

Abstract
Although traditionally recognized for maintaining extracellular matrix integrity during morphogenesis, the function of matrix metallo-proteinases (MMPs) and their inhibitors, the tissue inhibitors of metalloproteinases (TIMPs), in the mature nervous system is essentially unknown. Here, we report that TIMP-2 induces pheochromocytoma PC12 cell-cycle arrest via regulation of cell-cycle regulatory proteins, resulting in differentiation and neurite outgrowth. TIMP-2 decreases cyclins B and D expression and increases p21Cip expression. Furthermore, TIMP-2 promotes cell differentiation via activation of the cAMP/Rap1/ERK (extracellular signal-regulated kinase) pathway. Expression of dominant-negative Rap1 blocks TIMP-2-mediated neurite outgrowth. Both the cell-cycle arrest and neurite outgrowth induced by TIMP-2 was independent of MMP inhibitory activity. Consistent with the PC12 cell data, primary cultures of TIMP-2 knock-out cerebral cortical neurons exhibit significantly reduced neurite length, which is rescued by TIMP-2. These in vitro results were corroborated in vivo. TIMP-2 deletion causes a delay in neuronal differentiation, as demonstrated by the persistence of nestin-positive progenitors in the neocortical ventricular zone. The interaction of TIMP-2 with α3β1 integrin in the cerebral cortex suggests that TIMP-2 promotes neuronal differentiation and maintains mitotic quiescence in an MMP-independent manner through integrin activation. The identification of molecules responsible for neuronal quiescence has significant implications for the ability of the adult brain to generate new neurons in response to injury and neurological disorders, such as Alzheimer's and Parkinson's diseases.
Keywords: cell cycle, integrin, MMP, neurogenesis, neurite outgrowth, proliferation, differentiation, TIMP
Introduction
During brain development, a population of morphologically homogeneous, mitotically active precursors located within the neuroepithelium gives rise to all the neuronal and glial cells of the adult brain. Neurogenesis involves progenitor proliferation and appropriately timed generation of postmitotic neurons and, thus, is linked to the cell cycle. To control the total number of cells produced, and therefore final brain size, the number of proliferating progenitors must be exquisitely regulated. Whereas mitogen reduction induces cell-cycle arrest, sustained mitogen/receptor expression throughout neurogenesis suggests that other antiproliferative signals participate in cell-cycle withdrawal. The diverse nature of cell intrinsic (e.g., p27Kip) (Miyazawa et al., 2000) and extrinsic (e.g., the neuroactive peptide pituitary adenylate cyclase activating polypeptide) (Suh et al., 2001) factors that participate in cell-cycle exit demonstrates the complexity of coordinating such a critical aspect of development. This may be attributable, in part, to the irrevocable nature of cell-cycle withdrawal in neuronal cells. Although many factors regulating cell-cycle control early in development have been described previously (Ohnuma and Harris, 2003), the mechanism by which neuronal quiescence is maintained is currently poorly understood.
Proteolysis of extracellular signaling molecules as a mechanism of regulating neuronal proliferation and differentiation has recently received considerable attention. Extracellular matrix (ECM) synthesis and degradation must be exquisitely regulated to preserve matrix boundaries that are essential to normal tissue structure and function. Matrix metalloproteinases (MMPs) are the main physiologically relevant mediators of ECM degradation (Nagase and Woessner, 1999). Because MMPs function in the turnover of a broad-spectrum of ECM (Nagase, 1997) and non-ECM (McCawley and Matrisian, 2001) proteins, their activity must be tightly regulated. Tissue inhibitor of metalloproteinases (TIMPs) are 20-30 kDa secreted proteins that form tight but relatively low selectivity 1:1 complexes with the active forms of MMPs. Thus far, four members of the TIMP family have been cloned. Although traditionally recognized for MMP inhibitory activity, TIMPs are multifunctional molecules with diverse functions distinct from MMP inhibition (for review, see Baker et al., 2002).
Although the role of MMPs and TIMPs in the morphogenesis of non-neural tissues has been investigated, to date few studies have analyzed their expression during CNS development. TIMP-2 expression increases throughout development and is maintained at high levels into adulthood (Fager and Jaworski, 2000; Young et al., 2002). In vivo, TIMP-2 expression is restricted to postmitotic neurons in divergent regions of the nervous system (Fager and Jaworski, 2000); thus, it likely regulates the acquisition of a neuronal phenotype.
Here, we show that TIMP-2 promotes neuronal differentiation in vitro by directing proliferating cells out of the cell cycle, resulting in neurite outgrowth through a mechanism that involves Rap1 and cdk5 (cyclin-dependent kinase 5) activity. Strikingly, TIMP-2 exerts its effect on neuronal differentiation via an MMP-independent mechanism. These in vitro observations appear to be corroborated in vivo in that deletion of the TIMP-2 gene leads to an incomplete neuronal differentiation. This report demonstrates for the first time a phenotype for the TIMP-2-/- mouse and details the mechanism of TIMP-2 action in neuronal differentiation.
Materials and Methods
Animals. Mice bearing a targeted disruption of the TIMP-2 gene have been described previously (Wang et al., 2000). Mice are maintained on a C57BL/6 background after 10 backcrosses. Offspring from homozygous matings were used for primary cultures. For all other analyses, TIMP-2-/- mice and wild-type littermates from heterozygous matings were used. Procedures that involved animals were in accordance with the institutional guidelines of the University of Vermont Animal Care and Use Committee.
Antibodies and reagents. Antibodies against actin (1:5000), cyclin B (1:100), cyclin D (1:500), phosphorylated extracellular signal-regulated kinase (pERK) (1:500), ERK2 (1:500), phosphorylated epidermal growth factor receptor (pEGFR) (1:500), EGFR (1:500), inhibitor of differentiation protein 2 (Id2) (1:500), p35 (1:200), Rap1 (1:500), and TIMP-1 (1:500) were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against β1 integrin (1:1000), neurofilament-145 (1:500), neurofilament-160 (1:100), proliferating cell nuclear antigen (PCNA) (1:200), and TIMP-2 (1:1500 for Western blots) were from Chemicon (Temecula, CA). Antibodies against TIMP-2 (1:200 for histology) and PGP 9.5 (protein gene product 9.5) (1:1000) were from Biogenesis (Kingston, NH). Antibodies against p21Cip1 (cyclin-dependent kinase inhibitor) (1:500) and α3 integrin (clone P1B5) were from Oncogene Research (San Diego, CA). Additional antibodies used were as follows: anti-Flag (1:150; Stratagene, La Jolla, CA), anti-pRb (retinoblastoma protein) Ser 807/811 (1:500; Cell Signaling Technology, Beverly, MA), anti-Ras (1: 500; Transduction Laboratories, San Jose, CA), anti-nestin (1:1; kindly provided by Dr. Susan Hockfield, Yale University, New Haven, CT), and biotinylated anti-bromodeoxyuridine (BrdU) (1:100; BD Biosciences, San Jose, CA). NGF was from BD Biosciences; H89 (N-[2-(p-bromocinnamylamino)-ethyl]-5-isoquinoline-sulfon-amide 2HCl), SQ 22536 [9-(tetrahydro-2′-furyl)adenine], and roscovitine were from Calbiochem (San Diego, CA). Protein A/G-Sepharose was from Santa Cruz Biotechnology.
Cell culture and transfection. PC12 rat pheochromocytoma cells (American Type Culture Collection, Manassas, VA) were cultured in RPMI medium supplemented with 10% FBS and 5% horse serum (S-RPMI). For NGF-mediated differentiation, 100 cells/mm2 were plated on poly-l-lysine (0.1 mg/ml)/laminin (10 μg/ml)-coated tissue culture dishes in RPMI medium containing N2 supplement. After 12 h, fresh RPMI-N2 medium was added containing NGF (100 ng/ml). Cultures were then maintained for 7 d in vitro (DIV) without media being replenished.
For transfection experiments, 2 × 105 PC12 cells were plated on coated 24-well plates in S-RPMI medium. Twenty-four hours after seeding, cells were transfected with 800 ng of DNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) incubated overnight, and then fresh S-RPMI medium was added. For synergy experiments, after transfection mix removal, fresh S-RPMI medium containing NGF (100 ng/ml) was added and incubated for 72 h.
Primary cultures from cerebral cortex were prepared from postnatal day 0 (P0) TIMP-2-/- and wild-type mice as described previously (Minturn et al., 1995). Cells (88 cells/mm2) were plated in DMEM supplemented with 10% FBS in the presence or absence of NGF (100 ng/ml). After 2 DIV, cultures were treated with 10 μm cytosine-D-arabinofuranoside. Cultures were maintained for 7 DIV at 37°C in a humidified air/5% CO2 incubator.
For rescue experiments, primary cultures from P0 TIMP-2-/- cortex were either (1) transfected with TIMP-2 or vector alone using the mouse neuron nucleofector kit (Amaxa Biosystems, Gaithersburg, MD) following the instructions of the manufacturer or (2) treated exogenously with recombinant TIMP-2 (rTIMP-2). Cultures treated with rTIMP-2 (50 nm) were maintained in Neurobasal medium supplemented with B27. Cultures were harvested at 7 DIV, and total neurite length was determined as described below.
Plasmid constructs. Full-length TIMP-2 cDNA was generated by reverse transcription (RT)-PCR from adult rat brain. The primers used were as follows: forward, 5′-ATTTAGAATTCATGGGCGCCGCGGCCCGC-3′; reverse, 5′-GTCTGCTCGAGCGGGTCCTCGATGTCAAG-3′. The PCR product was cloned into the EcoRI and XhoI sites in the pIRES-hrGFP-1a vector (Stratagene) and sequenced to verify a complete open reading frame. TIMP-2C72S mutant (substitution of cysteine 72 to serine) was generated using the QuickChange site-directed mutagenesis kit (Stratagene) using forward (5′-CCGACGCCAGCAGCTGCTC-3′) and reverse (5′-GAGCAGCTGCTGGCGTCGG-3′) primers with the base substitution underlined. The resulting construct was sequenced to verify mutagenesis.
pDsRed-RapN17 was constructed after PCR amplification using the pCMVRAP1b (Vossler et al., 1997) vector as the template. The PCR primers used were as follows: forward, 5′-AATATAAGCTTATGCGTGAGTATAAGCTAG-3′; reverse, 5′-TAATGAATTCGAAGCAGCTGACATGACGA-3′. The resultant amplification product was cloned into the HindIII and EcoRI sites in the pDsRed2-N1 vector (Clontech, Palo Alto, CA) and sequenced.
BrdU incorporation. Nineteen hours after transfection, cells were pulsed with 10 μm BrdU for 5 h at 37°C. Cells were then fixed with 1% formaldehyde (EM Polysciences, Warrington, PA) for 15 min at 4°C. After two washes with PBS, cells were incubated for 1 h at room temperature (RT) with blocking buffer (PBS, 10% horse serum, and 0.5% Triton X-100) and treated with DNase I (50 U/ml) for 15 min at 37°C. Incorporated BrdU was detected with biotinylated anti-BrdU antibody diluted in PBS containing 1% BSA overnight at 4°C. After two washes with PBS for 10 min each, cells were incubated for 30 min at 4°C with streptavidincyanine 3 (Cy3) diluted in PBS/1%BSA. Samples were then washed three times with PBS, fluorescence was detected using a Nikon (Micro Video Instruments, Avon, MA) inverted epifluorescence microscope, and digital images were captured with a Spot RT camera (Diagnostic Instruments, Sterling Heights, MI). The percentage of BrdU-positive cells was calculated relative to the total number of green fluorescent protein (GFP)-positive cells. A total of 12 randomly selected microscopic fields were examined (at 40×).
Cell-cycle analysis. The cell-cycle profile of vector and TIMP-2transfected PC12 cells was examined by propidium iodine (PI) staining. Briefly, 24 h after transfection, cells were trypsinized (0.25% trypsin-EDTA) and recovered by centrifugation at 450 × g for 3 min at RT. After two washes with PBS/1% FBS, cells were fixed with 1% paraformaldehyde (PFA) in PBS/1% FBS for 15 min on ice. After two washes, cells (106 cells/ml) were resuspended in low-salt staining solution (3 g/ml polyethylene glycol PEG 8000, 50 μg/ml PI, 180 U/ml RNase A, 0.1% Triton X-100, and 4 mm sodium citrate) and incubated at 4°C for 30 min. An equal volume of high-salt solution (3 g/ml polyethylene glycol PEG 8000, 50 μg/ml PI, 180 U/ml RNase A, 0.1% Triton X-100, and 400 mm NaCl) was added. PI incorporation was analyzed by flow cytometry (electronic programmable individual cell sorter; Coulter, Krefeld, Germany).
Fluorescence-activated cell sorting. PC12 cells (10 × 106) were plated the day before transfection on coated 100 mm dishes in S-RPMI medium. Cultures were transfected as described above. After 24 h, cells were trypsinized, washed, and resuspended in PBS/1% FBS (1.8 × 106 cells/ml), and GFP-positive and GFP-negative cells were sorted.
Immunoprecipitation and Western blot analysis. Cells were lysed in lysis buffer (25 mm HEPES, pH 7.7, 150 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 0.5% Triton X-100, 0.5 mm DTT, 20 mm β-glycerophosphate, 1 mm Na3VO4, 5 mm NaF, 4 mm PMSF, 1 μg/ml leupeptin, and 1 μg/ml aprotinin) for 30 min at 4°C. Lysates were spun at 14,000 rpm for 15 min at 4°C, and the supernatant was used for Western blot or immunoprecipitation assays.
For Western blot analysis, cellular lysates (20 μg) were fractionated by SDS-PAGE and transferred to Immobilon-P membranes (Schleicher & Schuell, Keene, NH). Membranes were blocked with 5% nonfat milk in Tris-buffered saline (TBS) (10 mm Tris, pH 7.5, and 150 mm NaCl), followed by overnight incubation at 4°C with the indicated antibody diluted in TBS with 0.05% Tween 20 (TBS-T). After three washes with TBS-T, membranes were incubated with the appropriate secondary antibody coupled to horseradish peroxidase, and immunocomplexes were visualized by enhanced chemiluminescence according to the instructions of the manufacturer (PerkinElmer Life Sciences, Boston, MA).
For immunoprecipitation assays, cellular lysates were incubated with anti-Flag, anti-α3(1 μg/sample), or anti-β1(2 μg/sample) integrin overnight at 4°C. Immune complexes were harvested with protein A-Sepharose for 1 h on ice and washed once with cold TNE-T [150 mm NaCl, 50 mm Tris, pH 7.5, 5 mm EDTA, and 1% Triton X-100 (w/v)], twice with TNE (150 mm NaCl, 50 mm Tris, pH 7.5, and 5 mm EDTA), and once with H2O. Immunoprecipitated proteins were resolved by SDS-PAGE and subjected to immunoblotting.
cAMP assay. Transfected PC12 cells were maintained in S-RPMI containing 50 μm Ro-20-1724 [4-(3-butoxy-4-methoxybenzyl) imidazoline-2-one], a selective inhibitor of cAMP-specific phosphodiesterase. At 1, 2, and 3 DIV, cultures were extracted in absolute ethanol containing 100 μm Ro-20-1724 and processed for cAMP using the Biotrak non-acetylation protocol of the 125I-cAMP radioimmunoassay (Amersham Biosciences, Piscataway, NJ) according to the instructions of the manufacturer.
Glutathione S-transferase pull-down assay. Ras-GTP and Rap1-GTP activities were determined as previously described (Rebhun et al., 2000). PC12 cells (10 × 106) were transfected with vector or TIMP-2 in S-RPMI medium. Total cell lysates (150 μg) were incubated overnight at 4°C with glutathione S-transferase (GST)-Ral, which binds the GTP-bound form of Rap1, or GST-Raf, which binds the GTP-bound form of c-Raf. Beads were washed four times with cold lysis buffer, and bound proteins were resolved by SDS-PAGE and subjected to immunoblotting for Rap and Ras expression.
To measure Rap1 activity including the inhibitors H89 or SQ 22536, PC12 cells were transfected as described previously. After 24 h, cells were incubated with H89 (10 μm) or with SQ 22536 (50 μm) and further incubated for 24 and 48 h. For the 48 h time point, fresh inhibitors were added 24 h before harvesting. Rap1-GTP-bound form was then determined as described above.
Northern blot analysis. Northern blot analysis was performed exactly as described previously (Jaworski et al., 1994).
Reverse zymography. Forty-eight hours after transfection, PC12 cells were serum starved overnight in OPTIMEM medium (Invitrogen). After 12 h, supernatants were harvested and concentrated 11× using a Centricon YM-10 (Millipore, Bedford, MA). Concentrated supernatants (300 ng) or cell lysates (20 μg) from TIMP-2-transfected PC12 cells were electrophoresed through a 12% nonreducing SDS-PAGE gel containing 1% gelatin and conditioned medium from baby hamster kidney (BHK) cells. After electrophoresis, the gel was washed once for 15 min and then overnight in wash buffer (2.5% Triton X-100, 50 mm Tris, pH 7.5, and 5 mm CaCl2) to remove SDS. The gel was then rinsed three times in water, followed by 24 h incubation at 37°C in incubation buffer (50 mm Tris, pH 7.5, and 5 mm CaCl2). The gel was stained for 4 h in Coomassie brilliant blue (0.05% Coomassie blue, 10% acetic acid, and 30% isopropanol) and destained (10% isopropanol and 10% acetic acid). Recombinant human TIMP-2 (Chemicon) was used as a positive control.
Fluorescence microscopy. For immunofluorescence, cells were fixed for 30 min at RT with 2% PFA. After two washes with PBS for 10 min each, cells were incubated for 2 h at RT with blocking buffer (DMEM, 5% FBS, 0.1% glycine, 0.1% lysine, and 0.2% Triton X-100). Primary antibody was diluted in blocking buffer and incubated overnight at 4°C. After three washes with PBS for 10 min each, cells were incubated for 1 h at RT with the appropriate secondary antibody diluted in blocking buffer without Triton X-100. Samples were then washed three times with PBS, fluorescence was detected using a Nikon inverted epifluorescence microscope, and digital images were captured with a Spot RT camera.
For TIMP-2 localization, NGF-induced PC12 cells were live labeled with TIMP-2 antibody for 10 min at 37°C, and cells were then fixed with 4% formaldehyde for 10 min at RT. After three washes with PBS, a Cy3-conjugated secondary antibody was applied for 30 min at RT. Cells were then incubated for 20 min with PBS containing 1% BSA and 0.25 μg/ml fluorescence isothiocyanate-conjugated phalloidin (Molecular Probes, Eugene, OR). Nuclei were localized by Hoechst staining. After washing with PBS, samples were analyzed by DeltaVision deconvolution microscopy (University of Vermont Neuroscience COBRE Image Analysis Facility).
Immunohistochemistry was performed exactly as described previously (Jaworski et al., 1999). For PCNA labeling, sections were incubated in biotinylated mouse IgG (1:400), washed three times, and incubated with avidin-biotin-peroxidase complex (1:200; Vector Laboratories, Burlingame, CA) before visualization with diaminobenzidine (Sigma, St. Louis, MO).
Neurite outgrowth. PC12 cells were harvested 72 h after transfection, and GFP-positive cells were observed with a Nikon inverted epifluorescence microscope. For primary neuronal cultures, cells were fixed in 2% PFA after 8 DIV and immunostained using a polyclonal anti-PGP 9.5 antibody. Total neurite length was determined using software provided with the Spot CCD camera, and photomicrographs were prepared using Adobe Photoshop (Adobe Systems, San Jose, CA).
Data presentation. Data corresponds to three to five experiments, each containing two to four dishes. The exception is the rescue of primary cultured neurite length that was performed in two independent experiments, each with two 24-well plates. Data are presented as mean ± SEM. Significant interactions were identified by t test. Criterion for statistical significance is p < 0.05.
Results
TIMP-2 inhibits PC12 proliferation and regulates expression of cell-cycle proteins
Previous studies have demonstrated that TIMP-2 mRNA is expressed in the nervous system by neurons coincident with the onset of neuronal differentiation, and its expression is maintained into adulthood (Vaillant et al., 1999; Fager and Jaworski, 2000). To determine whether TIMP-2 plays a role in neuronal differentiation, we initially used the PC12 pheochromocytoma cell line, a well accepted model system for investigating the molecular mechanism underlying neuronal differentiation. Proliferating PC12 cells were transfected with either TIMP-2 cDNA cloned into the pIRES-hrGFP-1a vector (TIMP-2) or empty vector (vector).
Because neurogenesis requires cell-cycle exit and TIMP-2 has been shown to have anti-mitogenic properties independent of MMP inhibition in other systems (Hoegy et al., 2001), we determined whether TIMP-2 could promote neuronal differentiation by preventing cell proliferation. Cells entering into S phase were quantitated by BrdU immunocytochemistry. TIMP-2 induced an 8.7-fold reduction of BrdU incorporation (vector, 52 ± 6.4%; TIMP-2, 6 ± 5%) (Fig. 1A). To determine whether this function requires MMP inhibitory activity, a TIMP-2 mutant unable to inhibit MMPs was generated. Substitution of cysteine 72 to serine (TIMP-2C72S) completely abrogates MMP inhibitory activity without affecting protein conformation and MMP-independent functions (Caterina et al., 1997; Bond et al., 2000). Similar to TIMP-2, expression of the mutant TIMP-2C72S resulted in a pronounced reduction in the number of BrdU-positive cells (vector, 52 ± 6.4%; TIMP-2C72S, 9 ± 2%). These data are indicative of an antiproliferative function for TIMP-2 in PC12 cells independent of MMP inhibitory activity. To further corroborate these results, the cell-cycle profile of TIMP-2-transfected PC12 cells was assayed by flow cytometry (Fig. 1B). The DNA content of GFP-positive and GFP-negative cells was determined using propidium iodine staining. Untransfected and vector-transfected cells showed similar cell-cycle profiles 24 h after transfection. In contrast to GFP-negative cells, TIMP-2 transfected GFP-positive cells displayed a strikingly different profile. The majority of the cells accumulated at the G0/G1 phase (79.18 ± 3.34%), with few cells present in S (6.72 ± 3.13%) or G2/M (9.94 ± 1.94%) phase. TIMP-2 was secreted into the medium (Fig. 1C), yet neighboring GFP-negative cells did not display cell-cycle arrest, suggesting a cell-autonomous function for TIMP-2 in PC12 cell proliferation.
Figure 1.
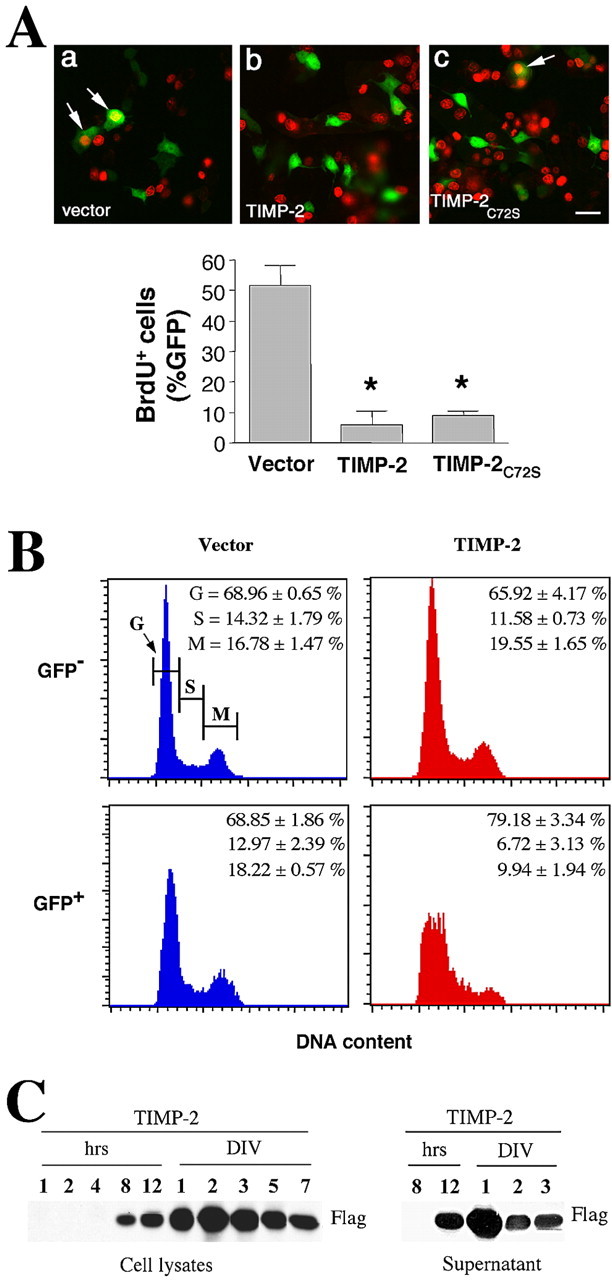
TIMP-2 inhibits PC12 proliferation. A, Immunofluorescence showing BrdU-labeled (Cy3, red) nuclei in transfected (GFP-positive) cells. After 19 h of transfection with TIMP-2 or TIMP-2C72S (a mutant TIMP-2 lacking MMP inhibitory activity), cells were pulsed with BrdU for 5 h, and the number of GFP-positive cells entering S phase was quantitated. PC12 cells transfected with TIMP-2 or TIMP-2C72S showed a significant reduction (*p < 0.001) in the number of BrdU-positive cells (arrows) when compared with vector-transfected cells (8.6- and 5.7-fold, respectively), indicating that TIMP-2 induced cell-cycle arrest independent of MMP inhibitory activity. TIMP-2 also induces neurite outgrowth via an MMP-independent mechanism (see Fig. 5). The reduction in the number of BrdU-positive cells is not attributable to either alterations in transfection efficiency or cell death. Transfection efficiency was comparable in TIMP-2-transfected (12.7 ± 1.7%) and TIMP-2C72S-transfected (22.7 ± 3.5%) cells relative to vector-transfected cells (15.6 ± 2.1%). Furthermore, flow cytometry revealed no increase in the number of nonviable cells. Scale bar, 20 μm. B, TIMP-2- and vector-transfected PC12 cells were stained with propidium iodine 24 h after transfection. Flow cytometry demonstrates the proportion of GFP-positive and GFP-negative cells at the G (G0/G1), S, and M (M/G2) phases of the cell cycle. Only TIMP-2-transfected cells underwent cell-cycle arrest, suggesting a cell-autonomous mechanism. C, The time course of cell-associated and secreted TIMP-2 in transfected cells was determined by Western blot analysis using an anti-Flag antibody.
To further characterize cell-cycle arrest in TIMP-2 transfected cells, the expression of cell-cycle regulatory proteins was examined by Western blot analysis. TIMP-2 GFP-positive cells were separated from GFP-negative cells by fluorescent activated cell sorting (FACS) 24 h after transfection. As a control, vector-transfected cells were also subjected to FACS analysis, whereas proliferating PC12 cells were used to establish the basal level of protein expression. Because cell growth arrest is characterized by a rapid dephosphorylation of Rb (Dailey et al., 2003), the levels of pRb in TIMP-2-transfected cells was examined. A significant decrease in pRb levels was observed in TIMP-2 GFP-positive cells compared with vector GFP-positive cells (Fig. 2). This decrement was even more evident with respect to proliferating cells. Consistent with this result, TIMP-2 GPF-positive cells contained lower levels of cyclins B and D. Furthermore, significant increased levels of the cyclin-dependent kinase inhibitor p21Cip1, which inhibits cyclin E-cdk2 kinase activity (Sherr and Roberts, 1999), was observed in TIMP-2 GPF-positive cells. Id helix-loop-helix (HLH) proteins inhibit differentiation by sequestering basic HLH differentiation factors into inactive complexes. The decreased expression of Id2 in TIMP-2 GPF-positive cells further substantiates the cyclin and pRb results and indicates that TIMP-2 overexpression induces cell-cycle arrest at G0/G1.
Figure 2.
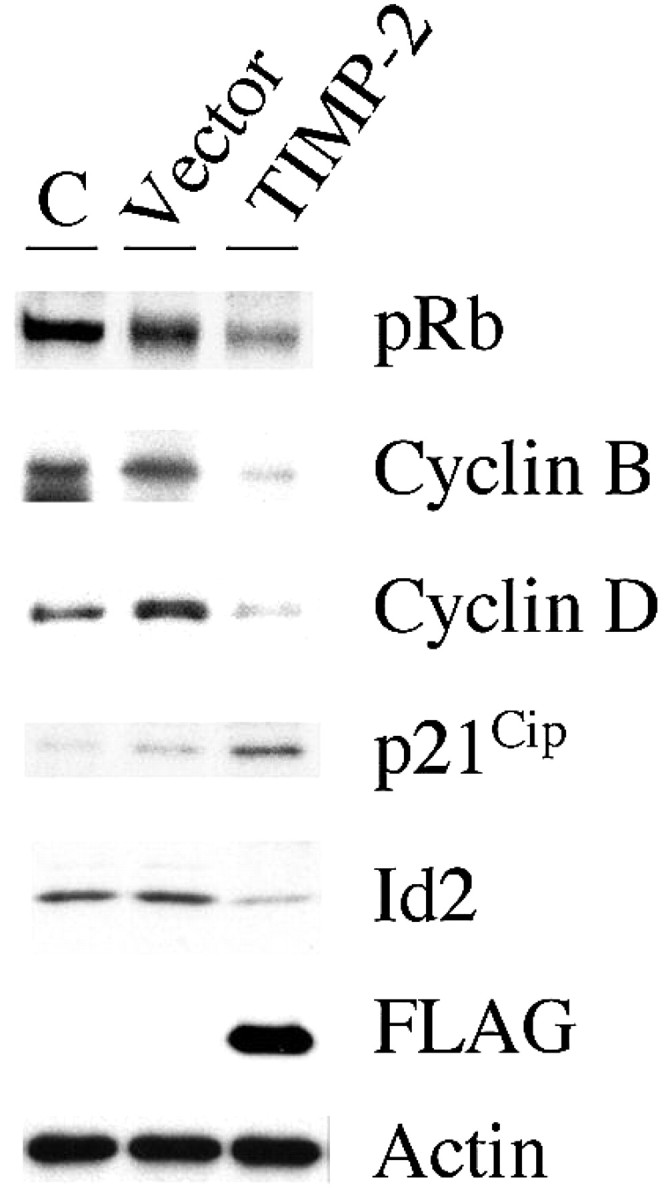
TIMP-2 regulates expression of cell-cycle proteins. The expression of cell-cycle checkpoint markers was analyzed by SDS-PAGE with whole-cell lysates (20 μg) from FACS-sorted GFP-positive TIMP-2- and vector-transfected PC12 cells. Proliferating PC12 cells were used as a control (C). TIMP-2 expression was verified by detection of the Flag epitope tag. Actin was used to verify equal loading of protein per lane. The decreased expression of pRb, cyclin B, cyclin D, and increased p21Cip1 expression in TIMP-2-transfected cells confirms the cell-cycle arrest at G0/G1 observed by flow cytometry.
TIMP-2 induces growth arrest via activation of integrin and cAMP/ERK pathways
We next sought to investigate the signaling pathways involved in TIMP-2-mediated cell-cycle arrest. Recently, it was reported that TIMP-2 inhibits endothelial cell proliferation via an MMP-independent mechanism that is mediated by α3β1 integrin (Seo et al., 2003). Because integrins play a role in all phases of neurogenesis, including cell-cycle arrest, we determined whether TIMP-2 interacts with integrins during neuronal differentiation. α3β1 and α1β1 have been identified as the principal integrins expressed in PC12 cells (Tomaselli et al., 1990; Arregui et al., 1994). Immunoprecipitation studies demonstrated that TIMP-2 binds to α3 integrin (Fig. 3A). No such interaction was detected with αv or α5 integrin (data not shown). A low level of α3 integrin-TIMP-2 interaction was detected in proliferating PC12 cells. A greater interaction was present in serum-starved cells and was further increased in differentiated cells. The increased interaction may simply reflect elevated surface integrin expression in response to NGF (Zhang et al., 1993) or a more specific association required to activate downstream signaling cascades.
Figure 3.
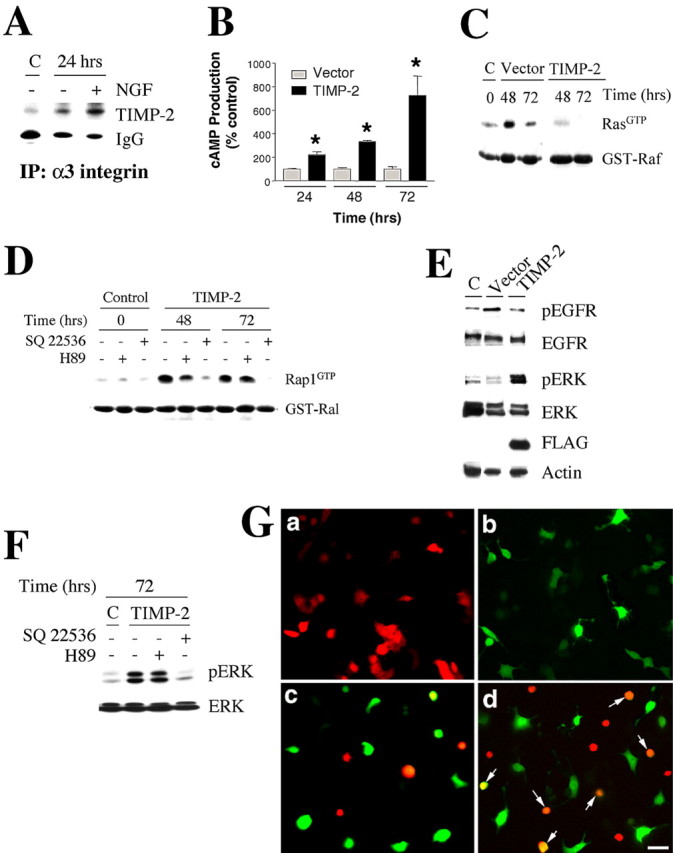
TIMP-2 activation of integrin and cAMP/ERK pathways occurs via a PKA-independent mechanism. A, Cell lysates from proliferating (C), serum-starved, or NGF-treated (24 h) PC12 cells were immunoprecipitated using anti-α3 integrin antibody, and association for TIMP-2 was determined by Western blot analysis. TIMP-2 binds to α3 integrin in PC12 cells to a greater extent in differentiated cells. B, cAMP levels were determined by radioimmunoassay in TIMP-2- and vector-transfected PC12 cells at the indicated time points. cAMP levels were first expressed in femtomoles and then normalized to 0 DIV at each time point. The time course of increased intracellular cAMP levels at 48 and 72 h after TIMP-2 overexpression correlates with increased Rap1-GTP activity (Fig. 3D). At each time point, cAMP levels are significantly greater in TIMP-2-transfected cells (*p < 0.001). C, Ras activity is reduced (48 h) or completely abolished (72 h) in TIMP-2-transfected cells. D, The effect of the PKA inhibitor H89 and the adenylate cyclase inhibitor SQ 22536 on Rap1 activity was determined by RBD (Ras-binding domain) affinity binding assay. Whereas SQ 22536 potently downregulated TIMP-2-mediated Rap1 activity, H89 had no effect relative to untreated cells, indicating that cAMP increased Rap1 activity via a PKA-independent mechanism. E, The expression the pEGFR and its downstream effector pERK was analyzed by SDS-PAGE with whole-cell lysates (20 μg) from FACS-sorted GFP-positive TIMP-2- and vector-transfected PC12 cells. TIMP-2 transfection reduces pEGFR levels and produces a sustained ERK activation. F, Western blot analysis was performed to confirm that the pharmacologic treatments not only inhibited Rap1 activity but also its downstream effector pERK. As expected, TIMP-2-induced pERK levels were dramatically decreased in response to SQ 22536 but not H89 treatment. G, a, pDsRed-transfected cells; b, TIMP-2-transfected cells; c, pDsRed and pIRES-hrGFP-1a vector-transfected cells; d, pDsRed-RapN17 and TIMP-2-transfected cells. As opposed to cells only expressing TIMP-2 (green), cells coexpressing dominant-negative Rap and TIMP-2 (yellow to orange cells indicated by the arrows) were not differentiated 72 h after transfection, demonstrating that Rap activity is required for TIMP-2-mediated neurite outgrowth. Refer to Figure 5B for quantitation of neurite length in TIMP-2-transfected cells. Scale bar, 20 μm.
The MAPK (mitogen-activated protein kinase) cascade is one of the principal intracellular signaling pathways involved in cellular proliferation and differentiation, linking the activation of cell surface receptors to cytoplasmic and nuclear effectors. In PC12 cells, EGF causes proliferation by transiently inducing Ras activity that activates c-Raf, resulting in a transient activation of the MAP kinases ERK1 and ERK2. In contrast, NGF treatment promotes neuronal differentiation by inducing sustained Rap1 activity that activates B-Raf, leading to a prolonged activation of ERK1 and ERK2 (Marshall, 1995; Kao et al., 2001).
Depending on the cellular context, TIMP-2 exerts proliferative (Hayakawa et al., 1994; Hoegy et al., 2001) or antiproliferative (Hoegy et al., 2001; Seo et al., 2003) functions by increasing intracellular cAMP levels. Because TIMP-2 induces cell-cycle arrest in PC12 cells (Figs. 1, 2), cAMP production in TIMP-2-transfected PC12 cells was measured. TIMP-2 expression increased cAMP production in a time-dependent manner (Fig. 3B). Within 24 h after transfection, TIMP-2 produced a 2.2-fold increase in cAMP relative to vector-transfected cells. Further increased levels were observed at 48 h (3.3-fold) and 72 h (7.2-fold) after transfection.
cAMP functions as an antiproliferative signal during cerebral cortical development (Suh et al., 2001). cAMP suppress Ras activity and, hence, proliferation by regulating the Rap1/ERK pathway (Cook and McCormick, 1993; Schmitt and Stork, 2002). To determine whether TIMP-2-mediated cAMP production regulates Ras activity, cell extracts from TIMP-2-transfected PC12 cells were subjected to pull-down assays. A reduced (8-48 h) or absent (72 h) Ras activity was detected in TIMP-2-transfected cells relative to vector-transfected or proliferating PC12 cells (Fig. 3C). Furthermore, a significantly increased and sustained Rap1 activity was detected in TIMP-2-transfected cells relative to vector-transfected or proliferating PC12 cells (Fig. 3D, lanes 1,4,7). In correlation with these results, the levels of pEGFR were significantly reduced in TIMP-2 GFP-positive cells (Fig. 3E). These data indicate that TIMP-2 promotes cell-cycle arrest by inhibiting pEGFR and Ras activity.
cAMP regulates Rap1 activity either through a protein kinase A (PKA)-dependent mechanism (Schmitt and Stork, 2002) or by binding the guanine-nucleotide-exchange factor Epac (exchange protein directly activated by cAMP) (de Rooij et al., 1998). To test whether PKA activity is required for TIMP-2-mediated Rap1 induction, the levels of active Rap1 was analyzed in TIMP-2-transfected PC12 cells treated with the PKA inhibitor H89. Cells were harvested at the time points with the highest Rap1 activity (48 and 72 h). Proliferating cells were included as a control for basal levels of Rap1 activity (Fig. 3D, lanes 1-3). H89 treatment did not affect TIMP-2-mediated Rap1 activity compared with untreated cells (Fig. 3D, lanes 4,5 and 7,8). However, treatment with SQ 22536, a selective inhibitor of adenylate cyclase, significantly reduced (48 h) or completely abolished (72 h) Rap1 activity relative to untreated cells (Fig. 3D, lanes 4,6 and 7,9). The reduction in Rap1 activity was not a result of cell death induced by SQ 22536 treatment because protein content of untreated (11.82 μg/μl) and treated (11.33 μg/μl) TIMP-2-transfected cells was similar to that of untreated (11.94 μg/μl) and treated (11.59 μg/μl) untransfected cells. Altogether, these results demonstrate that TIMP-2 induced cell-cycle arrest by a cAMP-dependent mechanism via activation of an integrin containing an α3 subunit. This effect is independent of PKA activity.
In correlation with the fact that TIMP-2 expression induced sustained Rap1 activity (Fig. 3D), the levels of pERK were significantly upregulated in TIMP-2 GFP-positive cells compared with vector GFP-positive cells or proliferating cells (Fig. 3E). These differences were not attributable to a change in total ERK protein. Finally, TIMP-2-transfected PC12 cells were treated with H89 or SQ 22536, and pERK expression was evaluated 72 h after transfection. TIMP-2-induced sustained ERK activity (Fig. 3E) was insensitive to H89 treatment (Fig. 3F, lanes 2, 3). However, SQ 22536 treatment completely abolished the sustained ERK activity in TIMP-2-transfected cells (Fig. 3F, lanes 2, 4). Altogether, these data indicate that TIMP-2-mediated cell-cycle arrest allows neurons to acquire a differentiated state by inducing a sustained Rap1 activity.
Because Rap1 is required for NGF action in PC12 cells (Wu et al., 2001), the role of Rap1 activity in TIMP-2-mediated neuronal differentiation was examined using a dominant-negative mutant of Rap1, RapN17 (Vossler et al., 1997). PC12 cells were transfected with empty vectors or TIMP-2 alone or together with pDsRed-RapN17, and the extent of neurite outgrowth, as a hall-mark of neuronal differentiation, was analyzed by fluorescence microscopy 72 h after transfection. Cultures transfected with the pDsRed vector alone (Fig. 3Ga) or together with pIRES-hrGFP (Fig. 3Gc) showed no change in morphology. Within 72 h after TIMP-2 transfection, differentiated PC12 cells with short neuritic processes were present, although media contained 15% serum (Fig. 3Gb), indicating that TIMP-2 expression was sufficient to induce PC12 cell differentiation. However, TIMP-2-mediated differentiation in PC12 cells was blocked by RapN17 coexpression, as demonstrated by the lack of neurites in the double-positive cells (yellow) (Fig. 3Gd). The presence of single TIMP-2-transfected (green) or RapN17-transfected (red) cells in the same well served as controls. These data demonstrate that TIMP-2-mediated cell-cycle arrest results in PC12 cell differentiation by a cAMP/Rap1/ERK-dependent pathway.
TIMP-2-mediated cell-cycle arrest induces PC12 neurite outgrowth independent of MMP inhibitory activity
After exiting the cell cycle, cells acquire a neuronal fate that is associated with the expression of neuronal markers and neurite outgrowth. To further investigate the role of TIMP-2 in neurite outgrowth, TIMP-2 expression was examined in NGF differentiated PC12 cells. Northern blot analysis revealed that TIMP-2 mRNA was upregulated in response to NGF treatment at all time points tested (Fig. 4A). A 40-fold increase in TIMP-2 mRNA was detected 24 h after NGF treatment. Additional increase (53-fold) was observed at 3 DIV, after which TIMP-2 mRNA levels gradually decreased. A similar regulation was observed for TIMP-2 protein (Fig. 4B). Expression was increased in response to NGF in a time-dependent manner, peaking at 3 DIV (threefold) and then decreasing to basal levels after 7 DIV (Fig. 4B). These results were corroborated immunocytochemically (Fig. 4C). Before NGF addition (0 DIV), a low level of TIMP-2 expression was detected. As soon as cells changed morphology from round, proliferating cells to flat cells with albeit small processes, TIMP-2 expression was detectable. As PC12 cell differentiation proceeded, TIMP-2 expression increased to peak at 5 DIV. Although the most intense expression was present on the cell soma, TIMP-2 was also detected on neuritic processes. In contrast to TIMP-1, whose growth effects are exerted directly within the nucleus (Zhao et al., 1998; Ritter et al., 1999), TIMP-2 expression is not detected in the nucleus (Fig. 4C). Live labeling experiments demonstrated that TIMP-2 is expressed in a punctate manner on the cell surface of PC12 cells treated with NGF for 5 d (Fig. 4D). Interestingly, TIMP-2 was enriched at neuritic branch points and growth cone-like processes. TIMP-2 colocalized with phalloidin at branch points (Fig. 4D) and with caveolin at growth cones (data not shown). Because TIMP-1 induces cell-cycle arrest and increased branching morphogenesis (Fata et al., 1999) similar to that of TIMP-2 demonstrated here, TIMP-1 regulation during PC12 differentiation was examined. Unlike TIMP-2, TIMP-1 expression is not upregulated during PC12 cell differentiation (Fig. 4E). Altogether, these results indicate a specific role for TIMP-2 in PC12 cell differentiation, and its expression at the branch points suggests a role in neurite outgrowth.
Figure 4.
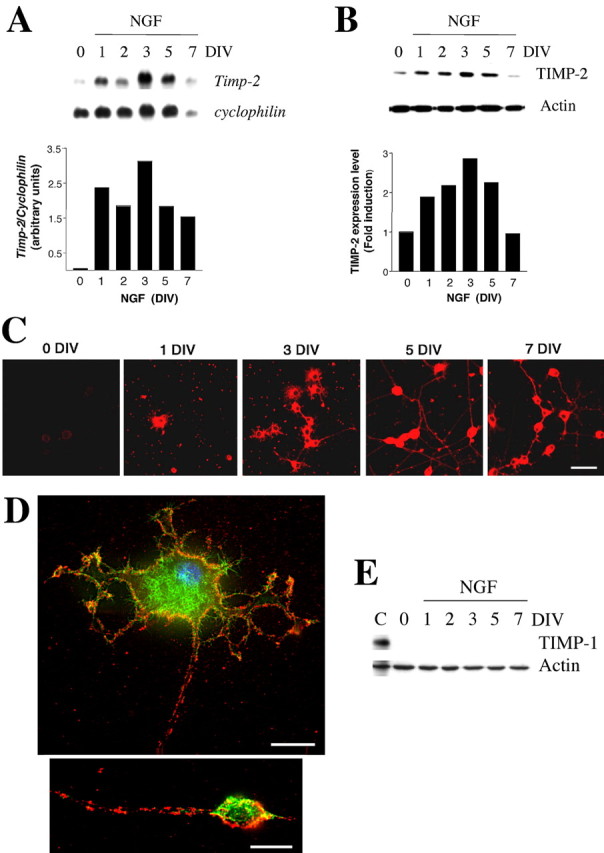
TIMP-2 expression is upregulated by NGF. A, Northern blot hybridization was performed with total RNA (25 μg) from PC12 cells treated with NFG for the indicated days. Although two TIMP-2 mRNA transcripts are present in vivo, only the 1.0 kb transcript was detected. TIMP-2 expression was normalized to the nondevelopmentally regulated gene cyclophilin. B, Western blot analysis was performed with PC12 whole-cell lysates (20 μg) treated with NGF. TIMP-2 expression was first normalized to actin at each time point, and then the extent of TIMP-2 upregulation was determined relative to undifferentiated PC12 cells (0 DIV). C, Immunocytochemistry confirms the upregulation of TIMP-2 expression coincident with PC12 differentiation observed by Northern and Western blot analyses. Scale bar, 20 μm. D, Live labeling of PC12 cells treated with NGF for 5 d. A 3 μm deconvolution section demonstrates punctate TIMP-2 surface labeling (red) suggestive of interaction with specific receptor(s). TIMP-2 binding sites on the cell soma, neuritic processes, as well as at growth cone-like processes colocalize with phalloidin (green). Scale bars: soma, 15 μm; growth cone-like process, 10 μm. E, Western blot analysis demonstrates that, unlike the upregulation of TIMP-2, TIMP-1 expression is only present in proliferating PC12 cells (C). Expression is dramatically downregulated during serum withdrawal (0 DIV) and is not detectable after NGF addition.
Because cell-cycle arrest is a measure of neuronal differentiation initiation and TIMP-2 was expressed coincident with NGF-induced PC12 cell differentiation, we determined whether TIMP-2 expression could induce neurite outgrowth, the hall-mark of terminal neuronal differentiation. PC12 cells were transfected with either TIMP-2 or vector in media containing 15% serum to maintain the cells in a proliferative environment. Differentiated PC12 cells with short neuritic processes were present, although media contained 15% serum and lacked NGF (Fig. 5Ac). Furthermore, TIMP-2-transfected cells bore processes and were Hu C/D and neurofilament-160 positive (data not shown), suggesting that TIMP-2 expression alone was sufficient to induce PC12 cell differentiation.
Figure 5.

TIMP-2-mediated PC12 neurite outgrowth is not dependent on its MMP function. A, Representative neurite outgrowth after 72 h in the presence or absence of NGF (a, e) or transfection with vector (b, f), TIMP-2 possessing MMP inhibitory activity (c, g), or mutant TIMP-2 lacking MMP inhibitory activity (TIMP-2C72S) (d, h). The short neuritic processes in control and vector-transfected cells is not surprising given that cells are maintained in 15% serum. Scale bar, 20 μm. B, Quantitation of total neurite length revealed that neurite length in TIMP-2-transfected cells is significantly increased (p < 0.05) in the absence (single asterisks) and presence (double asterisks) of NGF relative to vector-transfected cells. More strikingly, neurite length in response to the mutant TIMP-2C72S protein was comparable with active TIMP-2 in the absence (p = 0.2) and presence (p = 0.3) of NGF, clearly demonstrating that TIMP-2-mediated neurite outgrowth is MMP independent. C, Reverse zymography of transfected PC12 cell-conditioned media corroborates that the TIMP-2C72S protein lacks MMP inhibitory activity. The larger molecular weight of the wild-type active TIMP-2 protein, relative to human TIMP-2 (hTIMP-2), reflects the presence of the Flag tag. Western blot analysis with a Flag antibody was used to confirm that the TIMP-2C72S protein was indeed secreted into the media.
To determine whether terminal PC12 cell differentiation induced by TIMP-2 is independent of its MMP inhibitory activity similar to that observed for cell-cycle arrest (Fig. 1A), the ability of the TIMP-2 and the mutant TIMP-2C72S protein to induce neurite outgrowth was evaluated. To verify that TIMP-2 possessed MMP inhibitory activity, reverse zymography of conditioned media was performed. In reverse zymography, samples are electrophoresed on a nondenaturing SDS-PAGE containing gelatin and conditioned medium from BHK cells, which express MMPs. The MMPs degrade the gelatin in all regions of the gel except where there is TIMP activity. Reverse zymography verified that wild-type TIMP-2 possesses and TIMP-2C72S lacks MMP inhibitory activity (Fig. 5C, right). Western blot analysis was performed to confirm that both proteins were released into the medium (Fig. 5C, left panel). Total neurite length of TIMP-2 and TIMP-2C72S GFP-positive cells was determined 72 h after transfection (Fig. 5A,B). TIMP-2 expression induced a threefold increase (36 ± 5 μm), whereas TIMP-2C72S expression induced a 2.4-fold increase (29 ± 3 μm) in neurite length compared with vector-transfected cells (12 ± 1.3 μm), indicating that TIMP-2 mediated neurite outgrowth is independent of MMP inhibitory activity.
Because ERK activity is necessary and sufficient for neurite outgrowth in PC12 cells and is induced by TIMP-2 and NGF, we tested whether TIMP-2 could synergize with NGF to induce PC12 cell differentiation. TIMP-2- or vector-transfected cells were incubated in the presence or absence of NGF for 72 h, and the total neurite length of GFP-positive cells was determined. No differences in neurite outgrowth were observed in vector-transfected cells in the presence (11 ± 1.1 μm) or absence (12 ± 1.3 μm) of NGF relative to untransfected cells (10 ± 1 μm). The neurite outgrowth induced by TIMP-2 was 2.5-fold increased (92 ± 12 μm), whereas TIMP-2C72S was 3.7-fold increased (110 ± 12 μm) in the presence of NGF compared with vector-transfected, NGF-treated cells (Fig. 5A,B). From these results, we conclude that TIMP-2 activates a specific signal transduction pathway to act in concert with NGF to induce the expression of key molecules for neuronal differentiation and that this function does not require its MMP inhibitory activity.
NGF induces the expression of p35, the neuron-specific activator of cdk5, through activation of the ERK pathway (Harada et al., 2001). In addition, the cdk5-p35 complex is required for neurite outgrowth (Nikolic et al., 1996). To determine whether TIMP-2 regulates the cdk5-p35 complex, p35 expression in TIMP-2-transfected cells was examined. TIMP-2 induced a marked increase in p35 expression (Fig. 6A). The highest levels of p35 were detected 8 h after transfection (6.6-fold) (Fig. 6A, lanes 1, 3), which correlates with the earliest time point of detectable TIMP-2 expression (Fig. 1C). No change in p35 levels was observed in vector-transfected relative to untransfected cells (Fig. 6A).
Figure 6.

TIMP-2- and NGF-induced PC12 neurite outgrowth are synergistic and mediated by cdk5 and p35. A, p35 expression was upregulated in TIMP-2-transfected cells. Fold induction was calculated relative to nontransfected cells. B, Treatment of TIMP-2-transfected cells (48 h after transfection) with roscovitine, which blocks cdk5 activity, suggests that cdk5 activity is required for TIMP-2 to exert its neurite outgrowth-promoting effects. Roscovitine treatment not only blocked TIMP-2-mediated neurite outgrowth (compare c, g), it also blocked the synergistic effects of TIMP-2 with NGF (compare d, h). Scale bar, 20 μm.
To determine whether cdk5 activity in response to p35 expression plays a role in the neurite outgrowth mediated by TIMP-2, transfected PC12 cells were treated with a cdk5 inhibitor, roscovitine (Meijer et al., 1997). Proliferating PC12 cells were transfected with TIMP-2 in the absence or presence of NGF and incubated for 48 h with or without roscovitine. In the absence of roscovitine, TIMP-2-mediated neurite outgrowth was greatly enhanced in the presence of NGF (Fig. 6Bc,Bd). In the presence of roscovitine, however, the neurite outgrowth induced by either TIMP-2 alone or TIMP-2 plus NGF was severely impaired, with neurites no longer apparent (Fig. 6Bg,Bh). Given that PC12 cells were transfected in media containing 15% serum, cells transfected with vector alone and treated with NGF did not develop neurites, in either the absence or presence of roscovitine (Fig. 6Bb,Bf). No change in cell morphology was observed after roscovitine treatment (Fig. 6Ba,Be). These results show that TIMP-2- and NGF-induced PC12 neurite outgrowth are synergistic and mediated by cdk5 and p35.
TIMP-2 regulates CNS neurogenesis during development
TIMP-2 is expressed by neurons coincident with the onset of neuronal differentiation in vivo, and the data presented thus far demonstrate that TIMP-2 induces neuronal differentiation in vitro. Therefore, we sought to further characterize the role of TIMP-2 during nervous system development using mice carrying a targeted mutation in the TIMP-2 gene (Wang et al., 2000). No TIMP-2 mRNA was detected, indicating a null mutation. Although it was reported that the TIMP-2-/- mice lacked a gross phenotype, nervous system development was not analyzed. Thus, we examined whether the absence of TIMP-2 expression results in neuronal differentiation defects.
To determine whether TIMP-2 promotes neurite outgrowth in vitro as observed in PC12 cells, primary neuronal cultures were prepared from the cerebral cortex of newborn (P0) TIMP-2-/- and wild-type mice. TIMP-2-deficient mice showed a marked reduction in neurite outgrowth (62 ± 13 μm) compared with wild-type mice (351 ± 78 μm) (Fig. 7Aa,Ab and bottom panel). Because TIMP-2 acts synergistically with NGF to induce neurite outgrowth in PC12 cells (Fig. 5), we tested whether NGF treatment could rescue the neurite outgrowth phenotype in TIMP-2-/- neurons. NGF treatment only partially rescued the neurite length in cortical neurons (239 ± 68 μm) (Fig. 7Ac and bottom panel). These data indicate that TIMP-2 expression is necessary for neurite outgrowth in primary cultured cortical neurons.
Figure 7.

TIMP-2 knock-out mice display altered neuronal differentiation in vitro. A, Representative morphology of primary neuronal cultures of P0 cerebral cortex stained with PGP 9.5 after 8 DIV. Neurite length of TIMP-2-/- neurons was significantly reduced (5.6-fold) relative to wild-type neurons (*p < 0.001). Interestingly, NGF was able to partially rescue the TIMP-2 null phenotype (**p < 0.001); however, the increase did not attain wild-type lengths (p = 0.28). Scale bar, 25 μm. B, Primary P0 cortical cultures were stained with PGP 9.5 after 8 DIV. The reduced neurite length of TIMP-2-/- P0 cerebral cortical neurons was rescued by transfection with TIMP-2 (787 ± 117 μm) or by exogenous treatment with rTIMP-2 (293 ± 33 μm) when compared with vector-transfected (63 ± 11 μm) or untransfected cultures (43 ± 4 μm) (*p < 0.003). However, neurite length in response to rTIMP-2 was significantly less than that in response to transfection (**p < 0.001). Scale bar, 25 μm. ko, Knock-out; wt, wild-type.
To test the ability of TIMP-2 to rescue neurite outgrowth in the TIMP-2-/- neurons, primary cortical neurons were either transfected with TIMP-2 cDNA or treated with rTIMP-2. TIMP-2 transfection significantly increased total neurite length (787 ± 117 μm) compared with vector-transfected (63 ± 11 μm) or untransfected (43 ± 13 μm) cells (Fig. 7Ba,Bc,Bd and bottom panel). Exogenous application of rTIMP-2 also rescued the TIMP-2-/- phenotype (Fig. 7Bb and bottom panel). Although this treatment was less effective (293 ± 33 μm) than TIMP-2 transfection, it was sufficient to rescue neurite length comparable with wild-type neurons (351 ± 78 μm) (Fig. 7Aa and bottom panel). These results further demonstrate that TIMP-2 expression is necessary for proper neuronal differentiation.
To determine whether TIMP-2 deletion alters neurogenesis in vivo, cerebral cortical histogenesis was examined. At embryonic day 16 (E16), the size of the TIMP-2-/- cortical plate is reduced, whereas the ventricular zone is enhanced relative to wild-type littermates as revealed by Nissl stain (Fig. 8A). As observed at E16, the P0 cerebral cortex of TIMP-2-/- mice is thinner than wild-type littermates. This size reduction is not restricted to the cerebral cortex but is reflective of an overall decreased knock-out brain size (Fig. 8B). The reduction in brain size is not attributable to a reduced body size because the brain to body weight ratio of knock-out mice is also reduced. Reduced brain size is only apparent in the first postnatal week; afterward, there is no significant difference between TIMP-2-/- and wild-type mice. To determine whether the reduced cortical size resulted from fewer cells, cells of reduced size, decreased neuritic arborization, or increased cell death, immunohistochemistry was performed at P0 (Fig. 8C). PCNA labeling is reduced in the TIMP-2-/- cortex, in both the cortical plate and ventricular/subventricular zone. The reduction of PCNA at P0, well after neurogenesis occurs in the cerebral cortex, suggests that gliogenesis may be altered in TIMP-2-/- mice. Closer examination of the P0 neocortical ventricular zone reveals that TIMP-2-/- mice contains more nestin-positive progenitor cells than wild-type mice, in agreement with our hypothesis and in vitro data that TIMP-2 plays a role in neurogenesis. Moreover, neurofilament staining demonstrates that neuritic length and complexity is reduced in knock-out mice compared with wild-type mice. To test whether TIMP-2 interacts with integrins similar to that observed in PC12 cells, α3 and β1 integrins were immunoprecipitated, and Western blot analysis with TIMP-2 was performed from E16 cortex, a period of rapid neuronal proliferation (Fig. 8D). Analysis of embryos from two different litters clearly demonstrates the interaction of TIMP-2 with α3β1 integrin in vivo, consistent with our in vitro observation (Fig. 3A). This interaction is specific because immunoprecipitation with EGFR and blotting with TIMP-2 did not demonstrate interaction (data not shown). Together, these data suggest an incomplete or delayed initial and terminal neuronal differentiation in the absence of TIMP-2. It further suggests that TIMP-2 may exert its role on neuronal differentiation via activation of α3β1 integrin.
Figure 8.
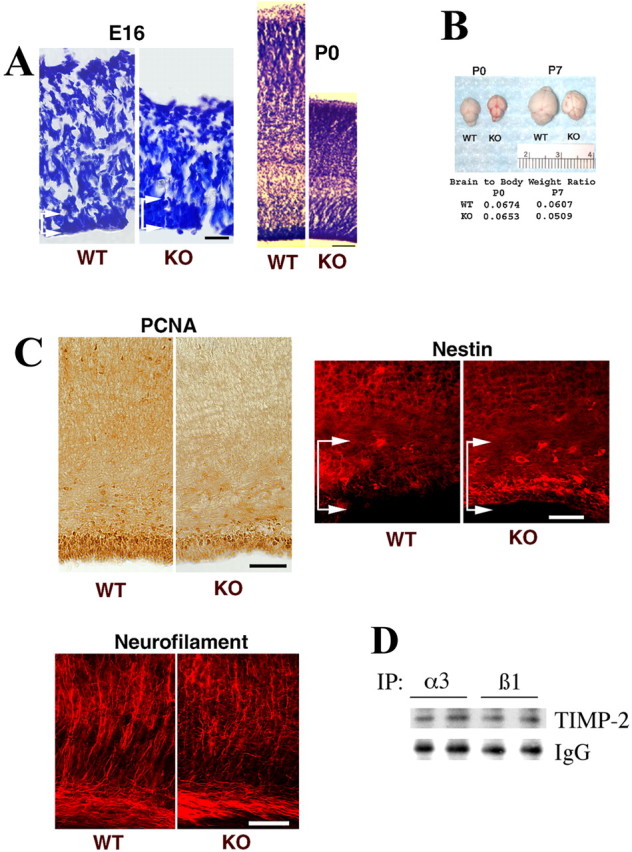
TIMP-2 knock-out mice display altered neuronal differentiation in vivo. A, Cresyl violet-stained sections at E16 and P0 cerebral motor cortex show a reduction in cortical size and increased ventricular zone size (area between arrows) in TIMP-2-/- (KO) mice relative to wild-type (WT) littermates. Scale bars: E16, 25 μm; P0, 100 μm. B, Gross specimens demonstrating reduced TIMP-2-/- brain size, which is reflected in a reduced brain to body weight ratio (n = 4). C, The reduction in cortical thickness is associated with decreased PCNA labeling throughout the cortical anlage. The presence of more nestin-positive progenitors in the cerebral cortical ventricular zone (area between arrows) in TIMP-2-/- mice suggests that neuronal differentiation is perturbed by TIMP-2 deletion. In addition, the number and organization of neurofilament-positive processes is altered in the TIMP-2-/- cortex. Scale bars, 50 μm. D, Cell lysates of E16 wild-type cortex from two different litters were immunoprecipitated (IP) using anti-α3 and anti-β1 integrin antibodies, and association for TIMP-2 was determined by Western blot analysis. This analysis clearly demonstrates that TIMP-2 interacts with α3β1 integrin in vivo.
Discussion
Soluble molecules, including growth factors and ECM molecules, play a key role in regulating neurogenesis. Here, we demonstrated that TIMP-2 promotes neuronal differentiation by inhibiting cell-cycle progression independent of its MMP inhibitory activity. In addition, neuronal differentiation in vivo is delayed in the absence of TIMP-2. This is the first report to demonstrate a phenotype for the TIMP-2-/- mouse and detail a mechanism for TIMP-2 action in neuronal differentiation.
TIMP-2 as a regulator of proliferation
The G1 restriction point is a critical regulator of neocortical neurogenesis (Caviness et al., 1999) and, hence, is controlled by multiple positive and negative signals. Although it is well accepted that mature neurons cannot reenter the cell cycle, the exact mechanism underlying mitotic quiescence is not well understood. Negative regulatory signals, such as TIMP-2, might serve as a G1 regulator to inhibit growth factor signals and, thus, promote cell-cycle arrest.
TIMP-2 can exert growth-inhibiting (Hoegy et al., 2001; Seo et al., 2003) and growth-promoting (Hayakawa et al., 1994) activities depending on the cellular context. Given that both functions are independent of MMP inhibition, TIMP-2 must activate distinct signaling pathways. Cell-cycle progression is regulated by growth factors and cell adhesion to the ECM (Renshaw et al., 1997). Growth factors and ECM molecules bind to receptor tyrosine kinases (RTKs) and integrins, respectively. RTKs and integrins cooperate to influence cell-cycle progression by regulating expression of cell-cycle regulatory proteins (for review, see Miranti and Brugge, 2002) and activation of ERK pathways. Here we showed that TIMP-2 induced cell-cycle arrest independent of MMP inhibitory activity by increasing the expression of the cdk inhibitor p21Cip1 and inhibiting cyclins B and D. Furthermore, TIMP-2 activates the cAMP/Rap1/B-Raf pathway. Because Rap1 can suppress Ras activity and, hence, EGFR-mediated proliferation (Cook and McCormick, 1993; Schmitt and Stork, 2002), the decreased EGFR activation detected in TIMP-2-transfected PC12 cells is likely a consequence of Rap1-mediated inhibition of the EGFR/Ras pathway and not a direct effect of TIMP-2 on the EGFR. Given that TIMP-2 binds to integrins, in both PC12 cells and the cerebral cortex in vivo, the cell-cycle arrest induced by TIMP-2 may be mediated via integrin activation.
In contrast to previous studies that used serum-starved quiescent cells, our studies were conducted in the presence of growth factors, conditions that are more representative of the neuroepithelium in which proliferating cells are juxtaposed with cells having just undergone differentiation. Interestingly, this permitted the observation that TIMP-2 expression induced PC12 cell-cycle arrest in a cell-autonomous manner in that GFP-positive TIMP-2-transfected cells, but not GFP-negative cells, in the same well underwent growth arrest. One possible explanation for this phenomenon is that only cells expressing specific cell surface receptors will respond to TIMP-2 signals. However, all PC12 cells express α3β1 integrin and, thus, should be responsive. A more plausible explanation for the apparent cell-autonomous effect is the presence of a TIMP-2 concentration gradient. Both mitogenic (Megason and McMahon, 2002) and morphogenetic (Cadigan, 2002) gradients play a role in neurogenesis. MMPs and their inhibitors regulate ECM molecule concentration gradients critical to bind cell surface receptors (Kanwar et al., 2004). For example, in our studies, α3β1 integrin may be activated by the elevated TIMP-2 concentrations present only around transfected cells. This hypothesis is supported by our rescue experiments in which addition of soluble TIMP-2 to primary cortical cultures did not rescue the TIMP-2-/- neurite outgrowth phenotype as effectively as TIMP-2 transfection.
If TIMP-2 plays a key role in the initiation of neuronal differentiation (e.g., cell-cycle arrest), one would predict neuronal hyperplasia in its absence. As observed for other cell-cycle regulatory molecules, in which results obtained from null mutations were enigmatic (Deng et al., 1995; Tong and Pollard, 2001), we observed a reduction in TIMP-2-/- brain size. The persistence of nestin-positive progenitors in the neocortical ventricular zone suggests that cell-cycle progression is indeed affected by the absence of TIMP-2. At present, we cannot rule out the possibility that TIMP-2-/- mice indeed display neuronal hyperplasia that is compensated for by reduced cell size, decreased process outgrowth, or increased apoptosis. Neurites of TIMP-2-/- neurons are significantly shorter than wild-type neurons in vitro, suggesting that the thinner cortices of TIMP-2-/- mice may stem from this neurite outgrowth defect. BrdU pulse labeling experiments during embryonic corticogenesis need to be performed to evaluate changes in neural progenitor cell-cycle dynamics in vivo.
TIMP-2 as a regulator of neurite outgrowth
Cell-cycle arrest is required for neuronal differentiation (Zhu and Skoultchi, 2001), and proliferation inhibition is correlated with the appearance of neurite outgrowth (Song et al., 2005), suggesting a causal relationship. However, these processes may merely be sequential in time and use separate and distinct pathways. Both NGF and integrins mediate growth arrest and subsequent PC12 cell neurite outgrowth. cAMP has been implicated in the regulation of integrin-mediated neurite outgrowth and growth cone guidance (Bos et al., 2003; Ivins et al., 2004). Furthermore, the cAMP/Epac pathway has been shown to synergize with NGF to promote PC12 cell neurite outgrowth (Christensen et al., 2003). Therefore, it is possible that the cAMP pathway regulating TIMP-2-mediated neurite outgrowth is distinct from that regulating cell-cycle inhibition.
Here, we present several lines of evidence that TIMP-2 expression negatively regulates cell-cycle progression, thereby resulting in neurite outgrowth. In addition, we showed that NGF acted synergistically with TIMP-2 to promote PC12 cell neurite outgrowth and partially rescued the reduced neurite length of TIMP-2-/- cortical neurons in vitro. The synergistic effect of NGF with TIMP-2 is not likely attributable to a direct interaction of TIMP-2 with TrkA because no change in TrkA expression was detected in TIMP-2-transfected PC12 cells. Furthermore, the partial rescue of neurite outgrowth of cultured TIMP-2-/- neurons by NGF cannot be a direct effect because cortical neurons do not express TrkA. However, at present, the participation of p75, the low-affinity neurotrophin receptor, cannot be excluded. Neurotrophin binding to the p75 receptor modulates axonal outgrowth (Yamashita et al., 1999), but p75-mediated outgrowth occurs via RhoA activation and not p35/cdk5, as observed for TIMP-2-transfected cells. Rather, cdk5 provides a point of convergence for NGF-mediated TrkA- and TIMP-2-mediated neurite outgrowth. Induction of the p35-cdk5 complex induces neurite sprouting in neuronal cell lines and during neuronal differentiation in vivo (Nikolic et al., 1996). NGF induces a sustained expression of p35 through activation of the ERK pathway (Harada et al., 2001). Integrin engagement also activates ERK (Short et al., 2000) and cdk5 (Li et al., 2000). Given that TIMP-2 upregulates p35 expression and TIMP-2-mediated neurite outgrowth requires cdk5 activity, it suggests that p35 is the major target for TIMP-2 in neurite outgrowth.
Why would TIMP-2 bind to integrin receptors directly when it is capable of regulating proteolysis and, thus, availability of integrin ligands? The temporal and spatial expression of TIMP-2 as well as the signaling mechanism activated likely plays a role. During early development, retinal neurons extend neurites in response to laminin-1 via α6β1 integrin (Calof et al., 1994). Late in development, they lose laminin-1 responsiveness attributable to decreased integrin activation (de Curtis and Reichardt, 1993). Because TIMP-2 expression is maintained at high levels into adulthood, it could overcome low expression levels of ECM molecules or changes in receptor affinity required for integrin activation. Alternatively, integrin receptor localization might allow TIMP-2 to exert multiple functions during neuronal differentiation. Distinct signaling pathways and biological responses can be activated by the same growth factor receptor depending on its cellular localization (Whitmarsh and Davis, 2001). TIMP-2 expression is enriched on the soma and at growth cone-like processes. Activation of an integrin receptor on the soma may elicit one cellular response, whereas activation at the nerve terminal may elicit another response. TIMP-2 expression at branch points, at which it colocalizes with phalloidin, suggests a role in modulating actin network dynamics to control branching morphogenesis, as observed for TIMP-2 in the metanephros (Barasch et al., 1999). Interestingly, it was reported recently that α3-/- integrin cortical neurons display aberrant actin cytoskeletal dynamics at the leading edge (Schmid et al., 2004).
Herein, we showed that TIMP-2 binds to α3β1 integrin and regulates cell-cycle progression of PC12 cells independent of MMP inhibitory activity. Our current model proposes that TIMP-2 is secreted into the extracellular space in which it binds to α3β1 integrin, which activates a cAMP/Rap1/Epac signaling pathway to maintain neuronal differentiation. Whether binding of TIMP-2 to α3β1 or other integrins exerts MMP-independent actions in vivo is yet to be determined. We cannot assume a priori that TIMP-2 binds to a single receptor (e.g., G-protein-coupled receptors or integrins) to exert its effect on cell-cycle arrest and neurite outgrowth. TIMP-2 may exert its diverse biological functions via interaction with multiple developmentally regulated receptors.
In addition to cell cycle and neurite outgrowth, integrins play a role in synaptic plasticity (Chan et al., 2003) and nerve regeneration (Werner et al., 2000; Vogelezang et al., 2001). TIMP-2-/- mice are deficient in the acquisition of fear-potentiated startle, a model of synaptic plasticity in the amygdala (D. M. Jaworski and W. A. Falls, unpublished observation). Whether TIMP-2 also plays a role in neuronal regeneration warrants additional investigation. A low state of integrin activation is associated with poorly regenerating neurons (Ivins et al., 2000). If TIMP-2 exerts effects on regeneration in an MMP-independent manner, it opens the possibility of using TIMP-2 therapeutically in response to injury.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS35874 and NS045225 [cofunded by the National Institute of Neurological Disorders and Stroke and National Center for Research Resources (NCRR)] and American Heart Association Grant 9950039N (D.M.J.). We thank Dr. Johanes L. Bos (Utrecht University, Utrecht, The Netherlands) for the kind gift of the RalGDS GST-RBD and Raf1 GST-RBD constructs and Dr. Philip J. S. Stork (Oregon Health Sciences University, Portland, OR) for the plasmid containing the dominant-negative RapN17 (pCMVRAP1b). TIMP-2-/- mice were kindly provided by Dr. Paul D. Soloway (Cornell University, Ithaca, NY). We also thank Dr.William G. Stetler-Stevenson (NIH, Bethesda, MD) for the kind gift of the recombinant TIMP-2. We are grateful to Dr. Victor May for insightful scientific discussion and Dr. Mercedes Rincon for graciously sharing reagents. Finally, we thank Dr. Gustavo Pedraza-Alva for critical review of this manuscript. We also thank the University of Vermont Neuroscience Center of Biomedical Research Excellence for the use of the Delta Vision Restoration Microscopy System (NIH-NCRR Grant IP20 RR16435). Densitometric analysis was performed in the Vermont Cancer Center DNA Analysis Facility and was supported in part by National Cancer Institute Grant P30CA22435. L.P.-M. was on research leave at the University of Vermont.
Correspondence should be addressed to Dr. Leonor Pérez-Martínez, Departamento de Genética del Desarrollo y Fisiología Molecular, Instituto de Biotecnología, Universidad Nacional Autónoma de México, Apdo Postal 510-3, Cuernavaca, Morelos 62210, México. E-mail: leonor@ibt.unam.mx.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254917-13$15.00/0
References
- Arregui CO, Carbonetto S, McKerracher L (1994) Characterization of neural cell adhesion sites: point contacts are the sites of interaction between integrins and the cytoskeleton in PC12 cells. J Neurosci 14: 6967-6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker AH, Edwards DR, Murphy G (2002) Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci 115: 3719-3727. [DOI] [PubMed] [Google Scholar]
- Barasch J, Yang J, Qiao J, Tempst P, Erdjument-Bromage H, Leung W, Oliver JA (1999) Tissue inhibitor of metalloproteinase-2 stimulates mesenchymal growth and regulates epithelial branching during morphogenesis of the rat metanephros. J Clin Invest 103: 1299-1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond M, Murphy G, Bennett MR, Amour A, Knauper V, Newby AC, Baker AH (2000) Localization of the death domain of tissue inhibitor of metalloproteinase-3 to the N terminus. J Biol Chem 275: 41358-41363. [DOI] [PubMed] [Google Scholar]
- Bos JL, De Bruyn K, Enserink J, Kuiperij B, Rangarajan S, Rehmann H, Riedl J, De Rooij J, Van Mansfeld F, Zwartkruis F (2003) The role of Rap1 in integrin-mediated cell adhesion. Biochem Soc Trans 31: 83-86. [DOI] [PubMed] [Google Scholar]
- Cadigan KM (2002) Regulating morphogen gradients in the Drosophila wing. Semin Cell Dev Biol 13: 83-90. [DOI] [PubMed] [Google Scholar]
- Calof AL, Campanero MR, O'Rear JJ, Yurchenco PD, Lander AD (1994) Domain-specific activation of neuronal migration and neurite outgrowth-promoting activities of laminin. Neuron 13: 117-130. [DOI] [PubMed] [Google Scholar]
- Caterina NC, Windsor LJ, Yermovsky A, Bodden M, Taylor KB, Birkedal-Hansen H, Engler JA (1997) Replacement of conserved cysteines in human tissue inhibitor of metalloproteinases-1. J Biol Chem 272: 32141-32149. [DOI] [PubMed] [Google Scholar]
- Caviness Jr VS, Takahashi T, Nowakowski RS (1999) The G1 restriction point as critical regulator of neocortical neuronogenesis. Neurochem Res 24: 497-506. [DOI] [PubMed] [Google Scholar]
- Chan CS, Weeber EJ, Kurup S, Sweatt JD, Davis RL (2003) Integrin requirement for hippocampal synaptic plasticity and spatial memory. J Neurosci 23: 7107-7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen AE, Selheim F, de Rooij J, Dremier S, Schwede F, Dao KK, Martinez A, Maenhaut C, Bos JL, Genieser HG, Doskeland SO (2003) cAMP analog mapping of Epac1 and cAMP kinase. Discriminating analogs demonstrate that Epac and cAMP kinase act synergistically to promote PC-12 cell neurite extension. J Biol Chem 278: 35394-35402. [DOI] [PubMed] [Google Scholar]
- Cook SJ, McCormick F (1993) Inhibition by cAMP of Ras-dependent activation of Raf. Science 262: 1069-1072. [DOI] [PubMed] [Google Scholar]
- Dailey L, Laplantine E, Priore R, Basilico C (2003) A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J Biol Chem 161: 1053-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Curtis I, Reichardt LF (1993) Function and spatial distribution in developing chick retina of the laminin receptor α6β1 and its isoforms. Development 118: 377-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396: 474-477. [DOI] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P (1995) Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82: 675-684. [DOI] [PubMed] [Google Scholar]
- Fager N, Jaworski DM (2000) Differential spatial distribution and temporal regulation of tissue inhibitor of metalloproteinase mRNA expression during rat central nervous system development. Mech Dev 98: 105-109. [DOI] [PubMed] [Google Scholar]
- Fata JE, Leco KJ, Moorehead RA, Martin DC, Khokha R (1999) Timp-1 is important for epithelial proliferation and branching morphogenesis during mouse mammary development. Dev Biol 211: 238-254. [DOI] [PubMed] [Google Scholar]
- Harada T, Morooka T, Ogawa S, Nishida E (2001) ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat Cell Biol 3: 453-459. [DOI] [PubMed] [Google Scholar]
- Hayakawa T, Yamashita K, Ohuchi E, Shinagawa A (1994) Cell growth-promoting activity of tissue inhibitor of metalloproteinase-2 (TIMP-2). J Cell Sci 107: 2373-2379. [DOI] [PubMed] [Google Scholar]
- Hoegy SE, Oh HR, Corcoran ML, Stetler-Stevenson WG (2001) Tissue inhibitor of metalloproteinases-2 (TIMP-2) suppresses TKR-growth factor signaling independent of metalloproteinase inhibition. J Biol Chem 276: 3203-3214. [DOI] [PubMed] [Google Scholar]
- Ivins JK, Yurchenco PD, Lander AD (2000) Regulation of neurite outgrowth by integrin activation. J Neurosci 20: 6551-6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivins JK, Parry MK, Long DA (2004) A novel cAMP-dependent pathway activates neuronal integrin function in retinal neurons. J Neurosci 24: 1212-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski DM, Kelly GM, Hockfield S (1994) BEHAB, a new member of the proteoglycan tandem repeat family of hyaluronan-binding proteins that is restricted to brain. J Cell Biol 125: 495-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski DM, Kelly GM, Hockfield S (1999) Intracranial injury acutely induces the expression of the secreted isoform of the CNS-specific hyaluronan-binding protein BEHAB/brevican. Exp Neurol 157: 327-337. [DOI] [PubMed] [Google Scholar]
- Kanwar YS, Wada J, Lin S, Danesh FR, Chugh SS, Yang Q, Banerjee T, Lomasney JW (2004) Update of extracellular matrix, its receptors, and cell adhesion molecules in mammalian nephrogenesis. Am J Physiol Renal Physiol 286: F202-F215. [DOI] [PubMed] [Google Scholar]
- Kao S, Jaiswal RK, Kolch W, Landreth GE (2001) Identification of the mechanisms regulating the differential activation of the mapk cascade by epidermal growth factor and nerve growth factor in PC12 cells. J Biol Chem 276: 18169-18177. [DOI] [PubMed] [Google Scholar]
- Li BS, Zhang L, Gu J, Amin ND, Pant HC (2000) Integrin α1β1-mediated activation of cyclin-dependent kinase 5 activity is involved in neurite outgrowth and human neurofilament protein H Lys-Ser-Pro tail domain phosphorylation. J Neurosci 20: 6055-6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CJ (1995) Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80: 179-185. [DOI] [PubMed] [Google Scholar]
- McCawley LJ, Matrisian LM (2001) Matrix metalloproteinases: they're not just for matrix anymore! Curr Opin Cell Biol 13: 534-540. [DOI] [PubMed] [Google Scholar]
- Megason SG, McMahon AP (2002) A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development 129: 2087-2098. [DOI] [PubMed] [Google Scholar]
- Meijer L, Borgne A, Mulner O, Chong JP, Blow J, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP (1997) Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem 243: 527-536. [DOI] [PubMed] [Google Scholar]
- Minturn JE, Fryer HJL, Geschwind DH, Hockfield S (1995) TOAD-64, a gene expressed early in neuronal differentiation in the rat, is related to unc-33, a C. elegans gene involved in axon outgrowth. J Neurosci 15: 6757-6766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranti CK, Brugge JS (2002) Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol 4: 83-90. [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Himi T, Garcia V, Yamagishi H, Sato S, Ishizaki Y (2000) A role for p27/Kip1 in the control of cerebellar granule cell precursor proliferation. J Neurosci 20: 5756-5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase H (1997) Activation mechanisms of matrix metalloproteinases. Biol Chem 378: 151-160. [PubMed] [Google Scholar]
- Nagase H, Woessner JF (1999) Matrix metalloproteinases. J Biol Chem 274: 21491-21494. [DOI] [PubMed] [Google Scholar]
- Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH (1996) The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev 10: 816-825. [DOI] [PubMed] [Google Scholar]
- Ohnuma S, Harris JA (2003) Neurogenesis and the cell cycle. Neuron 40: 199-208. [DOI] [PubMed] [Google Scholar]
- Rebhun JF, Castro AF, Quilliam LA (2000) Identification of guanine nucleotide exchange factors (GEFs) for the Rap1 GTPase. Regulation of MR-GEF by M-Ras-GTP interaction. J Biol Chem 275: 34901-34908. [DOI] [PubMed] [Google Scholar]
- Renshaw MW, Ren XD, Schwartz MA (1997) Growth factor activation of MAP kinase requires cell adhesion. EMBO J 16: 5592-5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter LM, Garfield SH, Thorgeirsson UP (1999) Tissue inhibitor of metalloproteinases-1 (TIMP-1) binds to the cell surface and translocates to the nucleus of human MCF-7 breast carcinoma cells. Biochem Biophys Res Commun 257: 494-499. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Shelton S, Stanco A, Yokota Y, Kreidberg JA, Anton ES (2004) α3β1 integrin modulates neuronal migration and placement during early stages of cerebral cortical development. Development 131: 6023-6031. [DOI] [PubMed] [Google Scholar]
- Schmitt JM, Stork PJS (2002) PKA phosphorylation of src mediates cAMP's inhibition of cell growth via Rap1. Mol Cell 9: 85-94. [DOI] [PubMed] [Google Scholar]
- Seo DW, Li H, Guedez L, Wingfield PT, Diaz T, Salloum R, Wei BY, Stetler-Stevenson WG (2003) TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell 114: 171-180. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13: 1501-1512. [DOI] [PubMed] [Google Scholar]
- Short SM, Boyer JL, Juliano RL (2000) Integrins regulate the linkage between upstream and downstream events in G protein-coupled receptor signaling to mitogen-activated protein kinase. J Biol Chem 275: 12970-12977. [DOI] [PubMed] [Google Scholar]
- Song JH, Wang CX, Song DK, Wang P, Shuaib A, Hao C (2005) Interferon gamma induces neurite outgrowth by upregulation of p35 neuron-specific cyclin-dependent kinase 5 activator via activation ERK1/2 pathway. J Biol Chem 280: 12896-12901. [DOI] [PubMed] [Google Scholar]
- Suh J, Lu N, Nicot A, Tatsuno I, DiCicco-Bloom E (2001) PACAP is an anti-mitogenic signal in developing cerebral cortex. Nat Neurosci 4: 1-2. [DOI] [PubMed] [Google Scholar]
- Tomaselli KJ, Hall DE, Flier LA, Gehlsen KR, Turner DC, Carbonetto S, Reichardt LF (1990) A neuronal cell line (PC12) expresses two β1-class integrins-α1β1 and α3β1-that recognize different neurite outgrowth-promoting domains in laminin. Neuron 5: 651-662. [DOI] [PubMed] [Google Scholar]
- Tong W, Pollard JW (2001) Genetic evidence for the interactions of cyclin D1 and p27(Kip1) in mice. Mol Cell Biol 4: 1319-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant C, Didier-Bazès M, Hutter A, Belin M-F, Thomasset N (1999) Spatiotemporal expression patterns of metalloproteinases and their inhibitors in the postnatal developing rat cerebellum. J Neurosci 19: 4994-5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelezang MG, Liu Z, Relvas J, Raivich G, Scherer SS, ffrench-Constant C (2001) α4 integrin is expressed during peripheral nerve regeneration and enhances neurite outgrowth. J Neurosci 21: 6732-6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ (1997) cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 89: 73-82. [DOI] [PubMed] [Google Scholar]
- Wang Z, Juttermann R, Soloway PD (2000) TIMP-2 is required for efficient activation of proMMP-2 in vivo J Biol Chem 275: 26411-26415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner A, Willem M, Jones LL, Kreutzberg GW, Mayer U, Raivich G (2000) Impaired axonal regeneration in α7 integrin-deficient mice. J Neurosci 20: 1822-1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ, Davis RJ (2001) Signal transduction by target-derived neurotrophins. Nat Neurosci 4: 963-964. [DOI] [PubMed] [Google Scholar]
- Wu C, Lai C-F, Mobley WC (2001) Nerve growth factor activates persistent Rap1 signaling in endosomes. J Neurosci 21: 5406-5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Tucker KL, Barde YA (1999) Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 24: 585-593. [DOI] [PubMed] [Google Scholar]
- Young DA, Phillips BW, Lundy C, Nuttall RK, Hogan A, Schultz GA, Leco KJ, Clark IM, Edwards DR (2002) Identification of an initiator-like element essential for the expression of the tissue inhibitor of metalloproteinases-4 (Timp-4) gene. Biochem J 364: 89-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Tarone G, Turner DC (1993) Expression of integrin α1β1 is regulated by nerve growth factor and dexamethasone in PC12 cells. Functional consequences for adhesion and neurite outgrowth. J Biol Chem 268: 5557-5565. [PubMed] [Google Scholar]
- Zhao WQ, Li H, Yamashita K, Guo XK, Hoshino T, Yoshida S, Shinya T, Hayakawa T (1998) Cell cycle-associated accumulation of tissue inhibitor of metalloproteinases-1 (TIMP-1) in the nuclei of human gingival fibroblasts. J Cell Sci 111: 1147-1153. [DOI] [PubMed] [Google Scholar]
- Zhu L, Skoultchi AI (2001) Coordinating cell proliferation and differentiation. Curr Opin Genet Dev 11: 91-97. [DOI] [PubMed] [Google Scholar]