a lower rate at 32°C than at 37°C raised the possibility that IncA secretion at 32°C is not completely blocked. Indeed, Van Ooij et al. detected increasing fusion of inclusions when 32°C cultures were incubated for more than 24 h (26). We therefore assayed the fusogenicity of inclusions over time by direct microscopic analysis of 32°C cultures (Fig. 6). HeLa cell monolayers were infected with C. trachomatis L2 at an MOI of 5, and cultures were fixed at 24, 48, and 72 h postinfection. Inclusions were visualized by staining preparations with anti-IncG and anti-Hsp60, and triplicate samples were counted directly. Observation of 37°C cultures at 24 h revealed that nearly 100% of the infected cells contained a single inclusion (data not shown). At 24 h, 32°C cultures contained very few cells (2.6%) harboring single inclusions. Interestingly, the percentage of single-inclusion-containing cells increased to 52.6% at 48 h and to 68.6% by 72 h postinfection. Since homotypic fusion of inclusions was occurring at these later times, we analyzed IncA localization in parallel cultures by indirect immunofluorescence. Merged images of IncA-specific signals (red) and Hsp60-specific signals (green) are shown in Fig. 6. As seen previously, IncA-specific signals colocalized with Hsp60-specific signals and were not detectable in the inclusion membrane after incubation for 24 h at 32°C. However, IncA staining resulted in the characteristic rim-like staining pattern indicative of inclusion membrane localization at both 48 and 72 h postinfection. These data indicate that incubation of cultures at 32°C slows, but does not inhibit, IncA deployment to the inclusion membrane. In addition, fusion of inclusions was detectable at times that correlated with the appearance of IncA in the inclusion membrane.
FIG. 6.
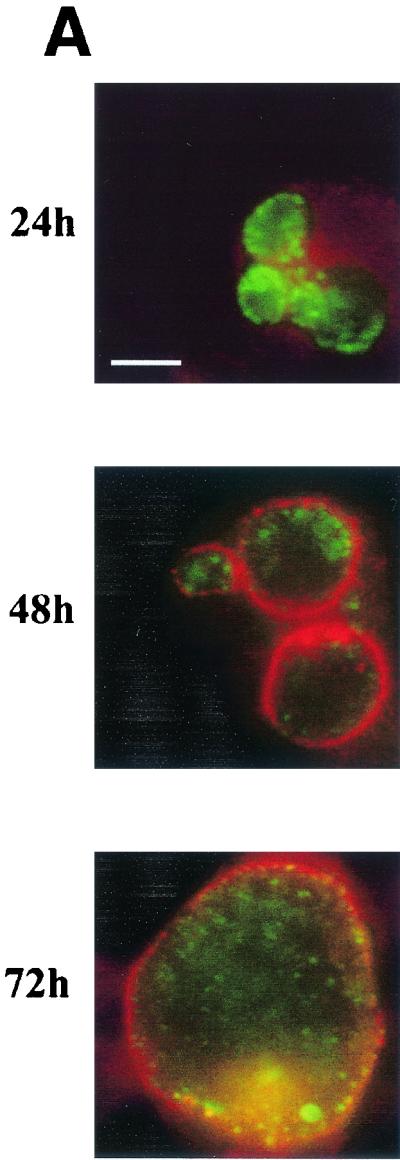

Homotypic fusion of inclusions and secretion of IncA occur at later times in 32°C cultures. HeLa cell monolayers were infected at an MOI of 5 with C. trachomatis L2, and the numbers of inclusions per cell were determined by direct microscopic counting of triplicate samples at 24, 48, and 72 h postinfection (pi). The values in panel B are the percentages of infected cells containing single inclusions. Parallel cultures were stained for IncA (red) with anti-IncA antibodies and for chlamydiae (green) with anti-GroEL antibodies. Only merged images are shown in panel A. Proteins were visualized by probing with FITC-conjugated (for GroEL) or Texas Red-conjugated (for IncA) secondary antibodies. Bar = 5 μm.
DISCUSSION
Functional data for Chlamydia-encoded proteins is difficult to acquire since a tractable genetic system has not been developed for Chlamydia spp. Despite this limitation, IncA has been functionally implicated in homotypic fusion of C. trachomatis inclusions. Microinjection of neutralizing IncA-specific antibodies blocks fusion (10), and nonfusing C. trachomatis clinical isolates lack detectable IncA (25). Although incubation of C. trachomatis-infected cultures at 32°C also impairs homotypic fusion (26), the mechanism by which this effect is manifested has not been elucidated. Here we present data that are consistent with the role of IncA in homotypic fusion of C. trachomatis inclusions and provide a plausible mechanism by which homotypic fusion is impaired in 32°C cultures. Although we cannot exclude the possibility that other chlamydia- or host cell-derived factors contribute to the impairment of homotypic fusion at 32°C, our data collectively support a role for IncA and show that exposure of IncA to the cytoplasmic face of an inclusion is necessary for fusion to occur.
IncA is expressed by individual chlamydiae in 32°C cultures but does not reach the inclusion membrane at detectable levels by 24 h postinfection. We believe that a direct effect of low temperature on IncA activity is not a likely explanation for impaired homotypic fusion since cultures incubated for prolonged times revealed that there is a correlation between the appearance of membrane-localized IncA and fusion of inclusions. Instead, we favor a model in which IncA accumulates within chlamydiae due to restricted export to the inclusion membrane in 32°C cultures and must reach a critical level in the inclusion membrane to efficiently mediate homotypic fusion of inclusions. The inability of inclusions to fuse at 32°C is not restricted to the C. trachomatis LGV biovar or to a host cell range (26). Suchland et al. (25) noted that C. trachomatis serovar J inclusions did not fuse in 32°C cultures but possessed detectable amounts of inclusion membrane-localized IncA. They therefore concluded that IncA was not associated with this temperature-dependent phenomenon. This observation, however, is consistent with our results since the localization of IncA was tested at late time points (40 h postinfection); thus, IncA would have had time to accumulate in inclusion membranes even with slowed secretion kinetics.
Analysis of RB lysates by immunoblotting indicated that IncG secretion may also be impaired, yet we were able to readily detect inclusion membrane-localized IncG in 32°C cultures at 24 h. The fact that IncG is expressed early in the developmental cycle, whereas IncA is not expressed until midcycle, may be one explanation for this difference since IncG would have had more time to accumulate by our 24-h time point. An alternate possibility for the observed delay in homotypic fusion of inclusions could be that the overall chlamydial developmental cycle is kinetically slowed at 32°C. Indeed, our analysis of cultures by TEM (Fig. 1) suggested that the chlamydial developmental cycle may be protracted in 32°C cultures since EBs were not readily visible by 24 h postinfection. Progeny EBs were detectable in 32°C cultures during one-step growth curve experiments, but numbers approaching those detected in 24-h cultures incubated at 37°C were not detected until 48 h in 32°C cultures (data not shown). These observations are in agreement with studies performed by Van Ooij et al. (26) in which progeny EBs were first detectable at later times in 32°C cultures than in 37°C cultures. We were unable, however, to detect a corresponding delay in the expression of selected early-, mid-, or late-cycle genes in 32°C cultures by RT-PCR analyses (data not shown). Although a delayed developmental cycle may contribute to slowing homotypic fusion, we believe that the predominant cause is the lack of membrane-localized IncA since readily detectable amounts of Chlamydia-localized IncA are present at 24 h postinfection. Also, Van Ooij et al. (26) treated 32°C cultures with chloramphenicol to prevent de novo chlamydial protein synthesis and subsequently shifted the cultures to 37°C, where they observed efficient homotypic fusion. Hence, the components required for this event (IncA) must have been present in sufficient quantities to mediate fusion of inclusions. According to our model, the Chlamydia-localized pool of IncA would have gained access to the inclusion membrane after the shift to 37°C.
The mechanism by which low temperature affects IncA and IncG secretion remains to be determined. The possible mechanisms include an inability to be efficiently recognized as secretion substrates or a negative impact on the ability to insert into inclusion membranes due to changes in membrane fluidity at 32°C. However, the observation that IncA and IncG secretion is less efficient at 32°C is of particular interest since recent evidence indicates that Incs are exported by the chlamydial type III apparatus (24). Expression of the core type III component CdsJ was not affected by incubation of cultures at 32°C (data not shown). If the negative effect of low temperature acts at the level of the type III apparatus, this culture condition could be useful for elucidation other type III secretion substrates.
Here we provide further evidence supporting a functional role of IncA in homotypic fusion of chlamydial inclusions. Our results indicate that restricted secretion of IncA, and not a lack of incA expression, is a likely explanation for the impaired homotypic fusion of inclusions in multiply infected host cells. A more detailed analysis is required to evaluate whether this effect is manifested by a direct effect on individual secretion substrates or the apparatus required to export these substrates from the bacterial cell.
Editor: D. L. Burns
REFERENCES
- 1.Baehr, W., Y. X. Zhang, T. Joseph, H. Su, F. E. Nano, K. D. Everett, and H. D. Caldwell. 1988. Mapping antigenic domains expressed by Chlamydia trachomatis major outer membrane protein genes. Proc. Natl. Acad. Sci. USA 85:4000-4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bannantine, J. P., R. S. Griffiths, W. Viratyosin, W. J. Brown, and D. D. Rockey. 2000. A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cell. Microbiol. 2:35-47. [DOI] [PubMed] [Google Scholar]
- 3.Bannantine, J. P., W. E. Stamm, R. J. Suchland, and D. D. Rockey. 1998. Chlamydia trachomatis IncA is localized to the inclusion membrane and is recognized by antisera from infected humans and primates. Infect. Immun. 66:6017-6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blyth, W. A., and J. Taverne. 1972. Some consequences of the multiple infection of cell cultures by TRIC organisms. J. Hyg. 70:33-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caldwell, H. D., J. Kromhout, and J. Schachter. 1981. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect. Immun. 31:1161-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fields, K. A., and T. Hackstadt. 2000. Evidence for the secretion of Chlamydia trachomatis CopN by a type III secretion mechanism. Mol. Microbiol. 38:1048-1060. [DOI] [PubMed] [Google Scholar]
- 7.Gerard, H. C., J. A. Whittum-Hudson, and A. P. Hudson. 1997. Genes required for assembly and function of the protein synthetic system in Chlamydia trachomatis are expressed early in elementary to reticulate body transformation. Mol. Gen. Genet. 255:637-642. [DOI] [PubMed] [Google Scholar]
- 8.Hackstadt, T., D. D. Rockey, R. A. Heinzen, and M. A. Scidmore. 1996. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. EMBO J. 15:964-977. [PMC free article] [PubMed] [Google Scholar]
- 9.Hackstadt, T., M. A. Scidmore, and D. D. Rockey. 1995. Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proc. Natl. Acad. Sci. USA 92:4877-4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hackstadt, T., M. Scidmore-Carlson, E. Shaw, and E. Fischer. 1999. The Chlamydia trachomatis IncA protein is required for homotypic vesicle fusion. Cell. Microbiol. 1:119-130. [DOI] [PubMed] [Google Scholar]
- 11.Hatch, G. M., and G. McClarty. 1998. Phospholipid composition of purified Chlamydia trachomatis mimics that of the eukaryotic host cell. Infect. Immun. 66:3727-3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heinzen, R. A., M. A. Scidmore, D. D. Rockey, and T. Hackstadt. 1996. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect. Immun. 64:796-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 14.Mathews, S. A., K. M. Volp, and P. Timms. 1999. Development of a quantitative gene expression assay for Chlamydia trachomatis identified temporal expression of σ factors. FEBS Lett. 458:354-358. [DOI] [PubMed] [Google Scholar]
- 15.Matsumoto, A., H. Bessho, K. Uehira, and T. Suda. 1991. Morphological studies of the association of mitochondria with chlamydial inclusions and the fusion of chlamydial inclusions. J. Electron Microsc. 40:356-363. [PubMed] [Google Scholar]
- 16.Ridderhof, J. C., and R. C. Barnes. 1989. Fusion of inclusions following superinfection of HeLa cells by two serovars of Chlamydia trachomatis. Infect. Immun. 57:3189-3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rockey, D. D., E. R. Fischer, and T. Hackstadt. 1996. Temporal analysis of the developing Chlamydia psittaci inclusion by use of fluorescence and electron microscopy. Infect. Immun. 64:4269-4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rockey, D. D., D. Grosenbach, D. E. Hruby, M. G. Peacock, R. A. Heinzen, and T. Hackstadt. 1997. Chlamydia psittaci IncA is phosphorylated by the host cell and is exposed on the cytoplasmic face of the developing inclusion. Mol. Microbiol. 24:217-228. [DOI] [PubMed] [Google Scholar]
- 19.Rockey, D. D., R. A. Heinzen, and T. Hackstadt. 1995. Cloning and characterization of a Chlamydia psittaci gene coding for a protein localized in the inclusion membrane of infected cells. Mol. Microbiol. 15:617-626. [DOI] [PubMed] [Google Scholar]
- 20.Scidmore, M. A., D. D. Rockey, E. R. Fischer, R. A. Heinzen, and T. Hackstadt. 1996. Vesicular interactions of the Chlamydia trachomatis inclusion are determined by chlamydial early protein synthesis rather than route of entry. Infect. Immun. 64:5366-5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scidmore, M. A., and T. Hackstadt. 2001. Mammalian 14-3-3 beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol. Microbiol. 39:1638-1650. [DOI] [PubMed] [Google Scholar]
- 22.Scidmore-Carlson, M. A., E. I. Shaw, C. A. Dooley, E. R. Fischer, and T. Hackstadt. 1999. Identification and characterization of a Chlamydia trachomatis early operon encoding four novel inclusion membrane proteins. Mol. Microbiol. 33:753-765. [DOI] [PubMed] [Google Scholar]
- 23.Shaw, E. I., C. A. Dooley, E. R. Fischer, M. A. Scidmore, K. A. Fields, and T. Hackstadt. 2000. Three temporal classes of gene expression during the Chlamydia trachomatis developmental cycle. Mol. Microbiol. 37:913-925. [DOI] [PubMed] [Google Scholar]
- 24.Subtil, A., C. Parsot, and A. Dautry-Varsat. 2001. Secretion of predicted Inc proteins of Chlamydia pneumoniae by a heterologous type III machinery. Mol. Microbiol. 39:792-800. [DOI] [PubMed] [Google Scholar]
- 25.Suchland, R. J., D. D. Rockey, J. P. Bannantine, and W. E. Stamm. 2000. Isolates of Chlamydia trachomatis that occupy nonfusogenic inclusions lack IncA, a protein localized to the inclusion membrane. Infect. Immun. 68:360-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Ooij, C., E. Homola, E. Kincaid, and J. Engel. 1998. Fusion of Chlamydia trachomatis-containing inclusions is inhibited at low temperatures and requires bacterial protein synthesis. Infect. Immun. 66:5364-5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wylie, J. L., G. M. Hatch, and G. McClarty. 1997. Host cell phospholipids are trafficked to and then modified by Chlamydia trachomatis. J. Bacteriol. 179:7233-7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan, Y., K. Lyng, Y. X. Zhang, D. D. Rockey, and R. P. Morrison. 1992. Monoclonal antibodies define genus-specific, species-specific, and cross-reactive epitopes of the chlamydial 60-kilodalton heat shock protein (hsp60): specific immunodetection and purification of chlamydial hsp60. Infect. Immun. 60:2288-2296. [DOI] [PMC free article] [PubMed] [Google Scholar]