

Abstract
Staphylococcus aureus produces four major extracellular proteases: staphylococcal serine protease (V8 protease; SspA), cysteine protease (SspB), metalloprotease (aureolysin; Aur), and staphopain (Scp). Several in vitro studies have suggested that these enzymes are important virulence factors. Here we analyzed the protease production of 92 S. aureus strains from infected human soft tissue. Twenty-one strains produced variable zones of proteolysis on casein agar plates, while the remaining 71 strains appeared to be protease negative. The major protease genes were present in all protease-positive (n = 5) and protease-negative (n = 12) strains analyzed. Northern blotting showed that transcription of the protease genes was suppressed due to increased sigma factor B (SigB)-dependent expression of the protease repressor SarA. Other SigB-dependent traits such as pigmentation and expression of asp 23 were also increased in protease-negative compared to protease-positive strains. Inactivation of sarA in three protease-negative strains resulted in increased transcription of all protease genes and increased protease production, while overexpression of sarA in a strain producing protease at high levels repressed protease production. Our results suggest that the protease genes are conserved among clinical S. aureus strains and that the level of SigB-dependent expression of the protease repressor sarA determines the level of protease production in each strain.
Staphylococcus aureus causes a variety of infections, ranging from superficial skin and wounds infections to deep abscesses and septicemia. Recent reports of the U.S. National Nosocomial Infections Surveillance System have ranked S. aureus as a leading cause of hospital-acquired bacteremia, pneumonia, and surgical wound infection (11).
The virulence of S. aureus is considered to be the result of the coordinated activity of several secreted toxins and digestive enzymes, as well as a large number of proteins on the bacterial surface that bind extracellular matrix and plasma proteins (2, 3, 24, 33). In vitro studies have shown that staphylococcal proteases can cleave and degrade a number of important host proteins, including the heavy chains of all human immunoglobulin classes, plasma proteinase inhibitor, and elastin (42-45), indicating that they are important virulence factors. Recent reports suggest that proteases also play a role in the transition of S. aureus cells from an adhesive to an invasive phenotype by degrading bacterial cell surface proteins, such as fibronectin binding protein and protein A (16, 27, 35, 46, 49).
S. aureus produces four major extracellular proteases: serine protease (V8 protease; SspA), a cysteine protease (SspB) encoded within the same operon, metalloprotease (aureolysin; Aur), and a second cysteine protease (Scp; also named staphopain) [2; B. Hofmann, D. Schomburg, and H. J. Hecht, abstract from the 16th Congress of the International Union of Crystallography 1993, Acta Crystallogr. 49(Suppl.):102, 1993]. All four proteases are secreted as proenzymes, which are proteolytically cleaved to generate the mature enzymes. The proenzyme form of the serine protease is enzymatically inactive and needs to be cleaved by aureolysin to become active (18). In the case of SspB, which is processed by SspA, the proenzyme form appears to be enzymatically active (47). However, which enzymes are involved in the processing of aureolysin and staphopain remains to be determined.
Synthesis of extracellular proteases is activated by agr (7, 26, 32) and repressed by sarA (14, 32) in such a way that the production of proteases takes place mainly during the late exponential and postexponential phases of growth.
We have observed that the level of protease production varies considerably among clinical isolates of S. aureus. To investigate the background for this variation, 92 fresh clinical isolates were analyzed for protease production on casein agar plates. Strains producing different amounts of protease were tested for the presence of protease genes by PCR, and the levels of expression of four major protease genes and of the protease regulators agr (RNAIII; activator) and sarA (repressor) were analyzed by Northern blotting. Our results suggest that the variation in protease production between strains of S. aureus depends on different levels of SigB-dependent sarA expression.
MATERIALS AND METHODS
Bacterial strains, plasmids, and cultivation conditions.
Ninety-two S. aureus strains from pyogenic soft tissue infections, isolated at Karolinska Hospital Clinical Microbiology Laboratory in Stockholm, Sweden, were collected at two different time points (1999 and 2000). Strains of S. aureus were identified by colony morphology, Gram staining, DNase, coagulase, and the MONOSTAPH slide test (BIONOR, Skien, Norway). Other bacterial strains and plasmids used in this study are listed in Table 1. Screening for protease production was carried out on casein agar plates (8). S. aureus strains were precultured in tryptic soy broth for 16 to 18 h. Cells from precultures were inoculated in 100 ml of brain heart infusion (BHI) in a 1-liter baffled flask to give an initial optical density at 600 nm (OD600) of 1.0 and were incubated on a rotary shaker (180 rpm) at 37°C.
TABLE 1.
Bacterial stains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s)a | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | E. coli strain used for propagation of all plasmid constructs | Promega Corp. |
| S. aureus | ||
| 8325-4 | Prototype “wild-type” strain; rsbU mutant | 40 |
| RN 4220 | Restriction-deficient mutant of 8325-4 | 29 |
| PC1839 | 8325-4 sarA::km (Kmr) | 14 |
| KT111 | 8325-4 with ssp promoter element from 8325-4, fused to lacZ, inserted in geh (Emr) | 50 |
| CYL316 | RN4220(pYL112Δ19) | 31 |
| NA1 | Clinical strain KS7 sarA::km (Kmr) | This study |
| NA2 | Clinical strain KS30 sarA::km (Kmr) | This study |
| NA3 | Clinical strain KS33 sarA::km (Kmr) | This study |
| KA1 | Clinical strain KS26 with plasmid pKT601 (Tcr) | This study |
| KS30.1 | 8325-4 with ssp promoter element from KS30, fused to lacZ, inserted in geh (Emr) | This study |
| KS30.2 | 8325-4 with mutated ssp promoter element from KS30, fused to lacZ, inserted in geh (Emr) | This study |
| Plasmids | ||
| pKT601 | S. aureus plasmid containing the sarA gene under the control of the xylA promoter (Tcr) | 50 |
| pKT1 | pRZ5202 containing ermB and attP (Ampr) | 50 |
| pKS30.1 | pKT1 with ssp promoter::lacZ from KS30 (Ampr) | This study |
| pKS30.2 | PKT1 with mutated (G→A) ssp promoter::lacZ from KS30 (Ampr) | This study |
Kmr, resistance to kanamycin and neomycin; Emr, resistance to erythromycin and lincomycin; Tcr, resistance to tetracycline.
Preliminary sequence data were obtained from the website of The Institute for Genomic Research (http://www.tigr.org) for S. aureus strain COL and from the University of Oklahoma genome sequencing project website (http://www.genome.ou.edu/staph.html) for S. aureus strain 8325-4.
PCR.
The presence of protease genes in S. aureus strains was analyzed by PCR by use of the primers listed in Table 2. Chromosomal DNA from S. aureus strains was prepared with the DNeasy Tissue Kit (Qiagen) and was used as a template. PCR fragments were purified with the QiaQuick PCR purification kit (Qiagen) and analyzed on 1% agar gels (Sigma) together with a 1-kb Plus size marker (Life Technologies).
TABLE 2.
Primers used for detection of S. aureus protease genes by PCR
| Primer | Sequence |
|---|---|
| sspA forward | 5′-GAC AAC AGC GAC ACT TGT GA-3′ |
| sspA reverse | 5′-AGT ATC TTT ACC TAC AAC TAC A-3′ |
| sspB forward | 5′-TGA AGA AGA TGG CAA AGT TAG-3′ |
| sspB reverse | 5′-TTG AGA TAC ACT TTG TGC AAG-3′ |
| aur forward | 5′-TAG TAG CAC ACG AAT TAA CAC ACG-3′ |
| aur reverse | 5′-TTC CCT ATT GCT TGA ATC ACG-3′ |
| scp forward | 5′-AAA TTA TTG CAT GCA CTG ATA ATG TGT AA-3′ |
| scp reverse | 5′-ATT ACC TTC AGA ATT CAA AAC TGG-3′ |
Northern blot analysis.
Total S. aureus RNA was prepared by using the FAST RNA-blue kit (Bio 101) according to the manufacturer's instructions. The concentration of RNA was determined by measuring the absorbance at 260 nm. Samples containing 10 μg of total RNA were analyzed by Northern blotting as described previously (37). Internal fragments of the 16S rRNA gene (nucleotides [nt] 11 to 1023; GenBank accession no. X68417), the gene coding for RNAIII (nt 1095 to 1578) (28), sarA (nt 843 to 1260; GenBank accession no. U46541), lacZ (nt 8100 to 8700; GenBank accession no. AE000141), asp23 (nt 27 to 349; GenBank accession no. S76213), ssp (889 bp), aur (1,197 bp), and scp (407 bp) were amplified by PCR, radiolabeled with [α-32P]dCTP (Amersham) by using a random prime labeling kit (Roche Molecular Biochemicals), and used as probes. Radioactivity was detected by a radioisotope imaging system (PhosphorImager 445SI; Molecular Dynamics).
Analysis of extracellular protein.
Total exoproteins from 50 μl of culture supernatant were precipitated with methanol-chloroform (4), dissolved in 10 μl of loading buffer, separated by sodium dodecyl sulfate-12% polyacrylamide gel electrophoresis (SDS-12% PAGE), and stained with Coomassie brilliant blue according to standard protocols (4). For Western blot analysis of serine protease, culture supernatants corresponding to a bacterial density of 0.12 OD600 unit were separated by SDS-12% PAGE and transferred to polyvinylidene difluoride-based membranes (Immobilon-P; Millipore) by using a Bio-Rad Mini Trans-Blot Electrophoretic Transfer Cell as recommended by the supplier. Polyclonal rabbit anti-serine protease antibodies (25) were used and were detected with horseradish peroxidase-conjugated sheep anti-rabbit antibodies (Amersham Life Science).
Inactivation and overexpression of sarA in clinical strains.
sarA was inactivated in clinical S. aureus strains KS7, KS30, and KS33 by transfer of the sarA::km mutation from S. aureus strain PC1839 by phage transduction using φ11 (39). Transductants (NA1, NA2, and NA3, respectively) were selected on kanamycin (25 μg ml−1) agar plates. Mutations were confirmed by PCR analysis using primers internal to the sarA gene and flanking primers in the kanamycin cassette.
Plasmid pKT601, carrying the sarA gene under the control of the inducible xylA promoter, was transferred to S. aureus strain KS26 by electroporation (48). PCR analysis and restriction mapping of the plasmid confirmed the presence of pKT601 in strain KA1. For induction of sarA, bacteria were grown on casein agar plates with 0.10% xylose, or in glucose-free BHI containing 0.05% xylose, in the presence of tetracycline (5 μg ml−1).
Construction of chromosomally encoded ssp promoter::lacZ fusions.
The transcription reporter gene vector pKT1, containing the phage L54a attachment site attPI (50), was used to generate different ssp promoter::lacZ fusions. PCR fragments encompassing the promoter region of ssp (positions −312 to +14 relative to the transcription start point [12]) were amplified from KS30 chromosomal DNA by using a forward primer (5′-TAA TTG ACT AGT AAA CTT AAG CAC TCA AAT AAT ATA TC-3′) with an added SpeI restriction site and a reverse primer (5′-AAA AAT GGA TCC ACA AGT TAA ATA TAA CAA TAA AAA TTT TTA-3′) with an added BamHI restriction site. For construction of pKS30.2, in which the G in position 4 of the −35 box (TTGGCT) was replaced by an A (TTGACT), the reverse primer 5′-AAA AAT GGA TCC ACA AGT TAA ATA TAA CAA TAA AAA TTT TTA AGT CAA-3′ was used together with the forward primer described above. The PCR fragments were ligated to pKT1 cut with SpeI and BamHI and were used to transform Escherichia coli cells. Plasmid constructs were confirmed by nucleotide sequencing using the ABI PRISM BigDye Terminator Cycle Sequencing kit, version 2 (Perkin-Elmer Applied Biosystems), and the Applied Biosystems 377 DNA sequencer. The correct plasmids were then electroporated into S. aureus CYL316. Strain CYL316 contains the integrase gene of phage L54a, which allows the plasmid to integrate into the attB site of the lipase gene (geh). Integrations were confirmed by PCR and were then transduced into S. aureus strain 8325-4 by using the transducing phage φ11 as described elsewhere (39) to generate strains KS30.1 and KS30.2, respectively. Proper integration of the promoter fusion constructs was confirmed by PCR analysis using a forward primer specific for geh and a reverse primer specific for the pKT1 vector.
RESULTS
Expression of extracellular proteases among S. aureus clinical isolates.
Ninety-two S. aureus strains from human soft tissue infections were cultivated on casein agar plates in order to study extracellular protease production. Twenty-one strains (23%) produced a zone of precipitation around the bacterial streak (Fig. 1). Since the quality of the precipitation was most typical for the V8 protease (1), all strains were tested for the presence of the V8 protease gene (sspA) by PCR. A PCR product of the expected size (292 nt) was obtained with all strains (data not shown). Twelve protease-negative and five protease-positive strains were also tested for the presence of the other major protease genes. All strains tested were positive for sspB (cysteine protease), aur (aureolysin), and scp (staphopain) (data not shown).
FIG. 1.

Zones of proteolysis around 50 clinical isolates of S. aureus grown on a casein agar plate.
Analysis of expression of the regulators sarA and agr in protease-negative and protease-positive S. aureus strains.
A possible explanation for the variation in protease production between strains could be that they express different levels of the protease gene regulators sarA (a repressor) and agr (RNAIII; an activator). To test this we analyzed the levels of expression of sarA, RNAIII, and ssp in two protease-positive strains (KS26, a high producer, and KS36, an intermediate producer) and three protease-negative strains (KS7, KS30, and KS33). The level of ssp mRNA in each strain correlated roughly with the zone of proteolysis on casein agar plates; it was highest in strain KS26, intermediate in strain KS36, and lowest in the protease-negative strains (Fig. 2). It should be noted that strains KS7 and KS33 produced significant amounts of ssp mRNA although they were protease negative on casein agar plates. Western blot analysis of culture supernatants confirmed that KS7 and KS33, as well as KS26, produced serine protease (SspA) (Fig. 3). SspA could not be detected in culture supernatants from strain KS30, which was consistent with the lack of ssp mRNA. Serine protease from strains KS7 and KS33 appeared as two bands of higher molecular mass than the corresponding bands from KS26. Most likely, the larger forms of SspA produced by the protease-negative strains represented inactive proforms of the enzyme. The incomplete processing of SspA was consistent with the lack of aur expression in KS7 and KS33 (see Fig. 6). Inactivation of aur in S. aureus strain 8325-4 resulted in complete absence of a zone of proteolysis (27) and in appearance of the larger proforms (data not shown).
FIG. 2.

Northern blot analysis of sarA, RNAIII, and ssp transcripts in protease-negative (KS7, KS30, and KS33) and protease-positive (KS26 and KS36) clinical isolates of S. aureus at different time points during growth. 16S rRNA was used as an internal control of the amount of total RNA loaded. The same filter was hybridized with each of the specific probes.
FIG. 3.

Western blot analysis of serine protease (SspA) in culture supernatants of S. aureus clinical strains. Samples were taken after 4 h of growth.
FIG. 6.

Northern blot analysis of sarA, RNAIII, ssp, scp, and aur in the protease-negative clinical S. aureus strains and their corresponding sarA mutants. 16S rRNA was used as an internal control of the amount of total RNA loaded. The same filter was hybridized with each of the specific probes. KS33 and its sarA mutant were on a separate membrane.
An inverse correlation between the levels of ssp mRNA and sarA mRNA was observed, while there was no obvious correlation between levels of protease production and RNAIII (Fig. 2), suggesting that the variation in protease production between strains might be due to different levels of sarA activity. sarA is transcribed from three promoters, P1, P2, and P3 (5). P1 and P2 are ordinary sigma factor A (SigA)-dependent promoters, which are expressed mainly during the early exponential phase of growth, while P3 is sigma factor B (SigB) dependent and is expressed during the postexponential and stationary phases of growth (5, 17, 34). Interestingly, the major sarA mRNA in strains KS7, KS30, KS33, and KS36 was the P3 transcript, while the P1 transcript dominated in KS26 (Fig. 2), suggesting reduced sigB activity in the latter strain. This was also supported by the observation that transcription of the alkaline shock protein gene asp23, which is activated by SigB (20, 22, 30, 36), was severely suppressed in KS26 compared to KS7 (data not shown). In addition, pigmentation, which is also sigB dependent (30), was reduced in KS26 compared to that in the protease-negative strains (data not shown). Since sigB and sarA have been reported to regulate the synthesis of several other secreted proteins in addition to the proteases (19, 21, 51), we analyzed the extracellular-protein patterns of protease-positive and protease-negative S. aureus strains by SDS-PAGE. Very different protein patterns were produced (Fig. 4), supporting the hypothesis that these S. aureus strains expressed different levels of the global exoprotein regulators sigB and sarA.
FIG. 4.

SDS-PAGE analysis of total extracellular proteins from equal numbers of S. aureus cells harvested at the indicated time points. Molecular masses are indicated on the left.
Inactivation and overexpression of sarA in clinical strains.
To test the hypothesis that the variation in protease production between clinical strains was due to different levels of sarA activity, the sarA knockout mutation from strain PC1839 was transferred to the protease-negative strains KS7, KS30, and KS33. The resulting sarA mutants (NA1 through NA3) produced large zones of proteolysis on casein agar, which were similar in size to that of the clinical isolate with the highest protease production (Fig. 5). The zone of precipitation produced by strain NA2 had a less dense appearance, which is consistent with the lack of V8 protease (see below). Except for the sspAB mRNA in strain NA2, levels of protease-specific mRNAs were significantly higher in the sarA mutants than in their corresponding wild-type strains (Fig. 6). Notably, transcription of the protease genes increased in the sarA mutants, in spite of decreased RNAIII levels. These results indicate that all major protease genes, except for sspAB in strain NA2 (see below), were functionally intact in the protease-negative clinical strains and that protease production was repressed due to high levels of sarA expression. The complete lack of aur mRNA in KS7, KS30, and KS33 indicates that the aureolysin gene is the most sensitive to repression by sarA. As seen in Fig. 6, inactivation of sarA did not result in constitutive expression of the protease genes. Maximum transcription of ssp and aur occurred during the postexponential phase of growth (4 h), while expression of scp peaked during the exponential phase of growth, indicating that the protease genes are independently regulated in a growth phase-dependent manner. To test the hypothesis that the high level of protease production in strain KS26 was due to a low level of sarA, we introduced sarA under the control of the inducible xylA promoter in strain KS26. Induction of sarA transcription with xylose (Fig. 7B) completely repressed protease production (Fig. 7A). Since the level of RNAIII was unaffected (data not shown), the decrease in protease production was most likely a direct effect of increased sarA transcription.
FIG. 5.

Protease production by the clinical S. aureus strains KS7, KS30, and KS33 and their corresponding sarA mutants (NA1, NA2, and NA3) grown on a casein agar plate. The protease-positive strains KS26 and KS36 were included as controls.
FIG. 7.
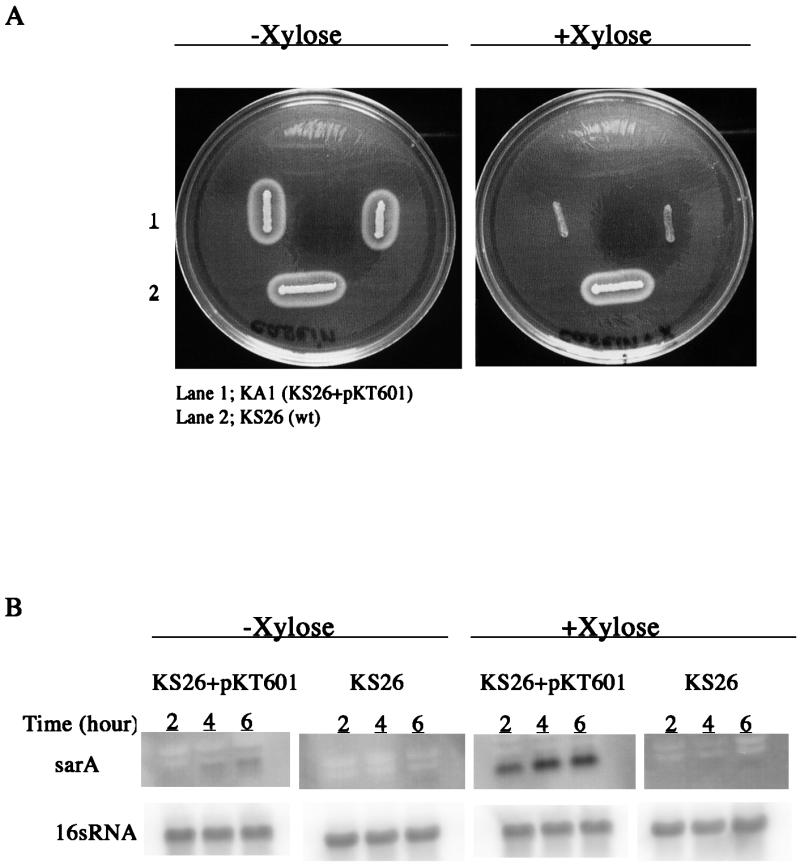
(A) Effect of induction of sarA expression in strain KA1, containing plasmid pKT601, on protease production analyzed on a casein agar plate. (B) Northern blot analysis of sarA transcription after induction with xylose in liquid culture.
Analysis of the ssp promoter in strain KS30.
To explain the lack of ssp transcription in the sarA mutant derived from strain KS30, we decided to study the ssp promoter of this strain in more detail. Nucleotide sequencing of the ssp promoter region in strain KS30 and 10 other protease-negative strains revealed a G at position 4 of the −35 promoter element (TTGGCT) in strain KS30 as opposed to an A (TTGACT) in all other strains, including four published sequences (The Institute for Genomic Research and University of Oklahoma websites [see Materials and Methods]) from protease-positive strains. This suggests that the point mutation in the −35 box of strain KS30 might be responsible for the lack of ssp transcription. However, minor differences in the nucleotide sequences upstream of the −35 box could also be responsible for the lack of ssp expression in strain KS30. To test this, ssp promoter DNA fragments (−312 to +14) from strain KS30 with either a G or an A in the −35 box were fused to a promoterless lacZ gene. These constructs were integrated as single copies into the lipase gene (geh) of the protease-positive prototype S. aureus strain 8325-4, and transcription of lacZ was analyzed. The corresponding ssp promoter fragment of strain 8325-4 was used as a control (50). As shown in Fig. 8, the original ssp promoter of KS30 was inactive, while the mutated promoter was almost as active as that of the control. These results suggest that the mutation in the −35 box of the ssp promoter in strain KS30 was responsible for the lack of ssp transcription and that nucleotide differences upstream of −35 were less important.
FIG. 8.

Northern blot analysis of lacZ in S. aureus 8325-4 containing different ssp promoter elements (nt −312 to +14) fused to lacZ as a single copy in the chromosomal lipase gene, geh. Samples were taken after 4 h of growth, and equal amounts of RNA were analyzed for each sample. Strain KT111 has the ssp promoter element from strain 8325-4 (−35 box, TTGACT), KS30.1 has the ssp promoter element from strain KS30 (−35 box, TTGGCT), and KS30.2 has the ssp promoter element from strain KS30 with a G-to-A substitution at position 4 in the −35 box (TTGACT).
DISCUSSION
In the present study we found that production of extracellular proteases varied considerably among clinical isolates of S. aureus. The presence of the major protease genes, sspA, sspB, aur, and scp, in all the protease-negative strains analyzed suggested that the lack of protease production was due to some regulating host cell factor.
Analysis of the protease regulators agr (an activator) and sarA (a repressor) in protease-negative and protease-positive strains indicated that protease production in vitro was primarily determined by the level of sarA expression. This was strongly supported by the demonstration that inactivation of sarA in three different protease-negative strains resulted in increased transcription of the protease genes, while overexpression of sarA in a protease-positive strain completely inhibited protease production. These experiments also show that the protease genes were functionally intact in the clinical S. aureus strains and responded normally to repression by sarA.
Although expression of all major protease genes was down-regulated in the protease-negative strains, transcription of aur seemed to be the most sensitive to repression by sarA (Fig. 6). Since aureolysin is required for activation of the staphylococcal serine protease, SspA (18), down-regulation of aur would lead to the loss of both aureolysin and serine protease activity. We have previously shown that inactivation of the aureolysin gene in the sarA mutant PC1839, which produces large amounts of all major proteases, resulted in a protease-negative phenotype (27). Accordingly, lack of aureolysin expression would explain the protease-negative phenotype of strain KS7 in spite of the fact that it has relatively high levels of ssp mRNA. The identification of nonprocessed serine protease in culture supernatants of KS7 supported this explanation.
The high sarA expression in protease-negative strains was due to significant transcription of sarA from its SigB-dependent promoter, P3, suggesting that protease-negative strains had higher SigB activity than protease-positive strains. This was supported by the observation that other SigB-dependent traits, such as expression of asp23 and pigmentation (20, 22, 30, 36), were also higher in protease-negative strains. Since protease production was repressed in the majority of clinical S. aureus strains one might assume that high SigB activity, and therefore high sarA activity, is the normal phenotype under in vitro growth conditions. Strains with high protease production seemed to have low SigB activity. In the protease-positive S. aureus strain 8325, SigB-dependent expression of sarA is decreased because of a deletion in the SigB activator gene rsbU (22, 30, 41). Whether the protease-positive clinical isolates have a similar SigB defect remains to be determined.
Interestingly, there was no correlation between protease production and the level of the activator RNAIII (agr) in the clinical strains, indicating that the negative effect of sarA on protease production is dominant over the stimulating effect of RNAIII. This was also supported by the observation that transcription of protease genes increased in the sarA mutants, in spite of reduced RNAIII levels (Fig. 6). The observed reduction of RNAIII levels in sarA mutants is consistent with previous reports suggesting that sarA is an activator of agr transcription (10, 15, 19, 23). However, on the other hand, overexpression of sarA in the protease-positive clinical strain KS26 had no significant effect on RNAIII production (data not shown), suggesting that other host cell factors modulate the regulation of RNAIII expression. Bischoff et al. (6) found that induction of sigB increased the expression of sarA but decreased the level of RNAIII. They suggested that SigB induced a repressor with a dominating effect over the sarA-dependent activation of agr transcription. An alternative explanation would be that SarA at very high concentrations acts as a repressor of agr. The function of SarA as a repressor has been demonstrated in an in vitro transcription system with S. aureus RNA polymerase (13).
All protease genes in the clinical S. aureus strains investigated seemed to be intact and reacted normally to SarA except for ssp in KS30, which was not up-regulated in response to inactivation of sarA. This was shown to be due to an A-to-G substitution at position 4 in the −35 hexanucleotide box (TTGACT) of the ssp promoter. The same base substitution in the Salmonella ant promoter resulted in a 30-fold reduction in promoter activity (38). As the ssp operon was silent in KS30, the zone of proteolysis produced by the sarA mutant NA2 is most likely due to staphopain and/or aureolysin. Together with the observation that an aureolysin mutant was protease negative on casein agar (27), this suggests that staphopain is activated by aureolysin.
Our results show that expression of the major protease genes is down-regulated in most clinical S. aureus strains under in vitro cultivation conditions due to high SigB-dependent sarA activity. Considering the potential role of the extracellular proteases in staphylococcal virulence, it must be assumed that they are produced at some point during infection. This means that SigB activity must be down-regulated under in vivo growth conditions or that an activator that has a dominating effect over the SarA-dependent repression of protease production must be produced. The same type of regulation would be needed for the in vivo expression of other virulence factors that are repressed by sarA and/or SigB, e.g., lipase, staphylokinase, alpha- and beta-hemolysins, leukotoxin, and collagen-binding protein D (9, 51). On the other hand, a number of virulence factors are activated by SigB and/or sarA (19, 51), which means that the bacteria must still be able to increase the levels of sigB and sarA expression during infection. Therefore, the ability to regulate the expression of virulence factors in a proper way is probably much more important than the virulence factor profile expressed in vitro. However, it remains to be proven whether protease-negative and protease-positive clinical S. aureus strains are equally virulent, although they were isolated from apparently similar type of infections.
Acknowledgments
We thank Lena Norenius and Agneta Wahlquist for skillful technical assistance. We are also grateful to the Department of Clinical Microbiology at Karolinska Hospital for providing the clinical S. aureus strains.
This work was supported by grant 4513 from the Swedish Medical Research Council.
Editor: E. I. Tuomanen
REFERENCES
- 1.Arvidson, A. 1973. Hydrolysis of casein by three extracellular proteolytic enzymes from Staphylococcus aureus, strain V8. Acta Pathol. Microbiol. Scand. 81:538-544. [DOI] [PubMed] [Google Scholar]
- 2.Arvidson, S. 2000. Extracellular enzymes, p. 379-385. In V. A. Fischetti, R. P. Novick, J. J. Ferretti, D. A. Portnoy, and J. I. Rood (ed.), Gram-positive pathogens. ASM Press, Washington, D.C.
- 3.Arvidson, S., and K. Tegmark. 2001. Regulation of virulence determinants in Staphylococcus aureus. Int. J. Med. Microbiol. 291:159-170. [DOI] [PubMed] [Google Scholar]
- 4.Ausubel, F. M., R. Brent, R. F. Kingston, D. D. Moore, J. G. Seidmann, J. A. Smith, and K. Struhl. 1987. Current protocols in molecular biology. John Wiley & Sons, Inc., New York, N.Y.
- 5.Bayer, M. G., J. H. Heinrichs, and A. L. Cheung. 1996. The molecular architecture of the sar locus in Staphylococcus aureus. J. Bacteriol. 178:4563-4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bischoff, M., J. M. Entenza, and P. Giachino. 2001. Influence of a functional sigB operon on the global regulators sar and agr in Staphylococcus aureus. J. Bacteriol. 183:5171-5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Björklind, A., and S. Arvidson. 1980. Mutants of Staphylococcus aureus affected in the regulation of exoprotein synthesis. FEMS Microbiol. Lett. 7:202-206. [Google Scholar]
- 8.Björklind, A., and S. Arvidson. 1977. Occurrence of an extracellular serine proteinase among Staphylococcus aureus strains. Acta Pathol. Microbiol. Scand. 85:277-280. [DOI] [PubMed] [Google Scholar]
- 9.Blevins, J., A. Gillaspy, T. Rechtin, B. Hurlburt, and M. Smeltzer. 1999. The staphylococcal accessory regulator (sar) represses transcription of the Staphylococcus aureus collagen adhesin gene (cna) in an agr-independent manner. Mol. Microbiol. 33:317-326. [DOI] [PubMed] [Google Scholar]
- 10.Blevins, J. S., K. E. Beenken, M. O. Elasri, B. K. Hurlburt, and M. S. Smeltzer. 2002. Strain-dependent differences in the regulatory roles of sarA and agr in Staphylococcus aureus. Infect. Immun. 70:470-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Booth, M. C., L. M. Pence, P. Mahasreshti, M. C. Callegan, and M. S. Gilmore. 2001. Clonal associations among Staphylococcus aureus isolates from various sites of infection. Infect. Immun. 69:345-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carmona, C., and G. L. Gray. 1987. Nucleotide sequence of the serine protease gene of Staphylococcus aureus, strain V8. Nucleic Acids Res. 15:6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chakrabarti, S., and T. Misra. 2000. SarA represses agr operon expression in a purified in vitro Staphylococcus aureus transcription system. J. Bacteriol. 182:5893-5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chan, P. F., and S. J. Foster. 1998. Role of SarA in virulence determinant production and environmental signal transduction in Staphylococcus aureus. J. Bacteriol. 180:6232-6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung, A. L., M. G. Bayer, and J. H. Heinrichs. 1997. sar genetic determinants necessary for transcription of RNAII and RNAIII in agr locus of Staphylococcus aureus. J. Bacteriol. 179:3963-3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung, A. L., K. Eberhardt, and J. H. Heinrichs. 1997. Regulation of protein A synthesis by the sar and agr loci of Staphylococcus aureus. Infect. Immun. 65:2243-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deora, R., T. Tseng, and T. K. Misra. 1997. Alternative transcription factor σB of Staphylococcus aureus: characterization and role in transcription of the global regulatory locus sar. J. Bacteriol. 179:6355-6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drapeau, G. R. 1978. Role of metalloprotease in activation of the precursor of staphylococcal protease. J. Bacteriol. 136:607-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunman, P. M., E. Murphy, S. Haney, D. Palacios, G. Tucker-Kellogg, S. Wu, E. L. Brown, R. J. Zagursky, D. Shalaes, and S. J. Projan. 2001. Transcription profiling-based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J. Bacteriol. 183:7341-7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gertz, S., S. Engelmann, R. Schmid, K. Ohlsen, J. Hacker, and M. Hecker. 1999. Regulation of σB-dependent transcription of sigB and asp23 in two different Staphylococcus aureus strains. Mol. Gen. Genet. 261:558-566. [DOI] [PubMed] [Google Scholar]
- 21.Gertz, S., S. Engelmann, R. Schmid, A. Ziebandt, K. Tischer, C. Scharf, J. Hacker, and M. Hecker. 2000. Characterization of the σB regulon in Staphylococcus aureus. J. Bacteriol. 182:6983-6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giachino, P., S. Engelmann, and M. Bischoff. 2001. σB activity depends on RsbU in Staphylococcus aureus. J. Bacteriol. 183:1843-1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heinrichs, J. H., M. G. Bayer, and A. L. Cheung. 1996. Characterization of the sar locus and its interaction with agr in Staphylococcus aureus. J. Bacteriol. 178:418-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iandolo, J. J. 1989. Genetic analysis of extracellular toxins of Staphylococcus aureus. Annu. Rev. Microbiol. 43:375-402. [DOI] [PubMed] [Google Scholar]
- 25.Janzon, L., and S. Arvidson. 1990. The role of the δ-lysin gene (hld) in regulation of virulence genes by the accessory gene regulator (agr) in Staphylococcus aureus. EMBO J. 9:1391-1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janzon, L., S. Löfdahl, and S. Arvidson. 1986. Evidence for a coordinate transcriptional control of α-toxin and protein A in Staphylococcus aureus. FEMS Microbiol. Lett. 33:193-198. [Google Scholar]
- 27.Karlsson, A., P. Saravia-Otten, K. Tegmark, E. Morfeldt, and S. Arvidson. 2001. Decreased amounts of cell wall-associated protein A and fibronectin-binding proteins in Staphylococcus aureus sarA mutants due to up-regulation of extracellular proteases. Infect. Immun. 69:4742-4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kornblum, J., B. N. Kreiswirth, S. J. Projan, H. Ross, and R. P. Novick. 1990. Agr: a polycistronic locus regulating exoprotein synthesis in Staphylococcus aureus, p. 373-402. In R. P. Novick (ed.), Molecular biology of the staphylococci. VCH Publishers, New York, N.Y.
- 29.Kreiswirth, B., S. Löfdahl, M. J. Betley, M. O'Reilly, M. P. Schleivert, M. S. Bergdoll, and R. P. Novick. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709-712. [DOI] [PubMed] [Google Scholar]
- 30.Kullik, I., P. Giachino, and T. Fuchs. 1998. Deletion of the alternative sigma factor σB in Staphylococcus aureus reveals its function as a global regulator of virulence genes. J. Bacteriol. 180:4814-4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee, C., S. Buranen, and Z. Ye. 1991. Construction of single-copy integration vectors for Staphylococcus aureus. Gene 103:101-105. [DOI] [PubMed] [Google Scholar]
- 32.Lindsay, J., and S. Foster. 1999. Interactive regulatory pathways control virulence determinant production and stability in response to environmental conditions in Staphylococcus aureus. Mol. Gen Genet. 262:323-331. [DOI] [PubMed] [Google Scholar]
- 33.Lowy, F. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339:520-532. [DOI] [PubMed] [Google Scholar]
- 34.Manna, A. C., M. G. Bayer, and A. L. Cheung. 1998. Transcriptional analysis of different promoters in the sar locus in Staphylococcus aureus. J. Bacteriol. 180:3828-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McGavin, M. J., C. Zahradka, R. Kelly, and J. E. Scott. 1997. Modification of the Staphylococcus aureus fibronectin binding phenotype by V8 protease. Infect. Immun. 65:2621-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyazaki, E., J. M. Chen, C. Ko, and W. R. Bishai. 1999. The Staphylococcus aureus rsbW (orf159) gene encodes an anti-sigma factor of SigB. J. Bacteriol. 181:2846-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morfeldt, E., L. Janzon, S. Arvidson, and S. Lofdahl. 1988. Cloning of a chromosomal locus (exp) which regulates the expression of several exoprotein genes in Staphylococcus aureus. Mol. Gen. Genet. 211:435-440. [DOI] [PubMed] [Google Scholar]
- 38.Moyle, H., C. Waldburger, and M. M. Susskind. 1991. Hierarchies of base pair preferences in the P22 ant promoter. J. Bacteriol. 173:1944-1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Novick, R. P. 1991. Genetic systems in staphylococci. Methods Enzymol. 204:587-636. [DOI] [PubMed] [Google Scholar]
- 40.Novick, R. P. 1967. Properties of a cryptic high-frequency transducing phage in Staphylococcus aureus. Virology 33:155-166. [DOI] [PubMed] [Google Scholar]
- 41.Palma, M., and A. L. Cheung. 2001. σB activity in Staphylococcus aureus is controlled by RsbU and an additional factor(s) during bacterial growth. Infect. Immun. 69:7858-7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Potempa, J., A. Dubin, G. Korzus, and J. Travis. 1988. Degradation of elastin by a cysteine proteinase from Staphylococcus aureus. J. Biol. Chem. 263:2664-2667. [PubMed] [Google Scholar]
- 43.Potempa, J., D. Fedak, A. Dubin, A. Mast, and J. Travis. 1991. Proteolytic inactivation of α1-anti-chymotrysin. Sites of cleavage and generation of chemotactic activity. J. Biol. Chem. 266:21482-21487. [PubMed] [Google Scholar]
- 44.Potempa, J., W. Watorek, and J. Travis. 1986. The inactivation of human α1-proteinase inhibitor by proteinases from Staphylococcus aureus. J. Biol. Chem. 261:14330-14334. [PubMed] [Google Scholar]
- 45.Prokesova, L., B. Potuznikova, J. Potempa, J. Zikan, J. Radl, L. Hachova, K. Baran, Z. Porwit-Bobr, and C. John. 1992. Cleavage of human immunoglobulins by serine proteinase from Staphylococcus aureus. Immunol. Lett. 31:259-265. [DOI] [PubMed] [Google Scholar]
- 46.Rice, K., M. Huesca, D. Vaz, and M. J. McGavin. 2001. Variance in fibronectin binding and fnb locus polymorphisms in Staphylococcus aureus: identification of antigenic variation in a fibronectin binding protein adhesin of the epidemic CMRSA-1 strain of methicillin-resistant S. aureus. Infect. Immun. 69:3791-3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rice, K., R. Peralta, D. Bast, J. de Azavedo, and M. J. McGavin. 2001. Description of staphylococcus serine protease (ssp) operon in Staphylococcus aureus and nonpolar inactivation of sspA-encoded serine protease. Infect. Immun. 69:159-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schenk, S., and R. A. Laddaga. 1992. Improved methods for electroporation of Staphylococcus aureus. FEMS Microbiol. Lett. 94:133-138. [DOI] [PubMed] [Google Scholar]
- 49.Tegmark, K., A. Karlsson, and S. Arvidson. 2000. Identification and characterization of SarH1, a new global regulator of virulence gene expression in Staphylococcus aureus. Mol. Microbiol. 37:398-409. [DOI] [PubMed] [Google Scholar]
- 50.Tegmark, K. 2000. The virulence gene regulator, SarA in Staphylococcus aureus, appears to be a non-specific DNA binding protein. Ph.D. thesis. Karolinska Institute, Stockholm, Sweden.
- 51.Ziebandt, A., H. Weber, J. Rudolph, R. Schmid, D. Höper, S. Engelmann, and M. Hecker. 2001. Extracellular proteins of Staphylococcus aureus and the role of SarA and σB. Proteomics 1:480-493. [DOI] [PubMed] [Google Scholar]