

Abstract
DAP12 is a signaling adaptor containing an immunoreceptor tyrosine-based activation motif (ITAM) that pairs with receptors on myeloid cells and natural killer cells. We examine here the responses of mice lacking DAP12 to stimulation through Toll-like receptors (TLRs). Unexpectedly, DAP12-deficient macrophages produced higher concentrations of inflammatory cytokines in response to a variety of pathogenic stimuli. Additionally, macrophages deficient in spleen tyrosine kinase (Syk), which signals downstream of DAP12, showed a phenotype identical to that of DAP12-deficient macrophages. DAP12-deficient mice were more susceptible to endotoxic shock and had enhanced resistance to infection by the intracellular bacterium Listeria monocytogenes. These data suggest that one or more DAP12-pairing receptors negatively regulate signaling through TLRs.
TLRs are pattern recognition receptors used by cells of the innate immune system to detect the presence of a wide variety of pathogens. The 11 TLRs known in mammals allow the innate immune system to respond to a range of pathogen products, including proteins, lipoproteins, polysaccharides and nucleic acids1. In macrophages, TLR ligation leads to the production of proinflammatory cytokines, including tumor necrosis factor (TNF), interleukin 6 (IL-6) and IL-12. This cytokine production is crucial to a productive innate and adaptive immune response, leading to pathogen clearance. Additionally, when cytokine production is not properly regulated, inflammatory disorders such as rheumatoid arthritis or septic shock may result.
The TLR signaling pathway differs somewhat for each TLR1. In common are a series of signaling proteins beginning with the adaptor protein MyD88, which associates with TLRs through homotypic interactions of Toll-IL-1 receptor (TIR) domains. When associated with a TLR, MyD88 recruits members of the IL-1 receptor–associated kinase (IRAK) family of serine-threonine kinases, which then phosphorylate and activate the ubiquitin ligase TNF receptor–associated factor 6 (TRAF6). Activation of TRAF6 leads to activation of the three mitogen-activated protein kinase (MAPK) pathways, p38 MAPK, ERK and JNK, and results in translocation of the transcription factor NF-κB into the nucleus. This occurs through the phosphorylation and degradation of IκBα, a protein that binds and retains NF-κB in the cytoplasm in resting cells. NF-κB is a key transcription factor in the induction of proinflammatory cytokines in macrophages, including TNF, IL-6 and IL-12 p40. Some TLRs also use other TIR-containing adaptor proteins, including TIRAP (also called Mal), TRIF (also called TICAM-1) and TRAM, which contribute to MyD88 signals or activate additional signaling pathways, such as those leading to interferon-β production.
The DAP12 signaling adaptor protein is expressed in cells that participate in innate immune responses, including macrophages, granulocytes and natural killer cells. DAP12 is a disulfide-linked, homodimeric transmembrane protein with a minimal extracellular domain, a charged aspartic acid in the transmembrane domain and an ITAM in its cytoplasmic tail2. DAP12 uses the acidic residue in its transmembrane domain to noncovalently associate with cell surface receptors that have a basic amino acid in their transmembrane region. Neither DAP12 nor many of its associated receptors can efficiently reach the cell surface alone, and DAP12 is required for their signaling abilities. In myeloid cells, several DAP12-associated receptors have been identified. These receptors fall into two categories: members of the immunoglobulin domain superfamily, such as TREM-1, TREM-2, TREM-3, MAIR-II, CD200RLa, SIRP-β and PILR-β; and members of the C-type lectin family, such as MDL-1 and mouse NKG2D-short3–14. Dendritic cell (DC) abnormalities have been observed in DAP12-deficient mice15,16.
The signaling cascade downstream of DAP12 and other ITAM-containing signaling adaptors, such as FcɛRIγ and CD3ζ, has been well characterized17,18. After ligation of a DAP12-associated receptor, the tyrosines within the ITAM are phosphorylated, presumably by Src family tyrosine kinases. In macrophages, these phosphotyrosines recruit the Syk tyrosine kinase, which promotes the recruitment and activation of adaptor complexes leading to the activation of the phospholipase C-γ, ERK and phosphatidylinositol-3 (PI3) kinase pathways. The best studied of these is the downstream phospholipase C-γ pathway, which leads to the activation of the transcription factor NFAT. In macrophages, ligation of DAP12-associated receptors results in secretion of cytokines and chemokines, including TNF, IL-6 and MCP-1 (ref. 3).
Two DAP12-pairing receptors, TREM-1 and TREM-2, are specifically implicated in responses to bacteria and bacterial products4,19. TREM-1 expression is induced on macrophages and neutrophils in response to lipopolysaccharide (LPS), a component of the cell wall of Gram-negative bacteria and a ligand for TLR4 (ref. 3). Additionally, a fusion protein consisting of the TREM-1 extracellular domain and the Fc portion of IgG reduces LPS-induced toxicity in a mouse model of septic shock4. Ligation of TREM-1 on monocytes and neutrophils synergizes with signals through several TLRs4,20,21. TREM-2, another DAP12-paired receptor, has been implicated as a pattern recognition receptor for a variety of bacteria and fungi19. These data suggest that DAP12 may be involved in the innate immune response to pathogens. We initiated these studies to further characterize the role of DAP12-associated receptors in TLR responses by investigating the responses of DAP12-deficient mice15 to pathogen products and bacterial infection. Unexpectedly, we found that DAP12-deficient mice show enhanced TLR responses in vitro and in vivo, suggesting that certain DAP12-associated receptors may function as negative regulators of TLR responses.
RESULTS
Increased cytokine production by DAP12-deficient macrophages
To investigate the role of macrophage DAP12-associated receptors in the responses to pathogenic stimuli, we grew bone marrow-derived macrophages from wild-type and DAP12-deficient mice15 and treated them with a variety of pathogen products that signal through different TLRs. Bone marrow macrophages from DAP12-deficient mice secreted higher concentrations of the proinflammatory cytokine TNF in response to TLR stimulation than do wild-type macrophages (Fig. 1a). TNF production was increased in response to all TLR stimuli tested (that is, LPS-TLR4, synthetic bacterial lipopeptide-TLR2/1, CpG DNA-TLR9, zymosan-TLR2, poly(I:C)-TLR3 and peptidoglycan-TLR2; Fig. 1a and data not shown). Increased TNF was not observed at all concentrations of TLR stimuli, but was most consistent and pronounced at doses that were below saturating amounts for TNF production. This is particularly evident for LPS stimulation, where concentrations that elicited no TNF from wild-type macrophages, such as 0.5 ng/ml, elicited maximal amounts from the DAP12-deficient macrophages.
Figure 1.
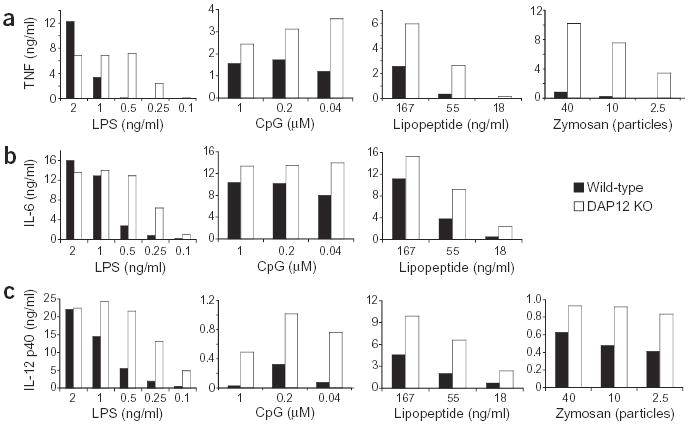
DAP12-deficient macrophages secrete increased amounts of cytokines after TLR stimulation. Bone marrow-derived macrophages from wild-type or DAP12-deficient (DAP12 KO) mice were incubated with the indicated concentrations of LPS, CpG DNA, synthetic bacterial lipopeptide or zymosan for 16 h. Supernatants were collected and the amounts of TNF (a), IL-6 (b) and IL-12 p40 (c) were measured. No IL-6 was detected after incubation of macrophages with zymosan. Data are representative of ten (LPS, zymosan; a), five (lipopeptide, CpG DNA; a) or four (b,c) independent experiments.
We investigated whether enhanced cytokine production in DAP12-deficient macrophages was specific to TNF or whether secretion of other proinflammatory cytokines was also elevated. Both IL-6 and IL-12 p40 secretion were higher in DAP12-deficient macrophages than in wild-type macrophages (Fig. 1b,c). As for TNF, cytokine production was more pronounced at lower concentrations of the TLR stimulus. Similarly, increased cytokine concentrations were also observed in response to stimulation with whole bacteria, including Escherichia coli and Staphylococcus aureus (data not shown). These data suggest that in the absence of DAP12, macrophages are more sensitive to signals through TLRs, and that, in wild-type macrophages, a DAP12 signal limits the TLR-induced response. TNF production was also assessed by intracellular TNF staining to determine whether the elevated TNF secretion was due to an increased percentage of cells producing TNF or increased TNF production on a per-cell basis. In response to LPS, CpG DNA and zymosan, both the percentage of cells that produced TNF (Fig. 2a) and the amount of TNF staining, as assessed by the mean fluorescence intensity (of the TNF-producing population), were increased in the DAP12-deficient macrophages (Fig. 2b). We also investigated the kinetics of the TNF secretion from wild-type and DAP12-deficient macrophages. Increased TNF production was observed as early as 4 h after activation with LPS and zymosan, and the difference between wild-type and DAP12-deficient macrophages increased over the course of the experiment (Fig. 2c).
Figure 2.
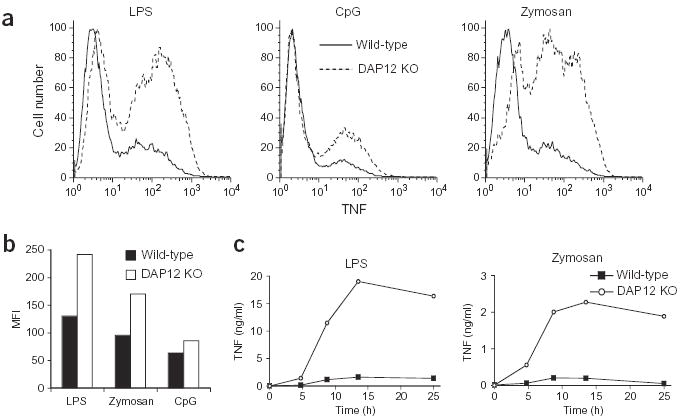
DAP12-deficient macrophages produce more TNF after TLR stimulation. (a,b) Bone marrow–derived macrophages from wild-type or DAP12-deficient (DAP12 KO) mice were incubated with LPS (0.4 ng/ml), CpG DNA (0.04 μM) or zymosan (ten particles per macrophage) for 4 h (CpG DNA and zymosan) or 14 h (LPS) in the presence of Brefeldin A for the final 4 h. TNF secretion was then assessed; data are represented by one-parameter histograms (a) or by the mean fluorescent intensity (MFI) of the TNF-producing population (b). Data are representative of four independent experiments. (c) Bone marrow–derived macrophages (day 7) were stimulated with LPS (0.5 ng/ml) or zymosan (ten particles per macrophage). Supernatants were removed at the indicated time points, and TNF concentrations were measured using ELISA. Data are representative of three independent experiments.
We examined whether DAP12 regulates the production of inflammatory cytokines induced by activating macrophage receptors distinct from TLRs. The low-affinity Fc receptor (FcR) for IgG (FcγRIII or CD16) associates with another ITAM-containing adaptor, the FcɛRIγ chain, and cross-linking of FcγRIII on macrophages results in phagocytosis and production of TNF and IL-6. Similar to what was observed for TLR stimulation, cross-linking of FcγRIII resulted in higher TNF and IL-6 secretion from DAP12-deficient macrophages compared with wild-type macrophages (Supplementary Fig. 1 online). Because DCs are closely related to macrophages and express several DAP12-associated receptors, we also investigated TLR-induced inflammatory cytokine production by DAP12-deficient DCs. The DAP12-deficient DCs responded identically to wild-type DCs in terms of TNF and IL-12 p40 production after stimulation with CpG DNA and in production of IL-12 p40 after stimulation with LPS (Supplementary Fig. 2 online). In contrast to DAP12-deficient macrophages, LPS-induced TNF production was actually somewhat lower in the DAP12-deficient DCs than in wild-type DCs. This suggests that TLR-induced cytokine production is regulated differently in macrophages and dendritic cells.
DAP12 reconstitution reduces TNF production
To confirm that the increase in intracellular TNF in response to TLR stimulation in DAP12-deficient macrophages was due to DAP12 deficiency, we used retroviral transduction of macrophages to introduce DAP12 into either wild-type or DAP12-deficient macrophages. After retroviral transduction of a control vector or DAP12-encoding vector, the macrophages were activated with zymosan or CpG DNA, and TNF production was assessed by flow cytometry. DAP12-deficient macrophages transduced with a control vector showed an increased percentage of cells producing TNF compared with transduced wild-type macrophages (Fig. 3), consistent with previous data (Fig. 2a). In contrast, reintroduction of DAP12 into DAP12-deficient macrophages caused a reduction in the percentage of TNF-producing cells to an amount equivalent to wild-type macrophages. Expressing additional DAP12 protein in wild-type macrophages did not change the percentage of TNF-expressing cells (Fig. 3a). This experiment showed that DAP12 deficiency caused the increased TNF production in the DAP12-deficient macrophages.
Figure 3.
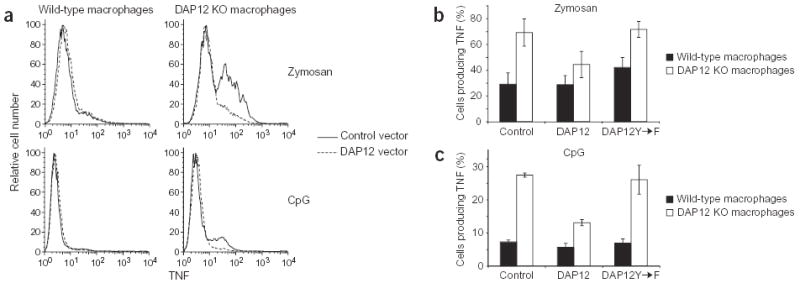
Reintroduction of DAP12 reduces the TNF production by DAP12-deficient macrophages in an ITAM-dependent manner. Bone marrow-derived macrophages from wild-type or DAP12-deficient (DAP12 KO) mice were transduced with an empty vector (control) retrovirus or retroviruses encoding wild-type DAP12 or a DAP12 ITAM mutant (DAP12Y→F). The macrophages were activated with zymosan (ten per macrophage) or CpG DNA (0.01 μM) in the presence of Brefeldin A for 4 h and TNF production was assessed by flow cytometry. Transduced cells were gated based on GFP fluorescence and the percentage of TNF-producing cells was determined. (a) Histograms of TNF expression in wild-type and DAP12-deficient macrophages transduced with control or DAP12-expressing retrovirus from one representative experiment. (b,c) The percentage of transduced cells producing TNF represented as the mean ± s.e.m. of three independent experiments (zymosan) or two independent experiments (CpG DNA).
This reconstitution experiment ruled out the possibility that DAP12 deficiency had caused a developmental defect in the bone marrow precursors used to generate the macrophages, thereby altering their ability to respond to TLR signals. DAP12-deficient macrophages derived from bone marrow grew similarly to wild-type macrophages and uniformly expressed F4/80 and Mac1 (CD11b) on their surface (data not shown). Additionally, DAP12-deficient macrophages were identical to wild-type macrophages in their surface expression of TLR4 as assessed by flow cytometry (data not shown), suggesting that at least this component of the LPS response was not different in DAP12-deficient macrophages.
Enhanced TNF production requires ITAM and Syk
We next investigated the signaling mechanism by which the lack of DAP12 resulted in increased cytokine production. Signaling through DAP12 is mediated through its ITAM, which relies on phosphorylation of the two tyrosines within the ITAM for propagation of a signal. To determine whether these tyrosines are required for DAP12-mediated negative regulation of TNF production, we transduced a retrovirus encoding a mutant DAP12 protein in which both tyrosines in the ITAM were mutated to phenylalanines into wild-type and DAP12-deficient macrophages derived from bone marrow22. Mutant DAP12 cannot be phosphorylated and thus cannot activate the ITAM-dependent signaling cascade. The mutant DAP12 was not able to reduce the TNF production of DAP12-deficient macrophages in response to stimulation with zymosan or CpG DNA (Fig. 3b,c). This ITAM-mutant DAP12 behaved similarly to the empty control retrovirus and contrasted with wild-type DAP12, which caused a reduction in cytokine production. These data show that ITAM-mediated signaling by DAP12 is required for dampening TLR signaling.
In macrophages, the Syk tyrosine kinase is recruited to ITAMs in adaptor proteins after phosphorylation, and Syk is required for downstream signaling18. We therefore compared the TLR responses of Syk-deficient macrophages with those of DAP12-deficient macrophages. As Syk-deficient mice are embryonic lethal23, we generated bone marrow–derived macrophages from Syk-deficient fetal liver chimeras24. The cytokine production induced by TLR stimulation was compared in wild-type, Syk-deficient and DAP12-deficient macrophages derived from bone marrow. Syk-deficient macrophages behaved similarly to DAP12-deficient macrophages, as shown by increased secretion of proinflammatory cytokines (TNF, IL-6 and IL-12 p40) in response to LPS, CpG DNA and synthetic lipopeptide (Fig. 4). These data suggest that DAP12 uses the conventional ITAM signaling pathway to regulate TLR signaling.
Figure 4.
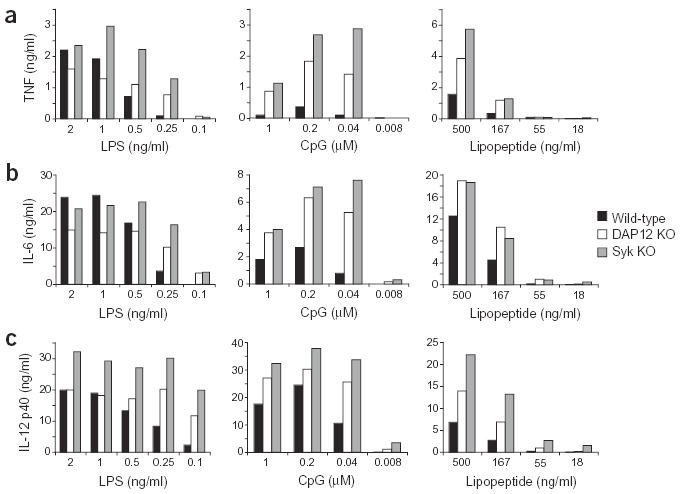
Syk-deficient macrophages secrete increased amounts of proinflammatory cytokines in response to TLR stimulation. Bone marrow-derived macrophages from wild-type mice, DAP12-deficient (DAP12 KO) mice or Syk-deficient (Syk KO) fetal liver chimeras were incubated with the indicated concentrations of LPS, CpG DNA, or synthetic bacterial lipopeptide for 16 h. Supernatants were collected and the amounts of TNF (a), IL-6 (b) and IL-12 p40 (c) were measured using ELISA. Data are representative of three independent experiments.
Anti-inflammatory cytokine production
Macrophages produce both inflammatory and anti-inflammatory cytokines in response to ligation of TLRs. Production of anti-inflammatory cytokines, particularly IL-10, is important in down-regulating secretion of inflammatory cytokines. We hypothesized that DAP12-deficient macrophages may produce reduced amounts of anti-inflammatory cytokines compared with that produced by wild-type macrophages, which may explain their increased secretion of TNF, IL-6 and IL-12 p40. To test this hypothesis, we treated wild-type and DAP12-deficient macrophages with TLR stimuli that induced TNF production and measured the IL-10 concentration in the supernatants. Rather than producing less IL-10, DAP12-deficient macrophages actually secreted more IL-10 than wild-type macrophages (Fig. 5a). To address the production of transforming growth factor-β or other anti-inflammatory cytokines, we activated wild-type and DAP12-deficient macrophages in a culture dish where these macrophages were separated by a 0.2-μm porous membrane. The production of TNF by the macrophages in the bottom chamber was then assessed by flow cytometry. Neither the wild-type nor the DAP12-deficient macrophages secreted soluble factors that influenced TNF production by the other macrophage population (Fig. 5b). This suggests that soluble factors produced by the TLR-activated wild-type macrophages are unable to diminish the hyper-responsive production of inflammatory cytokines by DAP12-deficient macrophages.
Figure 5.
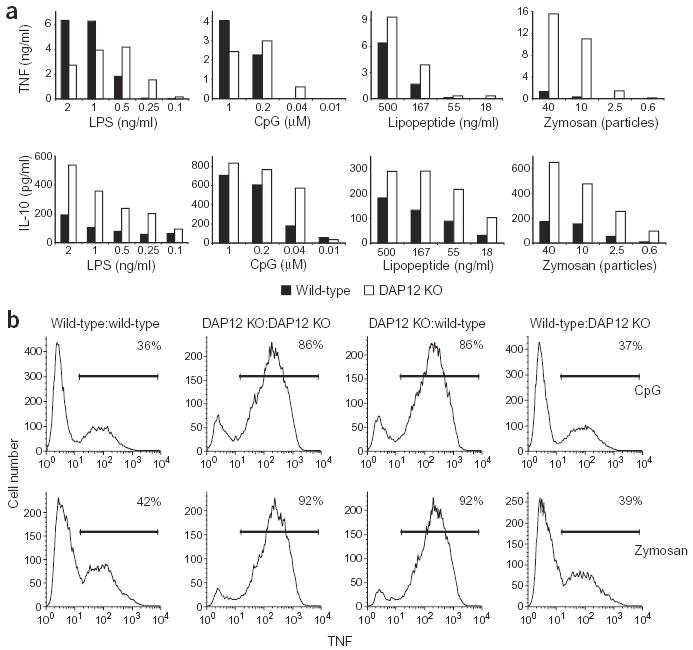
Soluble inhibitory factor production does not explain differences between wild-type and DAP12-deficient macrophages. (a) Bone marrow-derived macrophages from wild-type and DAP12-deficient (DAP12 KO) mice were stimulated with the indicated concentrations of LPS, zymosan, CpG DNA and lipopeptide for 16 h. Supernatants were then assayed for TNF or IL-10 using ELISA. (b) Wild-type and DAP12-deficient macrophages were cultured in the top and bottom chambers of culture plates separated by a porous membrane. Cells in both top and bottom chambers were treated with CpG DNA (0.01 μM) or zymosan (ten particles per macrophage) for 4 h, and TNF-producing cells in the bottom chambers were determined by using flow cytometry (Fig. 2a). In the histograms shown, the type of cells in the top and bottom chambers of the culture plates are indicated as cells in bottom chamber:cells in top chamber. The percentage of TNF-positive cells is indicated in the top right corner of each histogram. Data shown are representative of two independent experiments.
Enhanced ERK phosphorylation in DAP12-deficient macrophages
To determine where DAP12-initiated signals intersect the TLR signaling pathway, we examined the kinetics of activation of the three MAPKs and the NF-κB pathway, which are all downstream of TLR stimulation1. For these experiments, we used 0.5 ng/ml LPS, a concentration that gave us reproducible differences in cytokine secretion between the wild-type and DAP12-deficient macrophages. The kinetics and magnitude of p38 MAPK and JNK phosphorylation were similar in wild-type and DAP12-deficient macrophages (Fig. 6). In contrast, p42/44 ERK signaling was enhanced in the DAP12-deficient cells. In wild-type macrophages, no ERK phosphorylation was detected until 40 min after stimulation, whereas in DAP12-deficient macrophages, ERK phosphorylation was detected at 20 min (Fig. 6). Additionally, at all time points the magnitude of ERK phosphorylation in the DAP12-deficient macrophages was considerably greater than that in the wild-type macrophages. Although the amount of ERK phosphorylation was different, for both wild-type and DAP12-deficient macrophages ERK phosphorylation peaked at 40 min after stimulation and returned to baseline by 90 min (Fig. 6).
Figure 6.
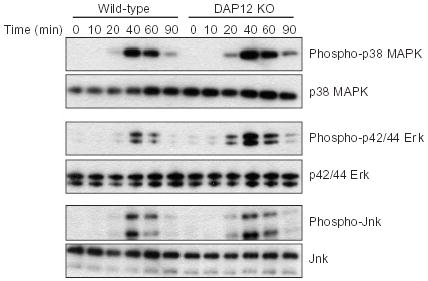
LPS-induced ERK phosphorylation is increased in DAP12-deficient macrophages. Bone marrow-derived macrophages from wild-type or DAP12-deficient (DAP12 KO) mice were stimulated with 0.5 ng/ml LPS for the indicated time (min), at which time cells were lysed. Cytoplasmic extracts were analyzed using antibodies specific for p38 MAPK, p42/44 ERK and JNK, or for phosphorylated versions of these proteins. Data are representative of four independent experiments.
We also measured activation of the NF-κB pathway by examining the degradation of the inhibitor IκBα, which serves to retain NF-κB in the cytoplasm, thereby preventing transcriptional activation by NF-κB. In both wild-type and DAP12-deficient macrophages, IκBα was degraded with similar kinetics and was present in similar amounts (data not shown). Therefore, the principal difference in TLR downstream signaling between wild-type and DAP12-deficient macrophages was in ERK phosphorylation.
Increased endotoxic shock in DAP12-deficient mice
Because DAP12-deficient macrophages produced more TNF in response to LPS in vitro, we investigated whether this happens in vivo as well. We first analyzed a model of endotoxic shock in which mice are injected intraperitoneally with a low dose (10 μg) of LPS and d-galactosamine, a hepatotoxic agent that makes mice more sensitive to TNF. In this model, mice usually succumb to shock between 5 and 10 h after injection; death is dependent on TNF25. DAP12-deficient mice were more susceptible to LPS-induced shock than wild-type mice. At 12 h after injection, 40% of the wild-type mice had survived, whereas none of the DAP12-deficient mice had survived (Fig. 7a). To determine whether this was due to elevated TNF production, the concentration of this cytokine in plasma was measured in mice injected with a higher dose of LPS (100 μg). A representative experiment shows that wild-type and DAP12-deficient mice had identical kinetics of TNF induction after LPS injection (with TNF concentrations peaking at 1 h and returning to baseline by 4 h), whereas DAP12-deficient mice often had approximately twofold more TNF in their plasma at the peak of the response (Fig. 7b). The average amount of TNF produced 1 h after LPS injection from five experiments with 25 mice showed DAP12-deficient mice to have significantly more TNF in their plasma compared with wild-type mice (P < 0.03; Fig. 7c). Additionally, in some experiments DAP12-deficient mice had increased plasma IL-6 concentrations at 4 h after injection of LPS (data not shown) and after intraperitoneal injection with 20 mg of zymosan (data not shown). Therefore, DAP12 negatively regulates TLR responses in vivo and in vitro.
Figure 7.
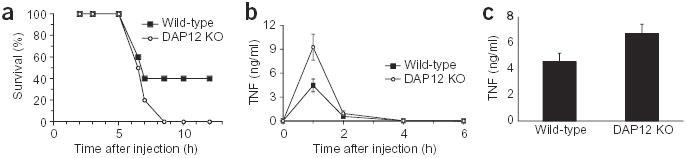
DAP12-deficient mice are more susceptible to endotoxic shock than wild-type mice. Wild-type (n = 10) or DAP12-deficient (DAP12 KO; n = 9) mice were injected intraperitoneally with LPS and D-galactosamine and then monitored for survival for 12 h (a). Data are representative of four independent experiments. Wild-type (n = 5) and DAP12-deficient (n = 5) mice were injected with 100 μg of LPS. At the indicated times, a sample of blood was drawn and plasma was analyzed for TNF using ELISA (b). The data are reported as mean ± s.e.m. and are representative of three independent experiments. Mice were treated as described in b and plasma TNF concentrations at 1 h postinjection are represented as the mean of 25 mice assayed in five independent experiments ± s.e.m (c). The difference between wild-type and DAP12-deficient mice is significant using Student’s t-test, P < 0.03 (two-tailed distribution).
Clearance of L. monocytogenes in DAP12-deficient mice
Because TNF is a critical cytokine in the innate immune response to bacterial infection, we examined whether DAP12-deficient mice have increased protection from infection with the intracellular bacterium L. monocytogenes. Wild-type and DAP12-deficient mice were infected intravenously with varying doses of L. monocytogenes, and the total colony forming units (CFU) in the spleens and livers were determined at day 3 after infection. At this time point, the response to the infection will be predominantly from the innate immune system, including macrophages, as there are no detectable T cell responses in this model26. At day 3 after infection, DAP12-deficient mice had substantially fewer bacteria in their spleens and livers than wild-type mice (Fig. 8). In some experiments, this difference could be seen as early as 1 d after infection (data not shown). These data are consistent with increased inflammatory cytokine production after infection, resulting in an enhanced innate immune response and increased bacterial clearance.
Figure 8.
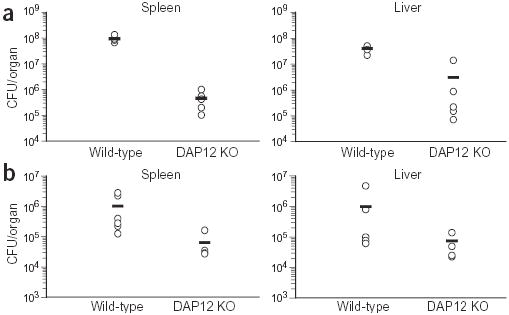
DAP12-deficient mice have an enhanced innate immune response to infection with L. monocytogenes. Wild-type and DAP12-deficient mice were infected intravenously with 5 × 104 (a) or 1 × 104 (b) CFU of L. monocytogenes strain 10403S. Three days after infection, the number of CFU in the spleens and livers of the mice were determined. The data are represented as the CFU in each individual mouse (circles) and the mean (horizontal line). In a, there were five mice per group; in b there were six wild-type mice and four DAP12-deficient mice per group. These data are representative of four independent experiments.
DISCUSSION
Here we report the unexpected finding that inflammatory cytokine production in response to TLR stimulation is elevated both in vitro and in vivo in DAP12-deficient mice. Our findings that TLR-induced cytokine production is increased, rather than decreased, in the absence of DAP12 or Syk suggest the outcome of DAP12-mediated signaling may depend on the context of receptor ligation. Signaling through another ITAM-dependent receptor, the T cell antigen receptor, can have different functional consequences depending on the nature of the ligand. During T cell development, strong T cell antigen receptor ligands cause T cell death (negative selection), whereas weak ligands favor survival (positive selection)27. Signaling through the IgA receptor (FcαRI), which pairs with the ITAM-containing FcɛRIγ adaptor, can also result in either positive or negative signaling28. Ligation of FcαRI with monomeric IgA (low avidity) results in negative regulation, whereas extensive cross-linking of the IgA receptor with multimeric IgA complexes (high avidity) causes cellular activation.
We propose that a similar situation applies to DAP12. DAP12 has previously been shown to induce inflammatory cytokine production3,7, and we have now reported that DAP12 can also give an inhibitory signal, thereby negatively regulating the production of these same cytokines in macrophages. As shown for the IgA receptor, our model predicts that a weak signal through one or more DAP12-paired receptors on macrophages results in basal Syk phosphorylation and the subsequent activation of a negative regulator of inflammatory cytokine production, thereby dampening ERK1/2 activation. Although we have not identified the critical substrate of Syk that functions as a negative regulator of ERK, phosphatases in the MAPK pathway upstream of ERK are candidates. Furthermore, we predict that the basal DAP12-induced signaling may emanate from the interaction between one or more DAP12-associated myeloid receptors on macrophages with low-affinity or low-avidity ligands that are also present on these macrophages. Although ligands for the DAP12-associated myeloid receptors have not been identified, if this basal DAP12 signaling is indeed ligand-induced then they must be expressed on macrophages themselves, because in the in vitro experiments these are the only cells present in the culture. In vivo, the ligands may be expressed on macrophages or other cells. For these DAP12-associated receptors to convert from negative to positive signaling, we propose that encounters with either higher-affinity or higher-avidity ligands would be necessary. In other words, only when the DAP12-associated receptors are cross-linked by a high-affinity or high-avidity ligand would this evoke stronger recruitment of Syk—with the net balance resulting in positive activation. Experiments showing that cross-linking DAP12-associated receptors on myeloid cells results in potent activation used antibodies against these receptors (that is, mimicking high-affinity or high-avidity engagement)3,7. In accordance with our findings, microglial cells lacking the DAP12-associated TREM-2 receptor have been reported to express higher amounts of inflammatory cytokines29, possibly causing the brain pathology observed in TREM-2–deficient humans30.
DAP12 negatively regulates signaling for macrophage inflammatory cytokine production by another ITAM-containing receptor, the low-affinity IgG receptor, FcγRIII or CD16. Perhaps any receptor that can induce ERK1/2 activation, such as TLRs or FcγRIII, can be inhibited by DAP12. Indeed, signaling through FcαRI was shown to inhibit the ITAM-associated IgG Fc receptors and IgE Fc receptor (FcɛRI)28. DAP12-deficient DCs did not show increased inflammatory cytokine production to the TLR stimuli LPS and CpG DNA. Although this may indicate that DAP12 cannot inhibit DC cytokine production, we favor an alternative hypothesis: the DCs used in these experiments do not express ligands for their DAP12-associated receptors, therefore, there is no basal DAP12 signaling in these cells.
In macrophages, antibody-mediated cross-linking of the DAP12-associated TREM-1 (ref. 3) and MAIR-II7 receptors leads to inflammatory cytokine production, demonstrating that a DAP12 signal alone can result in activation. Cross-linking of TREM-1 also can synergize with LPS for TNF and IL-1-β production4,20. Treatment of mice with a soluble TREM-1 fusion protein protects mice from LPS-induced shock4. Monocytes and macrophages produce a soluble form of TREM-1 in response to LPS treatment in vitro and in vivo, and a peptide mimic of this soluble receptor can protect mice from LPS-induced death31,32. Although it is proposed that the soluble TREM-1 protein or peptide acts as a TREM-1 receptor antagonist, an alternative possibility is that the soluble protein binds to an as-yet-undefined inhibitory receptor, which would also suppress an immune response. Indeed, many of the activating DAP12-paired receptors have related inhibitory receptors33.
Although the signaling pathways leading from activation through TLRs to inflammatory cytokine production have been well characterized, there is a growing literature on how these signals are dampened to prevent pathological consequences, such as septic shock and inflammatory disorders1. The negative regulators of TLR signaling include diverse proteins that intersect the TLR signaling pathways at different stages. Those expressed in macrophages include the cell surface receptors ST2 and TRAIL-R; the signaling molecules SOCS1, IRAK-M, PI3 kinase, MyD88s, SHIP and Tollip; and the ubiquitin-interacting proteins Triad3A and A2034–44. The mechanisms by which these proteins regulate TLR signaling are quite varied and most affect all TLRs, as does DAP12, whereas others, such as Triad3A and ST2, only affect a subset of the TLRs34,42. Triad3A acts at the level of receptor expression, ubiquitinating and therefore inducing the down-regulation of TLR4 and TLR9 (ref. 42). ST2 interferes with the association of TIR-domain containing adaptors with TLRs34. Further downstream, IRAK-M interacts with the IRAK-1/4 complex, preventing its release from MyD88 and therefore its binding to TRAF6 (ref. 44). MyD88s also acts at this level, preventing the phosphorylation of IRAK-1 (ref. 39). A20 acts directly on TRAF6, deubiquitinating it and thereby reducing activation of NF-κB43. It is unclear how many of the other negative regulators lead to inhibition of TLR responses, but deficiencies in these proteins result in a similar phenotype to the DAP12-deficient cells. However, none of these previously described negative regulators seem to work at the same level as DAP12.
Cells deficient in several of the described negative regulators have enhanced TLR signaling, as determined by increased activation of MAPK pathways after LPS stimulation. SOCS-1–deficient macrophages have increased p38 and JNK phosphorylation36, PI3 kinase-deficient DCs have increased p38 phosphorylation38, and IRAK-M–deficient macrophages show increased phosphorylation of p38, JNK and ERK44. DAP12-deficient macrophages showed increased ERK1/2 phosphorylation, whereas p38 and JNK phosphorylation was similar to that of wild-type macrophages. Although the amounts of ERK phosphorylation were increased at all time points after LPS stimulation, the kinetics of DAP12-deficient and wild-type macrophages were similar. DAP12 is known to specifically activate ERK45–47. Upstream of ERK activation in TLR signaling is the MAPK kinase kinase Tpl2; macrophages lacking Tpl2 secrete less TNF in response to LPS48. If DAP12 links to ERK through Tpl2, then Tpl2 activation may also be increased in DAP12-deficient cells.
Collectively, our data support a model whereby basal signaling through one or more DAP12-associated receptors causes a reduction in the amount of inflammatory cytokines produced by macrophages. This results in an increase in the susceptibility of DAP12-deficient mice to endotoxic shock and an enhanced resistance of these mice to infection with certain intracellular bacteria. Although other studies have suggested that DAP12 may induce proinflammatory signals that augment responses to bacterial infections, it is interesting to note that there have been no reports of increased susceptibility to infection in DAP12-deficient humans (a disease named Nasu-Hakola syndrome)49. Rather than an immune deficiency, Nasu-Hakola patients have bone defects and brain pathologies, similar to DAP12-deficient mice50,51. In fact, our data predict that these patients may have enhanced resistance to certain pathogens.
METHODS
Mice.
We purchased C57BL/6 mice from NCI Frederick or Charles River. DAP12-deficient mice15 were backcrossed to C57BL/6 mice for nine generations. Syk-deficient mice23 were backcrossed to C57BL/6 mice for six generations. Fetal liver chimeras were generated from Syk-deficient embryos as described24. All experiments were in accordance with protocols approved by the University of California San Francisco Institutional Animal Care and Use Committee.
Macrophages and dendritic cells.
Macrophages derived from bone marrow were grown as described52. After 6 d, nonadherent cells were removed and macrophages were replated for use in experiments. Dendritic cells derived from bone marrow were grown as described in medium containing 10 ng/ml recombinant GM-CSF (Peprotech)53. At day 8, nonadherent cells containing dendritic cells were removed and purity and maturation was assessed by flow cytometry. Cells were stained with antibodies to CD11c, CD86 and I-Ab (BD Pharmingen). The nonadherent cells were typically 60–75% CD11c+ dendritic cells, which were 80–90% immature (low/intermediate for CD86 and I-Ab).
TLR stimulation and cytokine measurement.
For all experiments, day 6 bone marrow-derived macrophages were plated and adhered overnight. Titrations of TLR stimuli were added at ×10 concentration. To exclude LPS contamination, all stimuli used, with the exception of LPS and E. coli, were treated with polymixin B (10 μg/ml; Sigma) before addition to cells. TLR stimuli were as follows: Salmonella minnesota R595 LPS (List Biological Laboratories); CpG DNA (ODN 1826) and peptidoglycan (Invivogen); zymosan, heat-killed E. coli and S. aureus (Molecular Probes); poly(I:C) (Amersham Pharmacia); and synthetic bacterial lipopeptide Pam3CSK4 (Roche). For cytokine secretion, the amounts of TNF, IL-6, IL-12 p40 and IL-10 in duplicate supernatants were measured by enzyme-linked immunosorbent assay (ELISA; TNF, eBioscience; IL-6, IL-12 p40 and IL-10, BD Pharmingen). For TNF measurement by intracellular cytokine staining, cells were stimulated for 4 h (zymosan and CpG DNA) or 14 h (LPS) in the presence of Brefeldin A (10 μg/ml) for the final 4 h. Macrophages were blocked with antibodies to FcR 2.4G2, and stained after fixation and permeabilization (CalTag) with a phycoerythrin-labeled antibody to TNF (BD Pharmingen). Day 8 dendritic cells were stimulated with LPS or CpG DNA for 4 h in the presence of Brefeldin A. Dendritic cells were surface stained with FITC-labeled antibody to CD11c before fixation, permeabilization and staining with phycoerythrin-labeled antibody to TNF or IL-12 p40 (BD Pharmingen). Cells were analyzed by flow cytometry using a FACScan (BD) running CellQuest software (BD) and analyzed with FlowJo software (TreeStar). In some experiments, wild-type and DAP12-deficient macrophages were cultured in Transwell dishes (Costar) separated by a 0.2 μM filter. In these experiments, TLR stimuli were added to both the top and bottom chambers.
Activation through Fc receptors.
We coated 96-well plates with a 1 mg/ml solution of DOTAP (Sigma) for 10 min at room temperature, washed with PBS and incubated overnight with monoclonal antibodies to FcκRII/III (hybridoma 2.4G2) in 0.1 M bicarbonate buffer (pH 9.0). Macrophage supernatants were collected at 16 h and tested for cytokines by ELISA.
Retroviral transduction of macrophages.
VSVg pseudotyped retroviruses were generated using the following constructs in the pMX-pie vector, in which the cDNA is followed by an IRES-GFP to identify infected cells: control empty vector, amino-terminus FLAG-tagged DAP12, and N-terminus FLAG-tagged mutant DAP12, in which both tyrosines in the ITAM have been changed to phenylalanine52. After 72 h, the cells were activated and TNF was measured using intracellular cytokine staining. Infected cells were gated based on GFP fluorescence and the percentage staining for TNF was determined.
Kinetics of MAPK phosphorylation.
Macrophages were activated with 0.5 ng/ml LPS, lysed at the indicated times in lysis buffer containing 1% Triton X-100, protease inhibitors and sodium orthovanadate (1 mM; Sigma). Cytoplasmic extracts were analyzed by SDS-PAGE and western blot using antibodies specific for phosphorylated and nonphosphorylated p38 MAPK, p42/44 ERK and JNK, and HRP-conjugated antibodies to rabbit IgG (Cell Signaling Technologies). Western blots were developed with ECL Plus (Amersham Pharmacia).
Endotoxic shock.
Mice were injected with 10 μg of S. minnesota LPS and 20 μg of D-galactosamine (Sigma) and were monitored for 12 h for survival or for failure to upright themselves, known as the righting response, at which point they were euthanized. To determine plasma TNF concentrations mice were injected with 100 μg of E. coli 055:B5 LPS (Sigma). At the indicated times, a sample of blood was taken and plasma TNF was determined by ELISA.
L. monocytogenes infection.
Mice were injected intravenously with the indicated amount of L. monocytogenes 10403S (a gift of E. Pamer) grown to mid-log phase (OD600 = 0.1) in tryptic soy broth (Fisher Scientific). At day 3 after infection, the livers and spleens were homogenized, serial dilutions were plated on tryptic soy broth plates and incubated overnight at 37 °C, and the CFU per organ was determined by colony count.
Supplementary Material
Acknowledgments
We thank M.B. Humphrey for the DAP12 ITAM mutant construct and helpful discussions; Y. Hu for help with the generation of Syk-deficient chimeras; S. Watson for assistance in endotoxic shock experiments; C. Chu for help with dendritic cell cultures; T. Nishiya and D. Underhill for helpful discussions; and A. DeFranco for reading the manuscript. This work was supported by a grant from the Irvington Institute for Immunological Research (J.A.H.) and NIH grants CA89294 (L.L.L.) and DK58066 (to C.A.L.). L.L.L. is an American Cancer Society Research Professor and C.A.L. is a scholar of the Leukemia and Lymphoma Society.
Footnotes
Note: Supplementary information is available on the Nature Immunology website.
COMPETING INTERESTS STATEMENT The authors declare that they have no competing financial interests.
References
- 1.Akira, S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 2.Lanier, LL, Bakker AB. The ITAM-bearing transmembrane adaptor DAP12 in lymphoid and myeloid cell function. Immunol Today. 2000;21:611–614. doi: 10.1016/s0167-5699(00)01745-x. [DOI] [PubMed] [Google Scholar]
- 3.Bouchon, A, Dietrich J, Colonna M. Cutting edge: inflammatory responses can Be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol. 2000;164:4991–4995. doi: 10.4049/jimmunol.164.10.4991. [DOI] [PubMed] [Google Scholar]
- 4.Bouchon, A, Facchetti F, Weigand MA, Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock . Nature. 2001;410:1103–1107. doi: 10.1038/35074114. [DOI] [PubMed] [Google Scholar]
- 5.Daws, MR, Lanier LL, Seaman WE, Ryan JC. Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur J Immunol. 2001;31:783–791. doi: 10.1002/1521-4141(200103)31:3<783::aid-immu783>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 6.Chung, DH, Seaman WE, Daws MR. Characterization of TREM-3, an activating receptor on mouse macrophages: definition of a family of single Ig domain receptors on mouse chromosome 17. Eur J Immunol. 2002;32:59–66. doi: 10.1002/1521-4141(200201)32:1<59::AID-IMMU59>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 7.Yotsumoto, K, et al. Paired activating and inhibitory immunoglobulin-like receptors, MAIR-I and MAIR-II, regulate mast cell and macrophage activation . J Exp Med. 2003;198:223–233. doi: 10.1084/jem.20021825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wright, GJ, et al. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J Immunol. 2003;171:3034–3046. doi: 10.4049/jimmunol.171.6.3034. [DOI] [PubMed] [Google Scholar]
- 9.Dietrich, J, Cella M, Seiffert M, Buhring HJ, Colonna M. Cutting edge: signal-regulatory protein β1 is a DAP12-associated activating receptor expressed in myeloid cells. J Immunol. 2000;164:9–12. doi: 10.4049/jimmunol.164.1.9. [DOI] [PubMed] [Google Scholar]
- 10.Tomasello, E, et al. Association of signal-regulatory proteins β with KARAP/DAP-12. Eur J Immunol. 2000;30:2147–2156. doi: 10.1002/1521-4141(2000)30:8<2147::AID-IMMU2147>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 11.Shiratori, I, Ogasawara K, Saito T, Lanier LL, Arase H. Activation of natural killer cells and dendritic cells upon recognition of a novel CD99-like ligand by paired immunoglobulin-like type 2 receptor. J Exp Med. 2004;199:525–533. doi: 10.1084/jem.20031885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bakker, AB, Baker E, Sutherland GR, Phillips JH, Lanier LL. Myeloid DAP12-associating lectin (MDL)-1 is a cell surface receptor involved in the activation of myeloid cells. Proc Natl Acad Sci USA. 1999;96:9792–9796. doi: 10.1073/pnas.96.17.9792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diefenbach, A, et al. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat Immunol. 2002;3:1142–1149. doi: 10.1038/ni858. [DOI] [PubMed] [Google Scholar]
- 14.Gilfillan, S, Ho EL, Cella M, Yokoyama WM, Colonna M. NKG2D recruits two distinct adapters to trigger NK cell activation and costimulation. Nat Immunol. 2002;3:1150–1155. doi: 10.1038/ni857. [DOI] [PubMed] [Google Scholar]
- 15.Bakker, AB, et al. DAP12-deficient mice fail to develop autoimmunity due to impaired antigen priming. Immunity. 2000;13:345–353. doi: 10.1016/s1074-7613(00)00034-0. [DOI] [PubMed] [Google Scholar]
- 16.Tomasello, E, et al. Combined natural killer cell and dendritic cell functional deficiency in KARAP/DAP12 loss-of-function mutant mice. Immunity. 2000;13:355–364. doi: 10.1016/s1074-7613(00)00035-2. [DOI] [PubMed] [Google Scholar]
- 17.Colonna, M, Facchetti F. TREM-1 (triggering receptor expressed on myeloid cells): a new player in acute inflammatory responses. J. Infect. Dis. 2003;187 (suppl 2):S397–S401. doi: 10.1086/374754. [DOI] [PubMed] [Google Scholar]
- 18.Turner, M, Schweighoffer E, Colucci F, Di Santo JP, Tybulewicz VL. Tyrosine kinase SYK: essential functions for immunoreceptor signalling. Immunol Today. 2000;21:148–154. doi: 10.1016/s0167-5699(99)01574-1. [DOI] [PubMed] [Google Scholar]
- 19.Daws, MR, et al. Pattern recognition by TREM-2: binding of anionic ligands . J Immunol. 2003;171:594–599. doi: 10.4049/jimmunol.171.2.594. [DOI] [PubMed] [Google Scholar]
- 20.Bleharski, JR, et al. A role for triggering receptor expressed on myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune response. J Immunol. 2003;170:3812–3818. doi: 10.4049/jimmunol.170.7.3812. [DOI] [PubMed] [Google Scholar]
- 21.Radsak, MP, Salih HR, Rammensee HG, Schild H. Triggering receptor expressed on myeloid cells-1 in neutrophil inflammatory responses: differential regulation of activation and survival. J Immunol. 2004;172:4956–4963. doi: 10.4049/jimmunol.172.8.4956. [DOI] [PubMed] [Google Scholar]
- 22.Mocsai, A, et al. The immunomodulatory adapter proteins DAP12 and Fc receptor γ-chain (FcRγ) regulate development of functional osteoclasts through the Syk tyrosine kinase. Proc Natl Acad Sci USA. 2004;101:6158–6163. doi: 10.1073/pnas.0401602101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turner, M, et al. Perinatal lethality and blocked B-cell development in mice lacking the tyrosine kinase Syk . Nature. 1995;378:298–302. doi: 10.1038/378298a0. [DOI] [PubMed] [Google Scholar]
- 24.Mocsai, A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity. 2002;16:547–558. doi: 10.1016/s1074-7613(02)00303-5. [DOI] [PubMed] [Google Scholar]
- 25.Pasparakis, M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF α-deficient mice: a critical requirement for TNF α in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184:1397–1411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pamer, E G. Immune responses to Listeria monocytogenes. Nat. Rev. Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 27.Starr, TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139–176. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 28.Pasquier, B, et al. Identification of FcαRI as an inhibitory receptor that controls inflammation: dual role of FcRγ ITAM. Immunity. 2005;22:31–42. doi: 10.1016/j.immuni.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi, K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2 . J Exp Med. 2005;201:647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paloneva, J, et al. CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts . Neurology. 2001;56:1552–1558. doi: 10.1212/wnl.56.11.1552. [DOI] [PubMed] [Google Scholar]
- 31.Gibot, S, et al. A soluble form of the triggering receptor expressed on myeloid cells-1 modulates the inflammatory response in murine sepsis . J Exp Med. 2004;200:1419–1426. doi: 10.1084/jem.20040708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knapp, S, et al. Cutting edge: expression patterns of surface and soluble triggering receptor expressed on myeloid cells-1 in human endotoxemia. J Immunol. 2004;173:7131–7134. doi: 10.4049/jimmunol.173.12.7131. [DOI] [PubMed] [Google Scholar]
- 33.Lanier, LL Face off—the interplay between activating and inhibitory immune receptors. Curr Opin Immunol. 2001;13:326–331. doi: 10.1016/s0952-7915(00)00222-3. [DOI] [PubMed] [Google Scholar]
- 34.Brint, EK, et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance . Nat Immunol. 2004;5:373–379. doi: 10.1038/ni1050. [DOI] [PubMed] [Google Scholar]
- 35.Diehl, GE, et al. TRAIL-R as a negative regulator of innate immune cell responses . Immunity. 2004;21:877–889. doi: 10.1016/j.immuni.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 36.Kinjyo, I, et al. SOCS1/JAB is a negative regulator of LPS-induced macrophage activation. Immunity. 2002;17:583–591. doi: 10.1016/s1074-7613(02)00446-6. [DOI] [PubMed] [Google Scholar]
- 37.Nakagawa, R, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–687. doi: 10.1016/s1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- 38.Fukao, T, et al. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat Immunol. 2002;3:875–881. doi: 10.1038/ni825. [DOI] [PubMed] [Google Scholar]
- 39.Burns, K, et al. Inhibition of interleukin 1 receptor/Toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med. 2003;197:263–268. doi: 10.1084/jem.20021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sly, LM, Rauh MJ, Kalesnikoff J, Song CH, Krystal G. LPS-induced upregulation of SHIP is essential for endotoxin tolerance. Immunity. 2004;21:227–239. doi: 10.1016/j.immuni.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 41.Zhang, G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. J Biol Chem. 2002;277:7059–7065. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- 42.Chuang, TH, Ulevitch RJ. Triad3A, an E3 ubiquitin-protein ligase regulating Toll-like receptors. Nat Immunol. 2004;5:495–502. doi: 10.1038/ni1066. [DOI] [PubMed] [Google Scholar]
- 43.Boone, DL, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi, K, et al. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 45.McVicar, DW, et al. DAP12-mediated signal transduction in natural killer cells a dominant role for the syk protein-tyrosine kinase. J Biol Chem. 1998;273:32934–32942. doi: 10.1074/jbc.273.49.32934. [DOI] [PubMed] [Google Scholar]
- 46.Snyder, MR, Lucas M, Vivier E, Weyand CM, Goronzy JJ. Selective activation of the c-Jun NH(2)-terminal protein kinase signaling pathway by stimulatory KIR in the absence of KARAP/DAP12 in CD4(+) T Cells. J Exp Med. 2003;197:437–449. doi: 10.1084/jem.20020383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bouchon, A, Hernandez-Munain C, Cella M, Colonna MA. DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. 2001;194:1111–1122. doi: 10.1084/jem.194.8.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dumitru, CD, et al. TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell. 2000;103:1071–1083. doi: 10.1016/s0092-8674(00)00210-5. [DOI] [PubMed] [Google Scholar]
- 49.Paloneva, J, et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet. 2000;25:357–361. doi: 10.1038/77153. [DOI] [PubMed] [Google Scholar]
- 50.Humphrey, MB, et al. The signaling adapter protein DAP12 regulates multinucleation during osteoclast development. J Bone Miner Res. 2004;19:224–234. doi: 10.1359/JBMR.0301234. [DOI] [PubMed] [Google Scholar]
- 51.Kaifu, T, et al. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J Clin Invest. 2003;111:323–332. doi: 10.1172/JCI16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nishiya, T, DeFranco AL. Ligand-regulated chimeric receptor approach reveals distinctive subcellular localization and signaling properties of the Toll-like receptors. J Biol Chem. 2004;279:19008–19017. doi: 10.1074/jbc.M311618200. [DOI] [PubMed] [Google Scholar]
- 53.Lutz, MB, et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.