

Abstract
Apical membrane antigen 1 (AMA-1) is a highly promising malaria blood-stage vaccine candidate that has induced protection in rodent and nonhuman primate models of malaria. Authentic conformation of the protein appears to be essential for the induction of parasite-inhibitory antibody responses. Here we have developed a synthetic gene with adapted codon usage to allow expression of Plasmodium falciparum FVO strain AMA-1 (PfAMA-1) in Pichia pastoris. In addition, potential N-glycosylation sites were changed, exploiting the lack of conservation of these sites in Plasmodium, to obtain high-level secretion of a homogeneous product, suitable for scale-up according to current good manufacturing procedures. Purified PfAMA-1 displayed authentic antigenic properties, indicating that the amino acid changes had no deleterious effect on the conformation of the protein. High-titer antibodies, raised in rabbits, reacted strongly with homologous and heterologous P. falciparum by immunofluorescence. In addition, purified immunoglobulin G from immunized animals strongly inhibited invasion of red blood cells by homologous and, to a somewhat lesser extent, heterologous P. falciparum.
Accumulated data, including those from nonhuman primate (2, 5) and rodent (1, 3, 16) studies, have indicated that the apical membrane antigen 1 (AMA-1) family of molecules are targets for antibody-mediated protective immune responses. In all Plasmodium species reported to date, with the exception of Plasmodium falciparum (19) and P. reichenowi (13) (two parasites that form a phylogenetic clade distinct from other malaria parasites), AMA-1 is synthesized de novo as a 66-kDa transmembrane protein. The protein contains a predicted N-terminal signal sequence, an ectodomain, a predicted transmembrane region, and a C-terminal cytoplasmic domain. The ectodomain is further divided into three domains defined by disulfide bonds (10). In P. falciparum and P. reichenowi the protein is expressed as an 83-kDa protein, having an N-terminal extension compared to the 66-kDa forms that has been referred to as the prosequence (10). AMA-1 is processed by proteolytic cleavage between the different domains (11). Intraspecies sequence polymorphism due to point mutations (13, 15, 18, 23) reveals clustering of mutations in particular domains of the molecule. Despite this, between species there is considerable conservation of primary and predicted secondary amino acid structures. Evidence to date indicates that protection invoked by AMA-1 is directed at epitopes dependent on the disulfide bonding (1-3, 6, 9, 16) located in the AMA-1 ectodomain. Immunization with reduced AMA-1 fails to induce parasite-inhibitory antibodies (1, 6, 9), and so far only those monoclonal antibodies (MAbs) that recognize reduction-sensitive AMA-1 epitopes have been shown elsewhere to inhibit parasite multiplication in vitro for P. knowlesi (4, 21) and P. falciparum (13, 14). This indicates that for an AMA-1 vaccine the correct conformation will be critical.
Recombinant expression of P. falciparum AMA-1 (PfAMA-1) in a conformationally relevant way that allows production of clinical-grade material has been notoriously difficult. Expression of the PfAMA-1 ectodomain in Escherichia coli followed by a refolding protocol has been successful (9), but scaling up this process has proven problematic. We have previously obtained high-level expression of conformationally relevant P. vivax AMA-1 (PvAMA-1) ectodomain in the methylotrophic yeast Pichia pastoris (12). Initial attempts to produce PfAMA-1 ectodomain by the same system were unsuccessful, due to premature transcription stops evoked by A+T-rich stretches within the gene (C. H. M. Kocken and A. W. Thomas, unpublished data). We therefore opted for the generation of a complete synthetic gene utilizing P. pastoris codon usage. A second problem for expression in eukaryotic systems is N glycosylation. PfAMA-1 contains six potential N-glycosylation sites but is not N glycosylated by the parasite (11). Secreted expression of PvAMA-1 ectodomain in Pichia showed heterogeneous hyperglycosylation of the recombinant product (12). We therefore developed a variant PfAMA-1 sequence that exploited the lack of conservation of N-glycosylation sites in Plasmodium AMA-1, as we successfully did for PvAMA-1 (12). In this study we show that the synthetic PfAMA-1 ectodomain is efficiently secreted from recombinant P. pastoris. In addition we show that the recombinant protein, despite deliberate changes to the primary structure, attains its natural conformation by demonstrating the authentic antigenic character and that antibodies induced by immunization with the protein are potent inhibitors of P. falciparum growth in vitro.
MATERIALS AND METHODS
Parasites.
Cryopreserved parasite stocks from P. falciparum strain FVO (a kind gift from S. Herrera, Cali, Colombia) were prepared from an infected Aotus lemurinus griseimembra monkey at the young ring stage of development. P. falciparum strains NF54 and FCR3 were cultured in vitro by standard P. falciparum culture techniques (24) in an atmosphere of 5% CO2, 5% O2, and 90% N2. FCR3 AMA-1 (accession no. M34553) differs by one amino acid in the prosequence from FVO AMA-1 (sequence determined in this study), while NF54 AMA-1 (accession no. for the 3D7 clone of NF54 is U33274) differs at 29 amino acid positions from the FVO sequence.
Development of a synthetic gene for P. falciparum FVO strain ama-1.
P. falciparum FVO strain DNA was isolated (Gentra Systems Inc., Minneapolis, Minn.) directly from a parasite stock according to the manufacturer's instructions. Pfama-1 was amplified by PCR with Pfu polymerase (Stratagene, Amsterdam, The Netherlands) and primers PF83A (5′-GGGGGATCCATGAGAAAATTATACTGCGTATT-3′; nucleotides [nt] 1 to 23 and additional BamHI restriction site) and PF83B (5′-ACGTGGATCCTTAATAGTATGGTTTTTCCATCAGAACTGG-3′; complementary to nt 1843 to 1869 and additional BamHI restriction site) containing BamHI restriction sites to facilitate cloning in pBluescript. A pool of four independent clones was used for sequence analysis with the ABI Prism 310 automated sequencer (PE Applied Biosystems, Foster City, Calif.) according to the manufacturer's instructions and with primers previously synthesized for sequencing of Pfama-1 (23).
The FVO ama-1 nucleotide sequence (accession no. AJ277646) was used to develop a synthetic gene utilizing the codon usage of P. pastoris with the aid of the CODOP program as described previously (25). Briefly, 92 40-mer oligonucleotides were prepared from both DNA strands with a 20-nt overlap between primers from both strands. Gene synthesis was performed by assembly PCR with Pfu polymerase, and blunt-ended products corresponding to each half of the gene were cloned into pMOSBlue (Amersham Pharmacia, Little Chalfont, Buckinghamshire, United Kingdom) and fully sequenced before subcloning to produce the complete synthetic gene FVO ama-1syn. In designing the synthetic gene the six potential N-glycosylation sites were changed to prevent unwanted glycosylation (162 N→K, 288 T→V, 373 S→D, 422 N→S, 423 S→K, and 499 N→E) by using substituent amino acids from other available AMA-1 sequences of malaria parasites (12).
Expression of FVO AMA-1 ectodomain in P. pastoris.
For secreted, methanol-inducible expression in P. pastoris strain KM71H (Muts phenotype) vector pPICZαA (Invitrogen, Groningen, The Netherlands) was used. Primers for PCR amplification of the Pfama-1 ectodomain were Pf83A (5′-GGAATTCCAGAACTACTGGGAGCATCC-3′; nt 73 to 92 and additional EcoRI restriction site) and Pf83H (5′-GCTCTAGAATGTTATCGTACGTAGGCTT-3′; complementary to nt 1615 to 1634 and additional XbaI restriction site). A 50-μl PCR mixture contained 10 ng of template DNA, 100 ng of each of the primers Pf83A and Pf83H, 0.2 mM deoxynucleoside triphosphates, 5 μl of 10× Pfu reaction buffer, and 1 U of Pfu polymerase. Amplification proceeded as follows: 1 min at 94°C, 1 min at 52°C, and 1.5 min at 72°C for 3 cycles; 1 min at 94°C, 1 min at 60°C, and 1.5 min at 72°C for 30 cycles; 5 min at 72°C; and then storage at 4°C. The resulting 1,578-bp PCR product was sequentially digested with EcoRI and XbaI, ligated into EcoRI/XbaI-digested pPICZαA, and transformed into E. coli DH5α. Plasmids from resulting colonies were isolated by standard miniprep methods (20) and analyzed by restriction enzyme digestion. One clone containing the correct insertion was used to isolate plasmid DNA for transformation of P. pastoris.
The expression construct was linearized with SstI, and 10 μg of DNA was used to transform 80 μl of P. pastoris KM71H cells by electroporation according to the Invitrogen protocols. One milliliter of 1 M sorbitol was added, and the cells were allowed to recover for 2 h at 30°C. Cells were plated on YPDS (1% yeast extract, 2% peptone, 2% dextrose, 1 M sorbitol) agar plates containing 100 μg of zeocin/ml and incubated for 4 days at 30°C. Colonies were picked and grown for 48 h at 30°C in 10 ml of BMGY (1% yeast extract, 2% peptone, 1.34% yeast nitrogen base, 1% glycerol, 0.4 mg of biotin/liter, 0.1 M K-phosphate, pH 6.0) in 50-ml Falcon tubes with vigorous shaking. Cells were harvested by low-speed centrifugation, resuspended in 4 ml of BMMY (BMGY with glycerol substituted for 0.5% methanol), and cultured for an additional 48 h. Cells were harvested, and the culture supernatants and pelleted yeast cells were analyzed for the presence of the PfAMA-1 ectodomain by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), Western blotting, and Coomassie brilliant blue staining as described previously (12). A positive clone was selected and designated Pf4mH.
Antigenicity of PfAMA-1 ectodomain.
Culture supernatants (4 μl) were spot blotted onto nitrocellulose membranes and incubated with rat MAb 58F8 (recognizing a linear epitope in the N-terminal region of PfAMA-1 [17]) or 4G2 (recognizing a conformational epitope in the ectodomain and capable of blocking parasite multiplication in vitro [14]) for 1 h at room temperature. After incubation with goat anti-rat immunoglobulin G (IgG), color was developed with nitroblue tetrazolium-5-bromo-4-chloro-3-indolylphosphate. Alternatively, culture supernatants or purified protein were subjected to SDS-PAGE under nonreducing conditions and then analyzed by Western blotting with MAb 4G2.
The antigenicity of Pf4mH was further evaluated by enzyme-linked immunosorbent assay (ELISA) with PfAMA-1-specific MAbs 4G2, 58F8, and 28G2 (recognizing a linear epitope at the C terminus of PfAMA-1 [17]) and human serum from Guinea-Bissau, where malaria is endemic (22). A quantitative estimate of reactivity of Pf4mH with MAb 4G2 was also obtained by binding purified protein to the antibody immobilized on Sepharose (11). Protein was diluted into phosphate-buffered saline, pH 7.2, containing 0.05% (vol/vol) Triton X-100 (PBS/T) to a concentration of approximately 100 μg ml−1, and 100 μl was incubated with 50 μl of 4G2-Sepharose on ice for 1 h with mixing. The beads were then washed five times with PBS/T, and bound protein was eluted with 100 μl of 0.2 M glycine-HCl-0.15 M NaCl, pH 2.7 (11). The degree of protein binding and elution was assessed by SDS-PAGE and Coomassie blue staining.
ELISA and IFA.
ELISA was performed in triplicate on serum samples in 96-well flat-bottomed microtiter plates (Greiner, Alphen a/d Rijn, The Netherlands), coated with 100 ng of purified Pf4mH/ml according to published methods (8). As a control, plates were coated with identically purified material from a P. pastoris clone that was transformed with the empty expression vector pPICZαC, to be able to measure responses against impurities in the Pf4mH preparation. Secondary antibodies were goat anti-rat, anti-human, or anti-rabbit IgG conjugated to alkaline phosphatase (Pierce, Rockford, Ill.). Cutoff values to determine endpoint titers are the averages of preimmune sera plus three times the standard deviation. Indirect immunofluorescence assay (IFA) was performed as previously described (4) with schizont-infected red cells isolated from in vitro culture (P. falciparum NF54 or FCR3). Secondary antibodies were fluorescein isothiocyanate-conjugated goat anti-rabbit IgG (H & L) (Kirkegaard & Perry Laboratories, Gaithersburg, Md.).
Protein production.
For mid-scale production of Pf4mH recombinant P. pastoris was cultured in 1-liter baffled flasks (400 ml of BMGY per flask) for 48 h at 29 to 30°C under vigorous shaking. Cells were harvested, resuspended in 100 ml of BMMY, and then cultured for 48 h at 29 to 30°C under vigorous shaking. Methanol was added (0.5% [vol/vol]) every 24 h. After low-speed centrifugation, the culture supernatant was harvested. Protein was precipitated with ammonium sulfate (70% final concentration) at 0°C, and the precipitate was stored at 4°C until use.
Protein purification.
The ammonium sulfate precipitate from 50 ml of culture supernatant was solubilized in 2 ml of binding buffer (20 mM Na-phosphate [pH 7.8], 0.5 M NaCl, 5 mM imidazole) and loaded on an 8-ml Ni-nitrilotriacetic acid Superflow column (Qiagen, Crawley, United Kingdom) at 0.2 ml/min. The column was washed at 1 ml/min with 25 ml of binding buffer and 20 ml of the same buffer (pH 5.5) and then eluted with a 20-ml linear gradient of 5 to 500 mM imidazole in 20 mM Na-phosphate (pH 6.0)-0.5 M NaCl. Elution was monitored at 280 nm, and peak fractions were analyzed by SDS-PAGE. Protein concentrations were measured with a protein assay kit (Pierce).
Rabbit immunization.
Rabbit polyclonal antisera were prepared (Eurogentec S.A., Seraing, Belgium) by injection of 100 μg of purified Pf4mH (rabbits 1, 2, and 3) with Freund's complete adjuvant, followed by three booster injections of 100 μg at days 14, 28, and 56 with Freund's incomplete adjuvant. Antisera obtained 4 weeks after the last injection were tested for reactivity by ELISA and IFA. Antibodies were purified on protein A columns (Sigma, St. Louis, Mo.) by standard protocols (8), extensively dialyzed against RPMI 1640, filter sterilized, and stored at +4°C until use in the assays of inhibition of invasion. IgG concentrations were measured with a protein assay kit.
In vitro assay of inhibition of P. falciparum invasion.
The effect of purified antibodies on parasite invasion was evaluated in triplicate with 96-well flat-bottomed plates (Greiner) with in vitro-matured P. falciparum schizonts at a starting parasitemia of 0.5 to 1%, a hematocrit of 3.0%, and a total volume of 150 μl containing 10% normal human serum and 20 μg of gentamicin ml−1 in RPMI 1640. After 17 h, 25 μl of RPMI 1640 containing 10% human serum and [3H]hypoxanthine (ICN Biomedicals Inc., Irvine, Calif.), to yield a final concentration of 40 μCi ml−1, was added to each well. Parasites were harvested 10 h later onto glass fiber filters with a Titertek cell harvester (ICN). Incorporation of [3H]hypoxanthine was determined by liquid scintillation spectrometry. Parasite growth inhibition, reported as a percentage, was determined as follows: 100 − [(average cpmexperimental/average cpmcontrol) × 100]. The incorporation for erythrocytes alone was subtracted from all averages prior to determining the percentage of inhibition. Control IgG was isolated from rabbits that had been immunized with adjuvant only.
Nucleotide sequence accession number.
The GenBank nucleotide sequence accession number for P. falciparum FVO ama-1 is AJ277646.
RESULTS
Pfama-1 sequence from the FVO strain.
The obtained unambiguous FVO ama-1 sequence was compared with the FVO ama-1 sequence available from GenBank (J. C. Aguiar and S. L. Hoffman, unpublished data; accession no. U84348). Ten nucleotide differences resulting in three amino acid changes were observed. Amino acid changes at residues 95 and 473 have been observed earlier during ama-1 sequence analysis of natural P. falciparum infections (13) and are thus at known polymorphic sites. The third amino acid difference, at residue 484, is a deletion that is unique to the FVO sequence accession no. U84348. This Pfama-1 encodes 621 residues whereas all other available PfAMA-1 sequences including the FVO sequence identified here comprise 622 residues.
Recombinant P. pastoris secretes high levels of PfAMA-1 ectodomain.
Our previous experience with expressing Pfama-1 in P. pastoris suggested that the high A+T content of the P. falciparum gene complicates expression in P. pastoris. There are several A+T-rich regions within the coding sequence that are recognized as transcription termination and/or polyadenylation sites in P. pastoris, resulting in truncated mRNAs and no protein production. Site-directed mutagenesis of such sequences resulted in longer, yet not full-length, mRNA and still no protein production (Kocken and Thomas, unpublished). This suggested that a completely synthetic gene with codon usage adapted to P. pastoris could be a successful strategy to obtain expression. The recodon strategy is outlined in Table 1 and incorporates the mutations of the six potential N-glycosylation sites.
TABLE 1.
Adaptation of Pfama-1 codon usage from P. falciparum to Pichia pastorisa
| Amino acid and codon | No. of codons in:
|
Amino acid and codon | No. of codons in:
|
|||
|---|---|---|---|---|---|---|
| P. falciparum | P. pastoris | P. falciparum | P. pastoris | |||
| Ala | ||||||
| GCA | 15 | 6 | ||||
| GCC | 1 | 9 | ||||
| GCG | 0 | 2 | ||||
| GCU | 18 | 17 | ||||
| Total | 34 | 34 | ||||
| Arg | ||||||
| AGA | 19 | 13 | ||||
| AGG | 2 | 5 | ||||
| CGA | 1 | 0 | ||||
| CGC | 0 | 0 | ||||
| CGG | 0 | 0 | ||||
| CGU | 1 | 5 | ||||
| Total | 23 | 23 | ||||
| Asn | ||||||
| AAC | 10 | 50 | ||||
| AAU | 40 | 0 | ||||
| Total | 50 | 50 | ||||
| Asp | ||||||
| GAC | 5 | 17 | ||||
| GAU | 36 | 24 | ||||
| Total | 41 | 41 | ||||
| Cys | ||||||
| UGC | 3 | 7 | ||||
| UGU | 14 | 10 | ||||
| Total | 17 | 17 | ||||
| Gln | ||||||
| CAA | 17 | 11 | ||||
| CAG | 3 | 9 | ||||
| Total | 20 | 20 | ||||
| Glu | ||||||
| GAA | 53 | 37 | ||||
| GAG | 4 | 20 | ||||
| Total | 57 | 57 | ||||
| Gly | ||||||
| GGA | 12 | 9 | ||||
| GGC | 0 | 0 | ||||
| GGG | 4 | 0 | ||||
| GGU | 13 | 20 | ||||
| Total | 29 | 29 | ||||
| His | ||||||
| CAC | 4 | 7 | ||||
| CAU | 15 | 12 | ||||
| Total | 19 | 19 | ||||
| Ile | ||||||
| AUA | 11 | 0 | ||||
| AUC | 3 | 29 | ||||
| AUU | 17 | 2 | ||||
| Total | 31 | 31 | ||||
| Leu | ||||||
| CUA | 0 | 0 | ||||
| CUC | 0 | 8 | ||||
| CUG | 1 | 9 | ||||
| CUU | 5 | 2 | ||||
| UUA | 26 | 0 | ||||
| UUG | 4 | 17 | ||||
| Total | 36 | 36 | ||||
| Lys | ||||||
| AAA | 50 | 1 | ||||
| AAG | 56 | 55 | ||||
| Total | 56 | 56 | ||||
| Met | ||||||
| AUG | 15 | 15 | ||||
| Phe | ||||||
| UUC | 3 | 25 | ||||
| UUU | 22 | 0 | ||||
| Total | 25 | 25 | ||||
| Pro | ||||||
| CCA | 23 | 12 | ||||
| CCC | 3 | 3 | ||||
| CCG | 1 | 0 | ||||
| CCU | 8 | 20 | ||||
| Total | 35 | 35 | ||||
| Ser | ||||||
| AGC | 3 | 0 | ||||
| AGU | 10 | 12 | ||||
| UCA | 19 | 0 | ||||
| UCC | 1 | 10 | ||||
| UCG | 1 | 0 | ||||
| UCU | 1 | 13 | ||||
| Total | 35 | 35 | ||||
| Stop | ||||||
| UAA | 1 | 1 | ||||
| UAG | 0 | 0 | ||||
| UGA | 0 | 0 | ||||
| Total | 1 | 1 | ||||
| Thr | ||||||
| ACA | 13 | 0 | ||||
| ACC | 0 | 5 | ||||
| ACG | 2 | 1 | ||||
| ACU | 9 | 18 | ||||
| Total | 24 | 24 | ||||
| Trp | ||||||
| UGG | 6 | 6 | ||||
| Tyr | ||||||
| UAC | 6 | 39 | ||||
| UAU | 33 | 0 | ||||
| Total | 39 | 39 | ||||
| Val | ||||||
| GUA | 16 | 0 | ||||
| GUC | 3 | 13 | ||||
| GUG | 1 | 7 | ||||
| GUU | 10 | 10 | ||||
| Total | 30 | 30 | ||||
The total number of codons for both species is 623. P. falciparum codons are 70% A/T and 30% C/G; P. pastoris codons are 54% A/T and 46% C/G.
The synthetic Pfama-1 gene was used as a template to amplify the full ectodomain encoding residues 25 to 544, and the PCR product was cloned in frame with the N-terminal signal sequence and C-terminal myc epitope-His tag in pPICZα. One recombinant plasmid preparation was used to transform P. pastoris KM71H to yield Muts transformants that were analyzed for PfAMA-1 ectodomain secretion. All clones analyzed expressed an approximately 75-kDa protein that proved to be the PfAMA-1 ectodomain, designated Pf4mH. The 75-kDa product was efficiently secreted from yeast cells and was reactive by Western blotting with the PfAMA-1-specific, conformation-dependent MAb 4G2 (Fig. 1A). As expected (14) reactivity with MAb 4G2 was lost upon reduction of the protein sample prior to SDS-PAGE (data not shown). Expression levels of Pf4mH in these small-scale cultures were estimated to be 50 mg/liter. Culture supernatant from mid-scale production was ammonium sulfate precipitated, and the precipitate was purified by nickel chelate affinity chromatography. The peak fractions from the imidazole gradient eluate contained the 75-kDa Pf4mH protein as demonstrated by SDS-PAGE analysis (Fig. 1B). Yields from this purification procedure are about 20 mg of purified protein per liter of culture supernatant as measured with a protein assay kit.
FIG. 1.

Analysis of expression of recombinant PfAMA-1 ectodomain in P. pastoris. Cultures were methanol induced for 48 h. (A) Secretion of Pf4mH was analyzed by Western blotting with the conformation-dependent MAb 4G2. One milliliter of the culture was centrifuged to pellet the yeast cells. The cell pellet was disrupted in a total of 1 ml of lysis buffer, and equal amounts of the culture supernatant (lane S) and cell pellet extract (lane P) were subjected to SDS-PAGE under nonreducing conditions and analyzed by Western blotting with MAb 4G2. The majority of the 4G2-reactive protein was secreted into the culture supernatant under these conditions. (B) Ni-agarose-purified Pf4mH from culture supernatant was analyzed by SDS-PAGE. Samples containing 1 μg of purified protein were applied to an SDS-polyacrylamide gel and stained with Coomassie blue. Lane 1, Pf4mH under reducing conditions; lane 2, Pf4mH under nonreducing conditions. Relative molecular masses are indicated in kilodaltons. Note the small increase in relative molecular mass upon reduction, indicative of formation of disulfide bond structures in the recombinant material.
Antigenic properties demonstrate authentic conformation of Pf4mH.
The observed reactivity of Pf4mH with MAb 4G2 (which recognizes a reduction-sensitive AMA-1 epitope) suggested that at least one functional region of AMA-1 (the 4G2 epitope) is recreated in the recombinant molecule and that altering the six N-glycosylation sites did not interfere with the conformation of this region. Antigenicity of Pf4mH was further evaluated by ELISA (Fig. 2). As expected, MAb 28G2, reactive with the extreme C-terminal end of AMA-1, is nonreactive with Pf4mH. The positive control MAb 58F8, reactive with a linear N-terminal epitope, displays strong reactivity. Pf4mH is well recognized by both the conformational, parasite-inhibitory MAb 4G2 and by human serum from a region where malaria is endemic, demonstrating an authentic antigenic profile. Human serum from uninfected European donors does not react with Pf4mH.
FIG. 2.

Antigenicity of Pf4mH was evaluated by ELISA with MAbs 28G2 (reactive with the extreme C terminus of PfAMA-1, not present in Pf4mH), 58F8 (reactive with the N terminus of PfAMA-1), and 4G2 (reactive with a conformational AMA-1 epitope), European control human serum (Hu −ve), and human serum from Guinea-Bissau, where malaria is endemic (Hu +ve). MAbs were incubated at 10 ng ml−1, and sera were diluted 1:1,000. OD, optical density.
MAb 4G2 reactivity was further investigated by a more quantitative analysis. Figure 3 shows that purified Pf4mH could be quantitatively bound to and eluted from 4G2 Sepharose, indicating that essentially every molecule possesses the 4G2 epitope. Minor products with a molecular mass slightly smaller than that of Pf4mH are visible in Fig. 1B and 3 (lanes 1 and 4). These minor products are highly likely to be Pf4mH N-terminal degradation products because (i) they are reactive with antihexahistidine MAb (data not shown), (ii) they copurify with the major 75-kDa band under two independent chromatography procedures (Ni affinity chromatography [Fig. 1B and 3, lane 1] and 4G2 immunoaffinity chromatography [Fig. 3, lane 4]), and (iii) the supernatant from Pichia that had been transformed with the same vector without the PfAMA-1 insert did not reveal similar banding on SDS-PAGE or on Western blotting with rabbit anti-Pf4mH antisera.
FIG. 3.
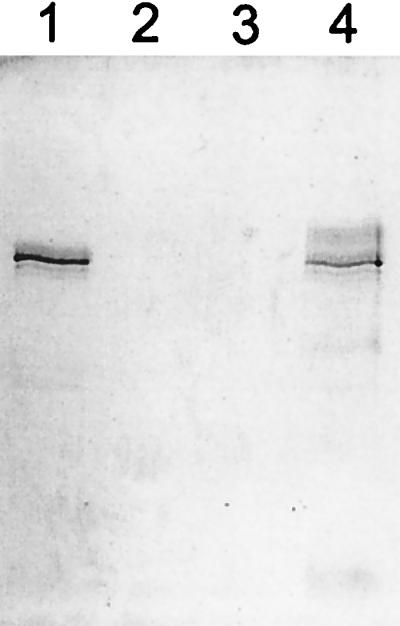
Quantitative recognition of Pf4mH by MAb 4G2. Purified Pf4mH in PBS/T (lane 1) was incubated with MAb 4G2-Sepharose for 1 h. A sample of the protein remaining in solution after this period was taken (lane 2), and then the 4G2 Sepharose was washed extensively and a sample of the final wash was taken (lane 3), prior to elution of bound protein (lane 4). Samples were analyzed by SDS-PAGE and Coomassie blue staining. Essentially all the Pf4mH could be bound to 4G2-Sepharose. The minor additional bands present in lane 4 compared to lane 1 are probably due to antibody leaching from the 4G2-Sepharose under the low-pH conditions used for elution.
Pf4mH induces parasite-inhibitory responses.
Rabbits were immunized with purified Pf4mH to evaluate its capacity to induce parasite-inhibitory antibodies. Pf4mH was highly immunogenic, giving ELISA endpoint titers up to 2.5 × 106 (Table 2). ELISA titers on P. pastoris control culture supernatant were at least 60-fold lower, indicative of the relative purity of the Pf4mH preparation that was used for immunization of the rabbits. More importantly, Pf4mH induces antibodies that react with mature P. falciparum schizonts by IFA (Table 2). IFA titers up to 256,000 were observed on FCR3 parasites, and somewhat lower titers were observed on NF54, which is more divergent in AMA-1 sequence. This difference is also reflected in the assays of inhibition of parasite invasion (Table 2), measuring the capacity of purified IgG at two concentrations to inhibit merozoite invasion of erythrocytes. FCR3 is inhibited to a greater extent than is NF54 in a concentration-dependent manner, indicating the presence of strain-specific as well as common epitopes in Pf4mH.
TABLE 2.
Analysis of anti-Pf4mH responses
| Rabbit no. | ELISA titer with vector:
|
IFA titer for parasite (105)
|
% Inhibition of invasion by parasite measured with purified IgG concn (mg ml−1)
|
|||||
|---|---|---|---|---|---|---|---|---|
| Pf4mH (106) | pPICZα (104) | FCR3 | NF54 | FCR3
|
NF54
|
|||
| 1.5 | 0.75 | 1.5 | 0.75 | |||||
| 1 | 2.5 | 4 | 2.5 | 1.3 | 85 | 55 | 55 | 39 |
| 2 | 2.5 | 4 | 2.5 | 0.6 | 75 | 43 | 58 | 34 |
| 3 | 1.3 | <1 | 1.3 | 0.3 | 50 | 36 | 44 | 20 |
DISCUSSION
Material suitable for current good manufacturing procedures (cGMP) production is required in order to proceed to phase I clinical testing of PfAMA-1 as a malaria blood-stage vaccine. We have developed the P. falciparum FVO strain AMA-1 for this purpose. The AMA-1 sequences from FVO and 3D7 (being developed for clinical testing elsewhere) differ by 28 amino acid residues, thus being two of the more divergent sequences reported to date. The availability of two extremes of diversity for clinical testing apart and in combination will be extremely informative. Since the FVO strain has not been adapted to in vitro culture, we have used FCR3 as a nearly homologous strain for in vitro assays. FCR3 AMA-1 differs by a single residue from FVO AMA-1 at amino acid 36 in the prosequence.
The initial lack of expression of P. falciparum AMA-1 ectodomain in P. pastoris prompted us to design a synthetic gene. In designing the synthetic gene we have made selective changes to the amino acid sequence to remove sites that are generally glycosylated by eukaryotic expression systems through the N-glycosylation pathway. The reasoning for this was threefold. Firstly, authentic PfAMA-1 is not detectably N glycosylated (11), and the presence and location of inappropriate N-linked glycosylation in a recombinant protein intended for vaccine development can have profound but unpredictable targeting and focusing effects on the immune response to it (7). Secondly, glycosylation is frequently highly heterogeneous (as demonstrated by expression of the native sequence PvAMA-1 ectodomain in Pichia [12]). Heterogeneous products may be difficult to reproducibly purify to acceptable standards under cGMP, and such heterogeneity may create batch-to-batch variation in the immunogenicity of the product. Thirdly, we wished to produce a protein with minimal heterogeneity in order to prepare crystals for crystallographic determination of structure.
High-level secreted expression of the PfAMA-1 ectodomain was observed by the outlined approach. The product is homogeneous with only minor degradation products present in the not-yet-optimized production process, as demonstrated by SDS-PAGE. It attains the native conformation and authentic antigenic profile as demonstrated by Western blotting, ELISA, and quantitative binding to immobilized MAb 4G2. The purified protein is highly immunogenic in rabbits, inducing high antibody titers that react with P. falciparum schizonts-merozoites. These results indicate that changing the N-glycosylation sites indeed results in a homogeneous product, as previously observed for PvAMA-1 (12), and that antibody responses are focused on native protein determinants. Quantitative reactivity with the conformation-dependent MAb 4G2 and reactivity with a human serum from a region where malaria is endemic both indicate that changing the primary structure following the strategy of exchanging residues for residues that exist in the AMA-1 repertoire from different Plasmodium species did not have an overall deleterious impact. This is further demonstrated by the capacity of the induced antibodies to inhibit erythrocyte invasion by merozoites at a concentration that is far below the concentration in serum obtained after immunization. This effect was observed on the homologous P. falciparum (FCR3) but also, to a lesser extent, on a P. falciparum strain (NF54) that has one of the more divergent AMA-1 sequences compared to FVO (26 amino acids different in the ectodomain). Similar results have been obtained by Hodder et al. (9), who immunized rabbits with E. coli-derived, refolded PfAMA-1 ectodomain and tested IgG in invasion inhibition assays on a divergent P. falciparum strain with a 23-residue difference in the AMA-1 ectodomain. These data demonstrate the presence of common protective epitopes that could possibly be further boosted by cross-immunization protocols, a strategy that we are currently developing.
Having shown that expression of PfAMA-1 ectodomain in P. pastoris as described here yields large amounts of a homogeneous product of authentic conformation that can easily be scaled up, we are now developing fermentation and downstream processing protocols under cGMP conditions. Immunogenicity of near-GMP-grade protein will be tested with clinically acceptable formulations as a prelude to phase I clinical testing in humans.
Acknowledgments
Clemens H. M. Kocken and Chrislaine Withers-Martinez contributed equally to this work.
This work was supported by EC DG XII KA2 program Quality of Life and Living resources under contract QLK2-CT-1999-01293 and by the Medical Research Council (United Kingdom).
We thank Augusto Valderrama for excellent technical assistance.
Editor: J. M. Mansfield
REFERENCES
- 1.Anders, R. F., P. E. Crewther, S. Edwards, M. Margetts, M. L. Matthew, B. Pollock, and D. Pye. 1998. Immunisation with recombinant AMA-1 protects mice against infection with Plasmodium chabaudi. Vaccine 16:240-247. [DOI] [PubMed] [Google Scholar]
- 2.Collins, W. E., D. Pye, P. E. Crewther, K. L. Vandenberg, G. G. Galland, A. J. Sulzer, D. J. Kemp, S. J. Edwards, R. L. Coppel, J. S. Sullivan, et al. 1994. Protective immunity induced in squirrel monkeys with recombinant apical membrane antigen-1 of Plasmodium fragile. Am. J. Trop. Med. Hyg. 51:711-719. [DOI] [PubMed] [Google Scholar]
- 3.Crewther, P. E., M. L. Matthew, R. H. Flegg, and R. F. Anders. 1996. Protective immune responses to apical membrane antigen 1 of Plasmodium chabaudi involve recognition of strain-specific epitopes. Infect. Immun. 64:3310-3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deans, J. A., T. Alderson, A. W. Thomas, G. H. Mitchell, E. S. Lennox, and S. Cohen. 1982. Rat monoclonal antibodies which inhibit the in vitro multiplication of Plasmodium knowlesi. Clin. Exp. Immunol. 49:297-309. [PMC free article] [PubMed] [Google Scholar]
- 5.Deans, J. A., and W. C. Jean. 1987. Structural studies on a putative protective Plasmodium knowlesi merozoite antigen. Mol. Biochem. Parasitol. 26:155-166. [DOI] [PubMed] [Google Scholar]
- 6.Deans, J. A., A. M. Knight, W. C. Jean, A. P. Waters, S. Cohen, and G. H. Mitchell. 1988. Vaccination trials in rhesus monkeys with a minor, invariant, Plasmodium knowlesi 66 kD merozoite antigen. Parasite Immunol. 10:535-552. [DOI] [PubMed] [Google Scholar]
- 7.Garrity, R. R., G. Rimmelzwaan, A. Minassian, W. P. Tsai, G. Lin, J. J. de Jong, J. Goudsmit, and P. L. Nara. 1997. Refocusing neutralizing antibody response by targeted dampening of an immunodominant epitope. J. Immunol. 159:279-289. [PubMed] [Google Scholar]
- 8.Harlow, E., and D. Lane. 1988. Antibodies: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 9.Hodder, A. N., P. E. Crewther, and R. F. Anders. 2001. Specificity of the protective antibody response to apical membrane antigen 1. Infect. Immun. 69:3286-3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hodder, A. N., P. E. Crewther, M. L. Matthew, G. E. Reid, R. L. Moritz, R. J. Simpson, and R. F. Anders. 1996. The disulfide bond structure of Plasmodium apical membrane antigen-1. J. Biol. Chem. 271:29446-29452. [DOI] [PubMed] [Google Scholar]
- 11.Howell, S. A., C. Withers-Martinez, C. H. Kocken, A. W. Thomas, and M. J. Blackman. 2001. Proteolytic processing and primary structure of Plasmodium falciparum apical membrane antigen-1 (PfAMA-1). J. Biol. Chem. 276:31311-31320. [DOI] [PubMed] [Google Scholar]
- 12.Kocken, C. H., M. A. Dubbeld, A. Van Der Wel, J. T. Pronk, A. P. Waters, J. A. Langermans, and A. W. Thomas. 1999. High-level expression of Plasmodium vivax apical membrane antigen 1 (AMA-1) in Pichia pastoris: strong immunogenicity in Macaca mulatta immunized with P. vivax AMA-1 and adjuvant SBAS2. Infect. Immun. 67:43-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kocken, C. H., D. L. Narum, A. Massougbodji, B. Ayivi, M. A. Dubbeld, A. van der Wel, D. J. Conway, A. Sanni, and A. W. Thomas. 2000. Molecular characterisation of Plasmodium reichenowi apical membrane antigen-1 (AMA-1), comparison with P. falciparum AMA-1, and antibody-mediated inhibition of red cell invasion. Mol. Biochem. Parasitol. 109:147-156. [DOI] [PubMed] [Google Scholar]
- 14.Kocken, C. H., A. M. van der Wel, M. A. Dubbeld, D. L. Narum, F. M. van de Rijke, G. J. van Gemert, X. van der Linde, L. H. Bannister, C. Janse, A. P. Waters, and A. W. Thomas. 1998. Precise timing of expression of a Plasmodium falciparum-derived transgene in Plasmodium berghei is a critical determinant of subsequent subcellular localization. J. Biol. Chem. 273:15119-15124. [DOI] [PubMed] [Google Scholar]
- 15.Marshall, V. M., L. Zhang, R. F. Anders, and R. L. Coppel. 1996. Diversity of the vaccine candidate AMA-1 of Plasmodium falciparum. Mol. Biochem. Parasitol. 77:109-113. [DOI] [PubMed] [Google Scholar]
- 16.Narum, D. L., S. A. Ogun, A. W. Thomas, and A. A. Holder. 2000. Immunization with parasite-derived apical membrane antigen 1 or passive immunization with a specific monoclonal antibody protects BALB/c mice against lethal Plasmodium yoelii yoelii YM blood-stage infection. Infect. Immun. 68:2899-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narum, D. L., and A. W. Thomas. 1994. Differential localization of full-length and processed forms of PF83/AMA-1, an apical membrane antigen of Plasmodium falciparum merozoites. Mol. Biochem. Parasitol. 67:59-68. [DOI] [PubMed] [Google Scholar]
- 18.Oliveira, D. A., V. Udhayakumar, P. Bloland, Y. P. Shi, B. L. Nahlen, A. J. Oloo, W. E. Hawley, and A. A. Lal. 1996. Genetic conservation of the Plasmodium falciparum apical membrane antigen-1 (AMA-1). Mol. Biochem. Parasitol. 76:333-336. [DOI] [PubMed] [Google Scholar]
- 19.Peterson, M. G., V. M. Marshall, J. A. Smythe, P. E. Crewther, A. Lew, A. Silva, R. F. Anders, and D. J. Kemp. 1989. Integral membrane protein located in the apical complex of Plasmodium falciparum. Mol. Cell. Biol. 9:3151-3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 21.Thomas, A. W., J. A. Deans, G. H. Mitchell, T. Alderson, and S. Cohen. 1984. The Fab fragments of monoclonal IgG to a merozoite surface antigen inhibit Plasmodium knowlesi invasion of erythrocytes. Mol. Biochem. Parasitol. 13:187-199. [DOI] [PubMed] [Google Scholar]
- 22.Thomas, A. W., J. F. Trape, C. Rogier, A. Goncalves, V. E. Rosario, and D. L. Narum. 1994. High prevalence of natural antibodies against Plasmodium falciparum 83-kilodalton apical membrane antigen (PF83/AMA-1) as detected by capture-enzyme-linked immunosorbent assay using full-length baculovirus recombinant PF83/AMA-1. Am. J. Trop. Med. Hyg. 51:730-740. [DOI] [PubMed] [Google Scholar]
- 23.Thomas, A. W., A. P. Waters, and D. Carr. 1990. Analysis of variation in PF83, an erythrocytic merozoite vaccine candidate antigen of Plasmodium falciparum. Mol. Biochem. Parasitol. 42:285-287. [DOI] [PubMed] [Google Scholar]
- 24.Trager, W., and J. B. Jensen. 1976. Human malaria parasites in continuous culture. Science 193:673-675. [DOI] [PubMed] [Google Scholar]
- 25.Withers-Martinez, C., E. P. Carpenter, F. Hackett, B. Ely, M. Sajid, M. Grainger, and M. J. Blackman. 1999. PCR-based gene synthesis as an efficient approach for expression of the A+T-rich malaria genome. Protein Eng. 12:1113-1120. [DOI] [PubMed] [Google Scholar]