

Abstract
A rise in intracellular Ca2+i (Ca2+) mediates various cellular functions ranging from fertilization to gene expression. A ubiquitous Ca2+ influx pathway that contributes significantly to the generation of Ca2+i signals, especially in non-excitable cells, is store-operated Ca2+ entry (SOCE). Consequently, the modulation of SOCE current affects Ca2+i dynamics and thus the ensuing cellular response. Therefore, it is important to define the mechanisms that regulate SOCE. Here we show that a rise in Ca2+i potentiates SOCE. This potentiation is mediated by Ca2+-calmodulin-dependent protein kinase II (CaMKII), because inhibition of endogenous CaMKII activity abrogates Ca2+i -mediated SOCE potentiation and expression of constitutively active CaMKII potentiates SOCE current independently of Ca2+i. Moreover, we present evidence that CaMKII potentiates SOCE by altering SOCE channel gating. The regulation of SOCE by CaMKII defines a novel modulatory mechanism of SOCE with important physiological consequences.
A rise in intracellular Ca2+ (Ca2+i)1 mediates various cellular responses ranging from exocytosis to cell death, often in the same cell. This is possible because of the specificity encoded in the spatial, temporal, and amplitude features of Ca2+ signals, which are generated because of either Ca2+ release from intracellular stores or Ca2+ influx from the extracellular space. The interplay between these two Ca2+ sources results in specific Ca2+i dynamics, which lead to a particular cellular response. A ubiquitous Ca2+ influx pathway activated downstream of phospholipase- coupled receptors is store-operated Ca2+ entry (SOCE) (1). SOCE is important for various cellular functions including store refilling (2), regulation of exocytosis (3), sperm capacitation (4), and T-cell activation (5, 6). Because SOCE contributes significantly to the generation of Ca2+i dynamics, it is important in specifying the ensuing cellular response. SOCE is defined as Ca2+ influx activated in response to intracellular Ca2+ store depletion. Although, store depletion is the primary required signal for SOCE activation, additional distinct mechanisms modulate SOCE activity (1). Characterizing such SOCE modulators is crucial to understanding the mechanisms by which SOCE contributes to defining specific cellular responses. In this paper, we show that both Ca2+i and Ca2+-calmodulindependent protein kinase II (CaMKII) positively modulate SOCE current (ISOC).
Both intracellular and extracellular Ca2+ have been shown to modulate SOCE activity in a complex fashion. This is best characterized for the Ca2+ release-activated Ca2+ current (ICRAC) (7), which is the first described SOCE current. Hallmarks of ICRAC include inward rectification, an ionic selectivity sequence of Ca2+ > Ba2+ > Mn2+ >> Na+, and a small unitary Ca2+ conductance estimated to be ~20 femtoSiemens (8, 9). Intracellular Ca2+ has been shown to negatively regulate ISOC by two mechanisms. 1) Fast Ca2+-dependent inactivation is due to inhibition of ISOC, arguably following Ca2+ binding to an intracellular site (8, 10). 2) Slow Ca2+-dependent inactivation is due in part to store refilling but also has a store-independent component that is poorly understood (11). In contrast, extracellular Ca2+ (Ca2+o) potentiates ISOC through a process termed Ca2+-dependent potentiation (CDP). CDP is attributed to a positive effect of extracellular Ca2+ on ISOC (12, 13) and probably results from Ca2+ binding to an extracellular site on the channel because it can be replicated by Ni2+ (12). Ni2+ does not permeate SOCE channels but potentiates the Ca2+ current through SOCE channels (12). Furthermore, CDP alters channel gating and not permeation (12), because current inactivation (which is dependent on permeation (8)) remains unchanged during CDP.
In this paper, we describe a novel positive regulation of ISOC by intracellular Ca2+ through CaMKII in Xenopus oocytes. Xenopus oocyte SOCE current has similar characteristics to ICRAC including high Ca2+ selectivity, inward rectification (14), and as shown here extracellular Ca2+-dependent potentiation. Allowing a Ca2+i rise during SOCE activation results in larger ISOC. Furthermore, expression of a constitutively active CaMKII (CaMKIIca) also leads to enhancement of ISOC. CaMKII potentiates ISOC by dramatically increasing the levels of CDP. Because CDP affects SOCE channel gating, our data argue that CaMKII potentiates SOCE by altering gating. CaMKII-mediated potentiation of ISOC has important physiological consequences because store depletion is physiologically accompanied by a rise in Ca2+i. Because CaMKII is able to decode Ca2+i dynamics into different levels of enzyme activity (15), CaMKII-mediated potentiation of ISOC provides a mechanism for Ca2+ to regulate its own entry into the cell depending on the levels and kinetics of Ca2+ release following receptor activation.
EXPERIMENTAL PROCEDURES
Oocyte and Electrophysiological Methods
Xenopus laevis oocytes were prepared as described previously (16). SOCE was activated by the depletion of intracellular Ca2+ stores with either ionomycin (10 μM) or thapsigargin (1 μm for ≥3 h unless otherwise indicated). Oocytes were injected with 7 nmol of BAPTA to buffer the Ca2+i rise and block the endogenous Ca2+-activated Cl− current (ICl,Ca) that would otherwise mask the SOCE current. Assuming an oocyte volume of 1 μl, this would result in a final concentration of ~ 7 mm BAPTA that completely blocks ICl,Ca. Oocytes were voltage-clamped with two microelectrodes by the use of a GeneClamp 500 (Axon Instruments). Electrodes were filled with 3 m KCl and had resistances of 0.5–2 megaohms. Voltage stimulation and data acquisition were controlled using a pClamp8 (Axon Instruments). Current data were filtered at 10 kHz, digitized, and analyzed using Clampfit9.0 (Axon Instruments) and Origin® software (Microcal Software Inc.). ISOC was typically measured in 30 Ca2+ solution (in mm: 55 NaCl, 30 CaCl2, 10 Hepes, pH 7.4), and Ca2+-free solution in ISOC-recording experiments was 70 Mg2+ (in mm: 70 MgCl2, 10 Hepes, pH 7.4). ISOC time course is plotted as the mean current 30–35 ms after the voltage step, and the leak current at the beginning of the experiment was subtracted.
Ca2+ activated Cl− currents were recorded in Ringer solution (in mm: 123 NaCl, 2.5 KCl, 1.8 CaCl2, 18 MgCl2, 10 Hepes, pH 7.4) or Ca2+-free Ringer solution that had the same composition as Ringer solution with the exception that CaCl2 was omitted, MgCl2 increased to 5 mm, and 0.1 mm EGTA was added.
cRNA for injection into oocytes was transcribed in vitro using the mMessage mMachine SP6 transcription kit (Ambion). Both pGEM3– 5HT1c and pSP-CaMKII(T286D) were linearized with EcoRI and transcribed with SP6 polymerase.
Ca2+ Imaging
Xenopus oocytes were injected with ~ 7.6 μm Ca2+- Green-1 coupled to 70 kDa of dextran and voltage-clamped as described above. The dye was allowed at least 30 min to equilibrate within the oocyte. Imaging experiments were performed in Ringer and Ca2+-free Ringer solutions. Confocal Ca2+ imaging was performed using an Olympus Fluoview confocal scanning system fitted to IX70 microscope using a ×10 (0.3 numerical aperture) objective. Images (256 × 256 pixels) were collected and analyzed using Olympus Fluoview software.
CaMKII Assay
CaMKII kinase activity was measured by lysing oocytes (20 μl/oocyte) in CaMKII extraction buffer (80 mm β-glycerophosphate, 20 mm Hepes, pH 7.5, 15 mm MgCl2, 1 mm sodium vanadate, 50 mm NaF, 1 mm dithiothreitol, 1× protease inhibitor mixture (Calbiochem)). Lysates were centrifuged at 500 × g for 15 min, and the supernatant was stored at − 70 °C until use in the kinase assay. The CaMKII kinase was performed using the SignaTECTTM kinase kit (Promega) according to manufacturer’s instructions.
RESULTS
Intracellular Ca2+ Potentiates SOCE
Physiological activation of SOCE is invariably preceded by a rise in Ca2+i because of Ca2+ release from stores. Although it is clear that Ca2+i is not required for SOCE activation, it is not known whether this Ca2+i modulates ISOC. To address this issue, we activated SOCE under conditions that either allow an intracellular Ca2+ rise or not. To activate SOCE in the absence of a Ca2+i rise, we injected cells with BAPTA and then depleted Ca2+ stores with ionomycin (BAPTA-Ion). This results in the activation of a characteristic SOCE current as previously described (Fig. 1B, filled squares) (14, 17). To allow a Ca2+i rise during SOCE activation, Ca2+ stores were depleted with ionomycin followed by repetitive hyperpolarization to induce Ca2+ influx (Ion- BAPTA). The cell was then injected with BAPTA, and ISOC was recorded (Fig. 1B, open circles). Treating cells according to the Ion-BAPTA protocol resulted in a significant (p = 1.6 × 10−7) potentiation of ISOC levels compared with control oocyte (BAPTA-Ion) (Fig. 1, B and C). Therefore, allowing a Ca2+i rise during SOCE activation leads to current potentiation.
Fig. 1. Intracellular Ca2+ rise potentiates ISOC.
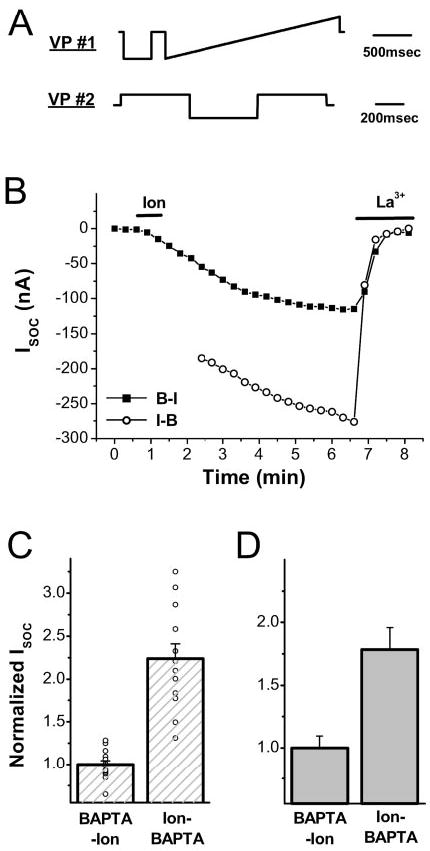
A, voltage protocols (VP) used to measure ISOC (#1) and to induce Ca2+ influx (#2). For VP#1, the cell was stepped to −140 mV followed by a ramp from −140 to 50 mV from a holding potential of −20 mV repeated once every 20 s. For VP#2, the cell was stepped to +40, −140, and +40 mV sequentially from a holding potential of −40 mV once every 30 s. B, time course of ISOC activation following the BAPTA-Ion (B-I) or Ion-BAPTA (I-B) protocols. For the BAPTA-Ion protocol, oocytes were injected with BAPTA (7 nmol/oocyte) followed by ionomycin (10 μm) treatment as indicated by the line. ISOC was measured as the average current 30–35 ms after stepping to − 140 mV in VP#1. For the Ion-BAPTA protocol, cells were treated with ionomycin and repetitively subjected to VP#2 for 10 min before ISOC recording (open circles). La3+ (100 μM) was added at the end of the experiment to block ISOC. C, summary of ISOC levels following the BAPTA-Ion (n = 14) and Ion-BAPTA (n = 12) protocols. The data are plotted as mean ± S.E. (p = 1.6 × 10−7). D, Ca2+i rise potentiates ISOC independently of hyperpolarization-induced Ca2+ influx. Cells (in 5 mM Ca2+) were exposed to ionomycin for 5 min and then injected with BAPTA (Ion-BAPTA), and ISOC was measured as described in B, BAPTA-Ion treatment was as described in B. The means are significantly different (p = 0.0044; n = 5).
The Ca2+-mediated potentiation of SOCE described above was obtained under conditions designed to maximize Ca2+i rise by hyperpolarization-induced Ca2+ influx. To determine whether such Ca2+ influx is required for ISOC potentiation, a similar experiment was performed without hyperpolarization using the ion-BAPTA protocol, that is Ca2+ stores were simply depleted in the absence of BAPTA injection. In this case, ISOC was also potentiated (Fig. 1D), showing that hyperpolarization per se is not required for the observed Ca2+i-mediated ISOC potentiation.
As discussed above, SOCE has been shown to be potentiated by extracellular Ca2+, which affects SOCE channel gating in a process referred to as CDP (12). To avoid confusion with CDP, we will refer to the intracellular Ca2+ effect on ISOC as Ca2+i-mediated potentiation (CMP). CMP provides a fitting feedback mechanism between Ca2+ release and SOCE. This is especially relevant because as shown by Parekh et al. (18), SOCE activation is an all or nothing process (18). That is even partial depletion of intracellular Ca2+ stores results in full activation of ISOC. To determine the relationship between store Ca2+ load and ISOC magnitude in Xenopus oocytes, SOCE was measured by Ca2+ imaging in voltage-clamped cells, which allows simultaneous recording of Ca2+i and SOCE in the same cell (Fig. 2) (19). Membrane potential was stepped repetitively to +40 mV (where Ca2+ influx is minimal) and −140 mV (to induce Ca2+ influx through SOCE). Under these conditions, the difference in Ca2+ fluorescence at −140 and +40 mV (Fig. 2, open triangles) provides a reliable measure of SOCE and correlates well with ISOC (19). To induce partial store depletion, cells were treated with thapsigargin (1 μm) for 1 h (complete store depletion requires ≥3 h), which results in SOCE activation as indicated by the increased fluorescence at −40 V (Fig. 2, open circles). Injection of IP3 leads to further Ca2+ release (Fig. 2, squares & circles), confirming that thapsigargin induced partial store depletion. However, the additional store depletion following IP3 injection does not augment SOCE (Fig. 2, open triangles), indicating that SOCE magnitude does not correlate well with the extent of store depletion. This shows that increased store depletion, at least in the range tested, does not translate into a larger SOCE, arguing that SOCE is modulated by other signaling pathways after store depletion. Fig. 1 shows that one such modulatory pathway is the Ca2+i levels during SOCE activation.
Fig. 2. Partial store depletion fully activates SOCE.

Oocyte was loaded with Ca2+-Green-1-dextran (7 μM) and incubated in thapsigargin (Thaps) (1 μM) for 1 h to partially deplete intracellular Ca2+ stores. SOCE was measured by clamping the cell using voltage protocol number 2 (Fig. 1A) as described by Machaca and Hartzell (19). Ca2+-Green- 1-dextran fluorescence at +40 mV (FCa(+40); close squares) provides basal Ca2+ levels because there is minimal Ca2+ influx at this voltage (19). In contrast, Ca2+ influx through SOCE channels is induced at −140 mV, resulting in increased Ca2+-Green-1-dextran fluorescence (FCa(−140); open circles). Therefore, the difference between CG1 fluorescence at −140 and +40 mV provides a measure of SOCE. Partial store depletion with a short thapsigargin incubation (1 h) activates SOCE. Injection of IP3 (~2 μM) leads to further Ca2+ release from stores as indicated by increased Ca2+-Green-1-dextran fluorescence at both voltages (squares and circles). However, the additional Ca2+ release leading to further store depletion does not enhance SOCE (open triangles). Switching the cell to Ca2+-free solution results in a loss of the Ca2+- Green-1-dextran fluorescence signal at −140 mV, confirming that the signal is because of Ca2+ influx. These data are representative of four similar experiments.
Correlation between Receptor-induced Ca2+ Mobilization and SOCE
To assess the physiological relevance of CMP, we sought to determine whether it could be observed following IP3-linked receptor stimulation. We were interested in inducing different levels of Ca2+i rise while minimizing experimental manipulation of Ca2+i (such as BAPTA injection or ionomycin treatment). Our approach was to express the G-protein-coupled serotonin receptor (5HT1c) and monitor changes in the endogenous Ca2+-activated Cl− currents as markers of Ca2+i (19). 5HT1c stimulation with serotonin (5HT) results in IP3 production through phospholipase-β activation (20). We have previously shown that Ca2+-activated Cl− currents (ICl1 and ICl1T) faithfully report Ca2+i changes below the plasma membrane in terms of amplitude and kinetics (19). ICl1 activates in response to Ca2+ release from internal stores, whereas ICl1T responds to Ca2+ influx from the extracellular space. During Ca2+ release, ICl1 activates as a sustained outward current upon depolarization (+ 40 mV, Fig. 3A, upper trace). Ca2+ release results in store depletion and SOCE activation. Ca2+ flowing through SOCE channels activates ICl1T as a transient current, only when the depolarization pulse is preceded by a hyperpolarization step, which induces Ca2+ influx (Fig. 3A, lower trace). ICl1T is transient because the Ca2+ that enters through SOCE channels during the −140-mV pulse dissipates rapidly during the subsequent 40-mV pulse, leading to ICl1T current decay (Fig, 3A, lower trace) (19) (for a more detailed description of the relationship between Ca2+ signals and Ca2+-activated Cl− currents, see Refs. 19, 21, and 22). Thus, monitoring ICl1 and ICl1T allows the real-time determination of Ca2+i (ICl1) and SOCE (ICl1T) levels following 5HT1c activation.
Fig. 3. Receptor mediated Ca2+ release levels correlate with SOCE.

Oocytes were injected with serotonin receptor 1c (5HT1c) RNA at either 10 or 30 ng/oocyte and were allowed to express for 2 days. The activity of the endogenous Ca2+-activated Cl− currents (ICl1 and ICl1T) were recorded. ICl1 and ICl1T provide endogenous reporters of Ca2+ release (ICl1) and SOCE (ICl1T) (see “Results” for details). A, voltage protocol and representative current traces of the Ca2+-activated Cl− currents. ICl1 is a sustained current recorded at +40 mV, whereas ICl1T is a transient current detected at +40 mV after the −140 mV hyperpolarization step. B and C, time course of ICl1 and ICl1T is cells injected with 10 ng (B) or 30 ng (C) 5HT1c RNA. The time course of ICl1 was plotted as the current at the end of the first +40-mV pulse (square in A), and that of ICl1T was plotted as the maximal current during the second +40-mV pulse (circle in A). C, higher 5HT1c expression results in increased levels of both ICl1 and ICl1T. D, mean ICl1 and ICl1T current levels (±S.E.) in cells expressing low (10 ng, hatched bars) and high levels (30 ng, filled bars) of 5HT1c. Increasing the expression of 5HT1c results in higher levels of ICl1 (indicative of Ca2+ release) and ICl1T (indicative of SOCE).
To generate graded Ca2+ release responses and determine the effect on SOCE, we injected cells with different amounts of 5HT1c cRNA (10 or 30 ng) and allowed them to express for 2 days. Cells were then stimulated with 5HT (10 μm) (Fig. 3, B and C), leading to Ca2+ release, which activates ICl1 (Fig. 3, B and C, squares) followed by ICl1T due to SOCE activation (Fig. 3, B and C, circles). ICl1T activates to a certain threshold and gradually returns to base line as a result of Ca2+ store refilling and SOCE inactivation (Fig. 3, B and C, circles). Cells injected with 10 ng of 5HT1c cRNA produced a smaller ICl1 (Fig. 3B) than those injected with 30 ng of the receptor (Fig. 3C). Furthermore, ICl1T activated to a lower threshold and inactivated more rapidly in cells injected with 10 ng of 5HT1c receptor cRNA (Fig. 3, compare B with C, circles). The responses of the Cl− currents summarized in Fig. 3D show that both Ca2+i (as indicated by ICl1) and SOCE (as indicated by ICl1T) were significantly enhanced at high 5HT1c (30 ng) expression. Therefore, enhanced Ca2+ release correlates with larger SOCE. A simple explanation for these data is that receptor stimulation in cells expressing high levels of 5HT1c leads to a more dramatic Ca2+ store depletion and larger SOCE. However, as shown in Fig. 2, the extent of Ca2+ store depletion does not linearly correlate with SOCE magnitude. Therefore, SOCE potentiation as reported by ICl1T in cells expressing more 5HT1c receptors is probably because of some mechanism other than the extent of store depletion. CMP of ISOC provides such a mechanism. High 5HT1c expression produces increased Ca2+i as indicated by ICl1, which would be expected to potentiate ISOC (ICl1T) through CMP. Note that store depletion provides a required signal for SOCE activation, but the extent of store depletion per se does not modulate SOCE magnitude. Rather, the data in Figs. 2 and 3 argue that it is the magnitude of the Ca2+i rise that modulates SOCE through CMP. Clearly the magnitude of Ca2+i rise can correlate with the extent of store depletion. These data reveal a subtle but important distinction in the mechanism of SOCE modulation and are consistent with the notion that CMP is a physiologically relevant modulator of SOCE.
The positive correlation between Ca2+i and SOCE described above is not limited to Xenopus oocytes. Gailly et al. (23) described a similar relationship in Chinese hamster ovary cells (23), arguing that CMP is a widespread mechanism of SOCE regulation.
CaMKII Potentiates SOCE
Ca2+i-mediated potentiation of ISOC could be either direct or indirect through the activation of Ca2+-dependent downstream effectors. A direct Ca2+-mediated potentiation of ISOC is unlikely because Ca2+ has been shown to inactivate ISOC, probably through a direct effect on SOCE channels (fast Ca2+-dependent inactivation) (8, 10). This finding suggests that CMP is the result of activation of Ca2+-dependent effectors, which in turn act on SOCE. A primary candidate for such an effector pathway is the Ca2+-CaM-activated protein kinase pathway. The most widespread Ca2+-CaM-dependent kinase is CaMKII, which is expressed in Xenopus oocytes (24). If Ca2+ potentiates ISOC through CaMKII, it is expected that ectopic activation of CaMKII would lead to increased ISOC levels.
We used a constitutively active CaMKII mutant (CaMKIIca) to determine whether CaMKII activation potentiates ISOC. The CaMKII holoenzyme is a multisubunit complex that is kept inactive by an autoinhibitory domain. The binding of Ca2+- CaM relieves autoinhibition and stimulates autophosphorylation at Thr-286, rendering the enzyme Ca2+-CaM-independent (25). CaMKIIca is a T286D mutation that mimics the effects of autophosphorylation by replacing Thr-286 with Asp, resulting in a Ca2+-CaM-independent and thus constitutively active CaMKII (26). We injected oocytes with CaMKIIca and measured ISOC (Fig. 4A). Expression of CaMKIIca results in an ~3-fold increase in ISOC (p = 1.1 × 10−7) (Fig. 4, A and B), consistent with CMP acting through CaMKII. In addition, CaMKIIca-mediated ISOC potentiation is expected to be independent of Ca2+i because CaMKIIca is Ca2+-CaM-independent. This prediction is confirmed in CaMKIIca-expressing cells, as ISOC is potentiated whether store depletion is preceded by a rise in Ca2+i (Ion-BAPTA) or not (BAPTA-Ion) (Fig. 4C). This finding further suggests that Ca2+-mediated potentiation is primarily through CaMKII activation, because no additive effect on ISOC is observed in CaMKIIca-expressing cells subjected to the Ion-BAPTA protocol (Fig. 4C).
Fig. 4. CaMKIIca potentiates ISOC.

Oocytes were injected with a constitutively active CaMKII (CaMKIIca, 1 ng/oocyte) and allowed to express for 12–16 h. SOCE was measured using voltage protocol number 1 in Fig. 1A with the exception that the voltage was stepped to −120 mV instead of −140 mV. A, time course of ISOC activation in control and CaMKIIca expressing cells. B, normalized ISOC levels (n as indicated; p = 1.1 × 10−7). C, normalized ISOC levels from control (Con.) and CaMKIIca-injected cells treated according to the BAPTA-Ion or Ion-BAPTA protocols as described in Fig. 1. The asterisks above the bars indicate the significantly different groups (n as indicated; p < 0.0164). D, basal CaMKII kinase activity from control and CaMKIIca-injected oocytes measured using an in vitro kinase assay without the addition of Ca2+-CaM (n = 5; p = 0.0469).
To directly confirm CaMKIIca expression, we measured CaMKII-specific activity in lysates from control and CaMKIIca-injected cells. Cells expressing CaMKIIca had higher levels of CaMKII activity (Fig. 4D), showing that CaMKIIca was expressed and functional in these cells. Because specific activity was measured in the absence of Ca2+-CaM (Fig. 4D), CaMKII activity data confirm the Ca2+-CaM independence of CaMKIIca.
It is important to note that the expression of CaMKIIca by itself is not sufficient to activate ISOC (Fig. 4A, circles). In CaMKIIca-expressing cells, store depletion is still required to activate SOCE because no ISOC is detected before ionomycin treatment (Fig. 4A, circles). However, ISOC levels reach a significantly larger maximal amplitude in CaMKIIca-expressing cells compared with control cells (Fig. 4, A and B). Therefore, CaMKII is not a component of the SOCE activation pathway induced in response to store depletion but rather positively modulates SOCE activity following SOCE activation.
If CMP is acting through CaMKII, CaMKII should be activated in cells where a Ca2+i rise is induced (Ion-BAPTA). We were unable to detect such an increase in CaMKII activity in oocytes treated according to the Ion-BAPTA protocol. This result argues that either Ca2+ and CaMKII potentiate ISOC by separate mechanisms or that CaMKII activation is transient and/or spatially localized, resulting in small changes in CaMKII activity that are difficult to detect in whole cell lysates. Furthermore, basal CaMKII activity was quite variable in oocytes donated from different females, making it difficult to reliably measure a small increase in CaMKII activity in different batches of cells. However, a Ca2+i rise has been shown to activate endogenous CaMKII in Xenopus oocytes using an in vivo CaMKII-specific kinase assay (27).
Nonetheless, if CMP is mediated by CaMKII, blocking endogenous CaMKII should inhibit CMP. Therefore, we blocked endogenous CaMKII and determined the effect on SOCE. Oocytes were injected with AIP, a specific inhibitory peptide of CaMKII that mimics the autoinhibitory domain and blocks CaMKII activity (28). Allowing a Ca2+i rise in control oocytes (Ion-BAPTA) potentiates ISOC by ~2-fold but not in cells injected with the CaMKII inhibitor AIP (Fig. 5A). Furthermore, AIP added to the CaMKII assay blocks kinase activity in a dose-dependent manner (Fig. 5B). These data show that inhibiting endogenous CaMKII activity blocks CMP, supporting the conclusion that Ca2+i potentiates SOCE through CaMKII activation.
Fig. 5. Inhibition of endogenous CaMKII blocks Ca2+ i-mediated SOCE potentiation.

Control and AIP-injected (Inj.) (10 μM) oocytes were treated according to the BAPTA-Ion and Ion-BAPTA protocols as indicated. A, normalized ISOC levels in the different treatment groups. The asterisk indicates the only significantly different group (p < 2.1 × 10−4). B, basal CaMKII activity in oocyte lysate without AIP (Con) and with 10 and 40 μM AIP as indicated (n = 6).
Mechanism of CaMKII Action on SOCE
Because SOCE is activated in response to store depletion, CaMKII could potentiate ISOC by targeting either the coupling mechanism between Ca2+ stores and SOCE or the SOCE channel. To differentiate between these possibilities and obtain a better understanding of the mechanism of action of CaMKII, we wanted to study the effects of CaMKII independently of store depletion. This was accomplished by irreversibly depleting Ca2+ stores with thapsigargin before CaMKIIca expression (Fig. 6). Oocytes were incubated in thapsigargin (1 μm) for 3 h to fully deplete Ca2+ store followed by CaMKIIca cRNA injection and incubation in nominally Ca2+-free (50 μm free Ca2+) solution. Control cells were treated with thapsigargin alone. Under these conditions, store depletion is complete before CaMKIIca expression and store Ca2+ load remains low throughout the experiment because thapsigargin irreversibly inhibits the ER Ca2+-ATPase. This allows us to study the effects of CaMKII on ISOC after store depletion and determine whether CaMKII is affecting the coupling mechanism or SOCE channel gating or permeation. In this experiment, SOCE was activated in the absence of the conducting ion (Ca2+) and therefore no SOCE current was observed at the beginning of the experiment (Fig. 6A). Switching control (thapsigargin-treated) oocytes to a Ca2+-containing solution produced an initial ISOC that was further enhanced over time, eventually saturating (Max ISOC) within ~6 min (Fig. 6A, squares). This behavior is the result of classical CDP where extracellular Ca2+ exerts a positive effect on SOCE channel gating (12, 13). CDP can be estimated as the ratio of maximal ISOC/initial ISOC (12) and is 3.11 ± 0.23 in thapsigargin-treated cells (Fig. 6B).
Fig. 6. CaMKII potentiates ISOC by increasing the levels of CDP.

Cells were incubated with thapsigargin (Thaps) (1 μM) in nominally Ca2+-free medium (50 μM) for 3 h to fully deplete Ca2+ stores. A subset of cells was then injected with 1 ng of CaMKIIca RNA (Thaps-CaMKca) and incubated in nominally Ca2+-free medium for 12–16 h. A, ISOC recorded from a representative control (Thaps) and CaMKIIca-injected oocyte (Thaps-CaMKca). Cells were incubated in Ca2+-free solution (70 Mg2+) before switching to Ca2+-containing solution (30 Ca2+) as indicated by the line. SOCE was measured using voltage protocol number 1 in Fig. 1A with the exception that the voltage was stepped to −120 mV instead of −140 mV. The addition of La3+ to block ISOC is also indicated. B, normalized ISOC levels showing initial ISOC (indicated by the asterisk in A) and maximal ISOC at the end of the experiment (indicated by the open square and filled circle in the Thaps and Thaps-CaMKca groups, respectively). The average levels of CDP calculated as maximal ISOC/initial ISOC are also shown. Maximal ISOC and CDP are significantly different between the two groups (p < 0.00132). C and D, current traces in response to a step voltage (−20 to −120 mV for 500 ms) (C) and a current ramp (−140 to +50 mV) (D) obtained at different time points during the experiment as indicated in A. E and F, superimposed current traces of maximal ISOC from Thaps and Thaps-CaMKIIca-treated cells in response to a step voltage pulse (E) and a voltage ramp (F).
Surprisingly, exposing CaMKIIca-expressing cells to Ca2+- containing solution results in an initial ISOC with a similar amplitude to initial ISOC in control cells (Fig. 6A, open circles). However, ISOC in CaMKIIca-expressing cells gradually increased to significantly higher levels than in control cells (Fig. 6A, open circles). This shows that CaMKIIca expression does not affect initial ISOC levels but results in a significantly (p = 1.46 × 10−5) larger maximal ISOC (Fig. 6B). It follows that CDP, calculated as the ratio of maximal/initial ISOC, was also augmented (5.72 ± 0.61) in CaMKIIca-expressing cells (Fig. 6B). Therefore, CaMKII potentiates ISOC by increasing the levels of CDP without affecting initial ISOC levels, even after prolonged expression of CaMKIIca.
These data provide important insights into the mechanism of action of CaMKII. The whole cell SOCE current is defined by the following equation: ISOC = NPoi, where N is the number of active channels, Po is the probability of opening, and i is the single channel conductance. Therefore, CaMKII can potentiate ISOC by increasing either N, i, or Po. If CaMKII was affecting the coupling mechanism, an increase in the number of active channels (N) is expected. The fact that CaMKIIca expression does not enhance initial ISOC provides evidence against an increase in channel number (N) or single channel conductance (i) (Fig. 6, A and B). This is because initial ISOC is due to current flowing through all of the open channels after Ca2+ addition. If either N or i was augmented by CaMKII, initial ISOC would be larger in CaMKIIca-expressing cells. This hypothesis argues that CaMKII potentiates ISOC by enhancing the probability of opening (Po) of SOCE channels.
The observed increase in CDP levels in CaMKIIca-expressing cells strongly supports the conclusion that CaMKII potentiates ISOC by increasing Po. This is because CDP has been shown to be the result of extracellular Ca2+ exerting a positive effect on SOCE channel gating (Po) (12, 13).
Representative current traces in response to a step pulse to −120 mV (Fig. 6C) and a voltage ramp from −140 to 50 mV (Fig. 6D) obtained from control (Thaps) and CaMKIIca-injected cells (Thaps-CaMKca) are shown. The time points during the experiments at which the traces were obtained are indicated in Fig. 6A (Thaps, open symbols; Thaps-CaMKca, filled symbols). Note the similar levels of initial ISOC in both treatments (Fig. 6C, open and filled stars). Initial ISOC traces from CaMKIIca-injected cells show a time-dependent increase in current amplitude (Fig. 6C, filled star) that is due to CDP occurring during the 500-ms duration of the voltage pulse. Zweifach and Lewis (8) have shown that the extent of Ca2+-dependent inactivation of ICRAC following a brief hyperpolarization depends on the single channel current (i) (8). That is, increased unitary current (i) results in a faster rate of ICRAC inactivation. Assuming that the same relationship holds for Xenopus oocyte ISOC, if CaMKII increases unitary conductance (i), we expect a faster ISOC inactivation rate shortly after the hyperpolarization pulse. However, the rate of ISOC inactivation shortly after (100 ms) hyperpolarization is similar between control and CaMKIIca-expressing cells (Fig. 6E). In fact, at steady-state, control cells exhibit a more marked ISOC inactivation (Fig. 6E). These inactivation kinetics argue that CaMKII does not enhance ISOC unitary conductance. Superimposed current-voltage relationships from control and CaMKIIca-injected cells were similar (Fig. 6F), indicating that CaMKII does not affect the voltage dependence of ISOC.
These results argue that CaMKIIca potentiates ISOC by targeting SOCE channel gating and not the coupling mechanism between Ca2+ stores and SOCE. Three pieces of evidences support the conclusion that CaMKII potentiates ISOC by affecting channel gating (Po) and not channel number (N) or unitary conductance (i): 1) similar levels of initial ISOC in control and CaMKIIca-expressing cells (Fig. 6, A and B) arguing against an effect of CaMKII on N or i. 2) enhanced CDP in CaMKIIca-expressing cells (Fig. 6B), suggesting that CaMKII enhances channel Po. 3) similar ISOC inactivation rates shortly after hyperpolarization in control and CaMKIIca-expressing cells (Fig. 6E) arguing against an increase in unitary conductance (i).
DISCUSSION
Ca2+i has been shown to negatively regulate ISOC by inducing channel inactivation either directly or through store refilling (8, 10, 11). Here we show that Ca2+i can in addition have a potentiating effect on SOCE. The Ca2+i effect on SOCE is probably the result of CaMKII activation, because Ca2+i, because inhibition of endogenous CaMKII activity blocks CMP and expression of CaMKIIca is sufficient to potentiate ISOC independently of Ca2+i. Although CaMKII potentiates ISOC, it is not sufficient by itself to activate SOCE independently of store depletion (Fig. 4A). This observation is consistent with the fact that a Ca2+i rise is not required for SOCE activation (1). Therefore, the CaMKII pathway modulates SOCE activity but is not an essential component of the SOCE activation pathway in response to store depletion. For CaMKII to exert its effects, SOCE has to be activated by store depletion.
Using pharmacological inhibitors, a role for CaMKII in skeletal muscle SOCE (29) and myosin light chain kinase (a Ca2+- CaM-dependent kinase related to CaMKII (25)) in endothelial SOCE (30) have been postulated. This finding argues that CaMKII modulation of ISOC is a widespread mechanism that is not cell type-specific. Nonetheless, a previous report by Matifat et al. (27) suggests a negative effect of CaMKII on SOCE in Xenopus oocytes. However, it is not clear from this study that the reported CaMKII effect was due to SOCE modulation because the Ca2+-activated Cl− current was used as an indicator of SOCE, making it impossible to differentiate between an effect of CaMKII on the Cl− current or SOCE. In contrast, we have directly measured the SOCE current while modulating CaMKII activity and show that CaMKII activation potentiates ISOC. This finding argues that the effects of CaMKII reported by Matifat et al. (27) are due to modulation of the Cl− currents or other Ca2+ influx pathways in the oocyte.
CaMKII provides an excellent modulator of SOCE activity because of its ability to decode spatial and temporal information encoded in Ca2+i signals into different levels of kinase activity. This capacity is attributable to spatial and structural features of CaMKII. CaMKII localizes to specific subcellular compartments such as the nucleus and cytoskeleton (31, 32), and its activity increases exponentially based on the number and frequency of Ca2+i oscillation (15, 33). These exceptional attributes allow CaMKII to provide specificity to Ca2+i signals by differentially activating effectors based on the kinetics of Ca2+i signals. CaMKII plays important roles in regulating various cellular functions such as gene expression, cell cycle progression, and learning and memory (25, 34). The data presented here show that SOCE can now be added to the list of cellular functions modulated by CaMKII.
In a similar fashion to the CaMKII effect on SOCE described here, CaMKII has been shown to augment both L-type (Cav1.x) (35) and T-type (Cav3.x) (36) Ca2+ currents. CaMKII regulation of SOCE described in this paper is reminiscent of Ca2+-dependent facilitation of inward L-type Ca2+ current. As Ca2+i increases, L-type Ca2+ current is enhanced in a process termed facilitation (37). This facilitation is mediated by CaMKII, which increases the open probability of L-type channels (35). Our data strongly argue that CaMKII potentiates ISOC in a similar fashion by altering channel gating (see Fig. 6). This similarity in the mechanism of action of CaMKII on both voltage- gated and store-operated Ca2+ channels argues for a common cross-talk between Ca2+i kinetics and Ca2+ influx pathways. It is attractive to postulate that CaMKII provides a mechanism for phospholipase-linked receptors to differentially modulate SOCE activity. That is, the spatiotemporal features of the Ca2+i signals downstream of receptor activation differentially activate CaMKII and thus SOCE.
In addition to the CaMKII-mediated regulation of SOCE described here, SOCE has been shown to be modulated by protein kinase C (38, 39). Both protein kinase C and CaMKII are downstream kinases that can be induced following activation of phospholipase-linked receptors. Therefore, the interplay between various modulatory mechanisms in a specific cellular context combines to generate different levels of Ca2+ influx through SOCE. This Ca2+ influx affects Ca2+i kinetics and thus Ca2+-dependent cellular responses. Consequently, CaMKII regulation of SOCE is likely to have broad and important physiological consequences.
Acknowledgments
I am grateful to H. Schulman and A. MacNicol for the constitutively active CaMKIIca mutant (T286D) and H. Lester for the 5HT1c clone. I thank Shirley Haun for performing some of the kinase activity experiments and Joe Stimers and Lou DeFelice for comments on the paper.
Footnotes
This work is funded by National Institutes of Health Grant GM- 61829 and a grant from the Tobacco settlement at University of Arkansas Medical Sciences. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: Ca2+i, intracellular Ca2+; SOCE, storeoperated Ca2+ entry; CaMKII, Ca2+-calmodulin-dependent protein kinase II; CaMKIIca, constitutively active CaMKII; ISOC, SOCE current; Ca2+o, extracellular Ca2+; ICRAC, Ca2+ release-activated Ca2+ current; CDP, Ca2+-dependent potentiation; AIP, autocamtide-2-related inhibitory peptide; IP3, inositol 1,4,5-trisphosphate; Ion, ionomycin; CMP, Ca2+i-mediated potentiation; BAPTA, 1,2-bis(2-aminophenoxy)ethane- N,N,N′,N′-tetraacetic acid.
References
- 1.Parekh AB, Penner R. Physiol Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- 2.Parekh AB, Foguet M, Lubbert H, Stühmer W. J Physiol (Lond) 1993;469:653–671. doi: 10.1113/jphysiol.1993.sp019836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fomina AF, Nowycky MC. J Neurosci. 1999;19:3711–3722. doi: 10.1523/JNEUROSCI.19-10-03711.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Toole CM, Arnoult C, Darszon A, Steinhardt RA, Florman HM. Mol Biol Cell. 2000;11:1571–1584. doi: 10.1091/mbc.11.5.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serafini A, Lewis RS, Clipstone NA, Bram R, Fanger C, Fiering S, Herzenberg LA, Crabtree GR. Immunity. 1995;3:239–250. doi: 10.1016/1074-7613(95)90093-4. [DOI] [PubMed] [Google Scholar]
- 6.Fanger C, Hoth M, Crabtree GR, Lewis RS. J Cell Biol. 1995;131:655–667. doi: 10.1083/jcb.131.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis RS. Adv Second Messenger Phosphoprotein Res. 1999;33:279–307. doi: 10.1016/s1040-7952(99)80014-7. [DOI] [PubMed] [Google Scholar]
- 8.Zweifach A, Lewis RS. J Gen Physiol. 1995;105:209–226. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoth M, Penner R. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 10.Hoth M, Penner R. J Physiol (Lond) 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zweifach A, Lewis RS. J Biol Chem. 1995;270:14445–14451. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]
- 12.Zweifach A, Lewis RS. J Gen Physiol. 1996;107:597–610. doi: 10.1085/jgp.107.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christian EP, Spence KT, Togo JA, Dargis PG, Patel J. J Membr Biol. 1996;150:63–71. doi: 10.1007/s002329900030. [DOI] [PubMed] [Google Scholar]
- 14.Yao Y, Tsien RY. J Gen Physiol. 1997;109:703–715. doi: 10.1085/jgp.109.6.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Koninck P, Schulman H. Science. 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- 16.Machaca K, Haun S. J Cell Biol. 2002;156:75–86. doi: 10.1083/jcb.200110059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Machaca K, Haun S. J Biol Chem. 2000;275:38710–38715. doi: 10.1074/jbc.M007887200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parekh AB, Fleig A, Penner R. Cell. 1997;89:973–980. doi: 10.1016/s0092-8674(00)80282-2. [DOI] [PubMed] [Google Scholar]
- 19.Machaca K, Hartzell HC. J Gen Physiol. 1999;113:249–266. doi: 10.1085/jgp.113.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quick MW, Simon MI, Davidson N, Lester HA, Aragay AM. J Biol Chem. 1994;269:30164–30172. [PubMed] [Google Scholar]
- 21.Hartzell HC. J Gen Physiol. 1996;108:157–175. doi: 10.1085/jgp.108.3.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuruma A, Hartzell HC. J Gen Physiol. 2000;115:59–80. doi: 10.1085/jgp.115.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gailly PH, Hermans E, Gillis JM. J Physiol (Lond) 1996;491:635–646. doi: 10.1113/jphysiol.1996.sp021245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stevens I, Rondelez E, Merlevede W, Goris J. J Biochem. 2001;129:551–560. doi: 10.1093/oxfordjournals.jbchem.a002890. [DOI] [PubMed] [Google Scholar]
- 25.Hook SS, Means AR. Annu Rev Pharmacol Toxicol. 2001;41:471–505. doi: 10.1146/annurev.pharmtox.41.1.471. [DOI] [PubMed] [Google Scholar]
- 26.Waldmann R, Hanson PI, Schulman H. Biochemistry. 1990;29:1679–1684. doi: 10.1021/bi00459a002. [DOI] [PubMed] [Google Scholar]
- 27.Matifat F, Fournier F, Lorca T, Capony JP, Brule G, Collin T. Biochem J. 1997;322:267–272. doi: 10.1042/bj3220267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishida A, Kameshita I, Okuno S, Kitani T, Fujisawa H. Biochem Biophys Res Commun. 1995;212:806–812. doi: 10.1006/bbrc.1995.2040. [DOI] [PubMed] [Google Scholar]
- 29.Vazquez G, de Boland AR, Boland RL. J Biol Chem. 2000;275:16134–16138. doi: 10.1074/jbc.C901008199. [DOI] [PubMed] [Google Scholar]
- 30.Norwood N, Moore TM, Dean DA, Bhattacharjee R, Li M, Stevens T. Am J Physiol. 2000;279:L815–L824. doi: 10.1152/ajplung.2000.279.5.L815. [DOI] [PubMed] [Google Scholar]
- 31.Srinivasan M, Edman CF, Schulman H. J Cell Biol. 1994;126:839–852. doi: 10.1083/jcb.126.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen K, Meyer T. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- 33.Hanson PI, Meyer T, Stryer L, Schulman H. Neuron. 1994;12:943–956. doi: 10.1016/0896-6273(94)90306-9. [DOI] [PubMed] [Google Scholar]
- 34.Hudmon A, Schulman H. Annu Rev Biochem. 2002;71:473–510. doi: 10.1146/annurev.biochem.71.110601.135410. [DOI] [PubMed] [Google Scholar]
- 35.Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Nat Cell Biol. 2000;2:173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- 36.Barrett PQ, Lu HK, Colbran R, Czernik A, Pancrazio JJ. Am J Physiol. 2000;279:C1694–C1703. doi: 10.1152/ajpcell.2000.279.6.C1694. [DOI] [PubMed] [Google Scholar]
- 37.McCarron JG, McGeown JG, Reardon S, kebe M, Fay FS, Walsh JV., Jr Nature. 1992;357:74–77. doi: 10.1038/357074a0. [DOI] [PubMed] [Google Scholar]
- 38.Petersen CC, Berridge MJ. J Biol Chem. 1994;269:32246–32253. [PubMed] [Google Scholar]
- 39.Parekh AB, Penner R. Proc Natl Acad Sci U S A. 1995;92:7907–7911. doi: 10.1073/pnas.92.17.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]