

Abstract
Interferon regulatory factor 3 (IRF3) mediates the transcriptional induction of interferon-stimulated genes (ISGs) in response to viral and bacterial infections. Here we show that the hydroxystilbene piceatannol inhibits the LPS-mediated activation of IRF3 and subsequent ISG induction. Consequently, piceatannol blocks the LPS-induced up-regulation of critical mediators of the inflammatory response such as interleukin 6 (IL-6), tumor necrosis factor-alpha (TNF-α), intercellular adhesion molecule 1 (ICAM-1), and macrophage chemoattractant protein (MCP-1). Furthermore, the LPS-mediated induction of tissue factor (TF), a cell surface protein responsible for initiating the coagulation cascade, is also inhibited by piceatannol. The effectiveness of piceatannol in blocking both the inflammatory response and the coagulation pathway is evidenced by its ability to confer protection against LPS-induced septic shock in a murine model. Thus, IRF3 appears to be a promising target for pharmacologic intervention in the prevention or treatment of septic shock syndrome.
Keywords: IRF3, LPS, sepsis, piceatannol, IL-6, TF, ISG, interferon
INTRODUCTION
The type I interferons, IFNα and IFN β, promote their biologic effects through the induction of immediate early response genes termed interferon-stimulated genes (ISGs) (1). Transcriptional induction of ISGs by IFNα/β is mediated by members of the SH2 domain-containing signal transducer and activator (STAT) family of transcription factors, whose activation depends on their COOH-terminal tyrosine phosphorylation (2–4). The kinases responsible for the phosphorylation events associated with IFNα/β receptor signaling are the Jak tyrosine kinase family members, Jak1 and Tyk2. Tyrosine-phosphorylated, activated STAT proteins dimerize via SH2 domain interaction and subsequently translocate to the nucleus where they complex with the DNA-binding adapter ISGF3γ (IRF9) to bind interferon-stimulated response elements (ISREs) in the promoters of ISGs (5).
In addition to the STAT proteins, members of the interferon regulatory factor (IRF) family of transcription factors are also able to bind to the ISRE (6). The nine members of the IRF family are characterized by a broad range of functions (7). For example, IRF1 acts as a tumor suppressor, and IRF2 functions to promote cell growth (8). IRF3 plays a critical role in the innate immune response. IRF3 was first identified as a key component in the induction of type I interferon genes during viral infection (9–11). In addition, IRF3 is also activated through the actions of Toll-like receptor (TLR) ligands such as lipopolysaccharide (LPS) or lipoteichoic acid (LTA) (12). Activation of IRF3 involves its phosphorylation on several serine residues, followed by its dimerization and nuclear translocation (10, 11, 13). In the nucleus, IRF3 binds to ISREs in the promoters of a subset of ISGs, many of which represent pro-inflammatory cytokines and chemokines.
Sepsis and its more severe form, septic shock, occur when a normal immune response to a localized bacterial infection becomes systemic and uncontrolled (14, 15). Continuous activation of the inflammatory system triggers both coagulation and the complement system, creating a positive feedback loop that leads to increased vascular permeability and disseminated intravascular coagulation (DIC), causing extensive tissue damage and ultimately multiorgan failure. Cell wall components of both gram-positive and gram-negative bacteria act as triggers of the sepsis shock syndrome. However, despite the prevalent occurrences of sepsis (approximately 100,000 lethalities in the United States each year), the intracellular signaling events contributing to the development of sepsis are still unclear.
Recently, Karaghiosoff et al. reported that Tyk2-deficient mice fail to up-regulate IFNα4 and IFNβ in response to LPS challenge and display reduced susceptibility to septic shock (16). Because IRF3 plays a predominant role in the LPS induction of these interferons (17), we hypothesized that IRF3 activation could contribute to the development or progression of septic shock and that abrogation of LPS-induced IRF3 activation might exert a protective effect.
We had previously characterized piceatannol (trans-3,3′,4,5′-tetrahydroxystilbene) as an inhibitor of Tyk2 (18). Piceatannol is a stilbene derivative found in numerous plants such as Rheum undulatum (Korean rhubarb) or red grapes, where it functions as a phytoalexin to protect the plant against fungal infection (19, 20). In mammalian systems, the compound exerts antioxidative as well as anti-inflammatory effects that interfere with cytokine production and function, although the molecular targets remain elusive.
Here we show that piceatannol inhibits IRF3 activation in response to LPS, prevents the production of proinflammatory cytokines and chemokines, and protects animals from septic shock after LPS challenge.
EXPERIMENTAL PROCEDURES
Cells
Human U373 astrocytoma cells (U-373 MG) stably transfected with human CD14 were generously provided by Dr. Peter Tobias, and murine monocytic cell line, RAW 264.7 (ATCC) were maintained in Dulbecco’s Modified Media supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin (Irvine Scientific). Murine peritoneal macrophages were collected 5 days after intraperitoneal injection of thioglycolate.
Reagents
Piceatannol was purchased from A.G. Scientific. Tetramethoxystilbene (TMS) was manufactured custom-synthesized by Syngene, Inc. (India).
Electrophoretic mobility shift assay
Electrophoretic mobility shift assays (EMSAs) were performed using a 32P-end-labeled probes corresponding to the IFN-stimulated response element (ISRE) of the ISG15 promoter (5′-GATCCATGCCTCGGGAAAGGGAAACCGAAACTGAAGCC-3′). Equal amounts of protein were incubated with labeled oligonucleotides in binding buffer (40 mM KCl, 20 mM Hepes, pH 7.0, 1 mM MgCl2, 0.1 mM EGTA, 0.5 mM DTT, 4% Ficoll, 0.02% Nonidet P-40). Supershifts for IRF3 were done using anti-IRF3 antiserum described previously (9). Electrophoresis was performed on 6% nondenaturing TBE PAGE, and the gels were dried and subjected to autoradiography.
RNase protection assays
Total RNA was isolated using TRIzol Reagent (InVitrogen). 32P-Labeled anti-sense riboprobes for ISG54, ICAM-1, MCP-1, and GAPDH were generated by in vitro transcription of the linearized plasmid using T7 or SP6 RNA polymerase (Promega). Labeled riboprobe and 10 μg of RNA were incubated in hybridization buffer (4:1 formamide and 5× stock; 5× stock was 200 mM PIPES, pH 6.4, 2 M NaCl, 5 mM EDTA) overnight at 56°C before digestion with T1 RNase (Ambion) for 1 h at 37°C. After phenol extraction and ethanol precipitation, protected fragments were solubilized in RNA loading buffer (98% formamide, 10 mM EDTA, pH 8), boiled for 2 min, and subjected to electrophoresis on a 4.5% polyacrylamide/urea gel.
RT-PCR
cDNA was prepared from total RNA by reverse transcription using InVitrogen’s SuperScript First-Strand Synthesis System. cDNAs for RANTES, TNF-α, TF, and β-actin were amplified by using the Taq PCR Core Kit (Qiagen) with primers (5′-GCTGTCATCCTCATTGCTAC-3′) and (5′-TCTCCATCCTAGCTCATCTC-3′) for RANTES, (5′-AGCCTCTTCTCCTTCCTGATCG-3′) and (5′-TATCTCTCAGCTCCACGCC ATT-3′) for TNF-α, (5′-CGGGTGCAGGCATTCCAGAG-3′) and (5′-CAGGAGAGACAGGG TGCCTC-3′) for TF, and (5′-AAGAGAGGCATCCTCACCCT-3′) and (5′-TACATGGCTGG GGTGTTGAA-3′) for β-actin.
Enzyme-linked immunosorbent assay (ELISA)
IL-6 levels were measured using supernatants derived from RAW cells cultured under the indicated conditions. ELISA was performed according to the manufacturer protocol (eBioscience).
LPS-induced septic shock
NIH Swiss mice (Charles River Laboratories) were injected intraperitoneally (i.p.) with 33 μL solvent control (DMSO), or 1200 μg piceatannol or TMS 1 h before administration of 1000 μg LPS in 200 μL PBS. Subsequently, mice were given either 600 μg piceatannol or TMS, respectively, or solvent only subcutaneously every 12 h for 3 days following the initial injection. Surviving mice were monitored for 30 days after LPS administration. The animal experiments were performed in accordance with the National Institutes of Health guidelines for the care and handling of laboratory animals. All animal experiments were performed in accordance with UCSD Animal Subjects Program regulations. Statistical analysis (Dunnett’s test) was performed using SigmaStat.
Histologic analysis
Livers and spleens obtained from moribund mice 36 h after LPS administration were fixed in 10% formalin. Tissues were embedded in paraffin, sectioned, stained with hematoxylin and eosin (H&E), and analyzed under blinded conditions by the UCSD Pathology Core Facility.
RESULTS
We had previously reported that the p38-mediated activation of IRF3 is necessary for LPS to induce ISGs such as ISG54 or RANTES in a protein synthesis-independent manner (12). In addition, the virus- or LPS-mediated induction of several type I interferon genes depends on IRF3 (17). Therefore, in light of the observation that Tyk2 deficiency results in a failure to up-regulate IFNα4 and IFNβ in response to LPS (16), we decided to determine whether piceatannol, which we had previously identified as a Tyk2 kinase inhibitor (18), would exert an inhibitory effect on the signaling cascade leading to IRF3 activation and ISG induction. As shown in Figure 1A, pretreatment of cells with piceatannol resulted in a dramatic inhibition of LPS-induced RANTES expression. The promoter of RANTES contains an ISRE as well as an NF-κB site; however, although IRF3 binding to the ISRE is absolutely crucial for gene expression, engagement of the NF-κB site only increases the level of induction (21). ISG induction by LPS is clearly mediated by IRF3 rather than by STAT1 activated through an IFNα/β loop, as illustrated by an unimpaired LPS-induced RANTES expression in STAT1-deficient macrophages (Fig. 1B).
Fig. 1. Piceatannol inhibits LPS-induced ISG expression.
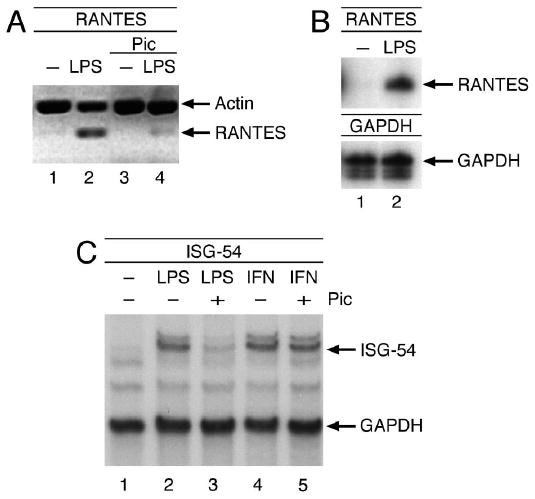
(A) U373 cells were treated with 100 μM piceatannol or solvent in the presence of 50 μg/mL cycloheximide before incubation with 1 μg/mL LPS for 6 h. (B) Similarly, STAT1-deficient peritoneal macrophages were stimulated with 1 μg/mL LPS for 6 h. RANTES mRNA levels were analyzed by RT-PCR or RNase protection assay, respectively. (C) Same as A, except cells treated with 1000 U/mL IFNβ for 6 h were included. Total RNA was prepared and analyzed for ISG54 mRNA levels by RNase protection assay.
To ensure that piceatannol was targeting IRF3 activation specifically, we also analyzed its effects on ISG54 expression (Fig. 1C) because the only enhancer element present in the promoter of ISG54 is an ISRE, making this gene solely dependent on IRF3 for its transcriptional activation by LPS. As anticipated, LPS-mediated ISG54 induction was abrogated as a result of piceatannol treatment. Importantly, ISG54 induction in response to IFN β, which is elicited through STAT1/2-heterodimer binding to the ISRE, is unaffected by the presence of piceatannol. Thus, the inhibition of ISG-induction by piceatannol is restricted to the IRF3 pathway and does not extend to STAT-regulated expression of these early response genes.
Transactivation of ISGs in response to LPS requires the ISRE binding of IRF3 as well as interaction of IRF3 with the transcriptional coactivator CBP/p300 (12). To determine whether piceatannol prevented IRF3 from binding DNA, we stimulated cells with LPS in the presence and absence of piceatannol and performed EMSAs using the ISG15-ISRE as a probe. Indeed, although LPS treatment alone clearly prompted IRF3 to bind DNA, the addition of piceatannol before LPS exposure reduced this response in a dose-dependent manner (Fig. 2). The identity of the band was confirmed to contain IRF3 by supershifting the band with anti-IRF3 antibodies (data not shown). These results demonstrate that piceatannol prevents ISG induction by LPS by exerting an inhibitory effect on the signaling cascade leading to IRF3 binding to the ISRE.
Fig. 2. Inhibition of IRF3 DNA binding by piceatannol.
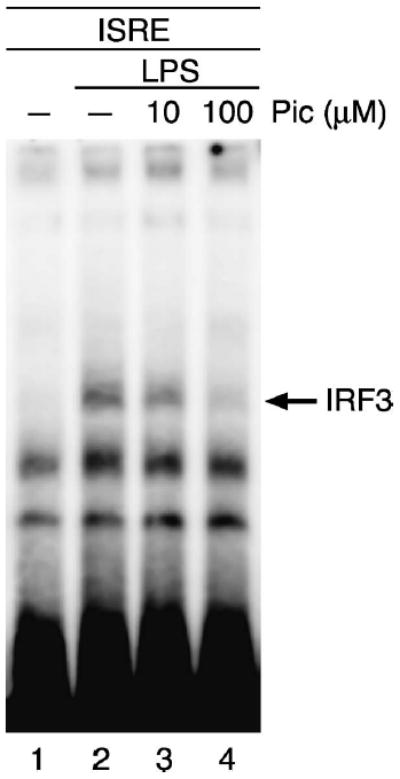
U373 cells were treated with 10 μM or 100 μM piceatannol as described in Figure 1. Whole-cell lysates were prepared, and IRF3 binding analyzed by EMSA using the ISG15-ISRE as a probe.
LPS exposure of cells leads to the transcriptional activation of a myriad of immediate early response genes, many of which contribute to the nonspecific inflammatory reaction that represents the innate immune response (22, 23). We were particularly interested in LPS-induced cytokines and chemokines, which have been implicated in the pathologic processes of sepsis and septic shock.
Increased serum concentrations of IL-6 are a hallmark of septic events, and the correlation between IL-6 levels and the rate of sepsis-associated morbidity make this cytokine the most promising prognostic marker to date (24). To test the effect of piceatannol on LPS-induced IL-6 production, we exposed peritoneal macrophages for 3, 6, or 12 h to LPS and determined the IL-6 concentration in the supernatants by ELISA. As shown in Figure 3, piceatannol was indeed able to prevent of IL-6 up-regulation in response to LPS stimulation.
Fig. 3. Piceatannol inhibits LPS-induced IL-6 up-regulation.
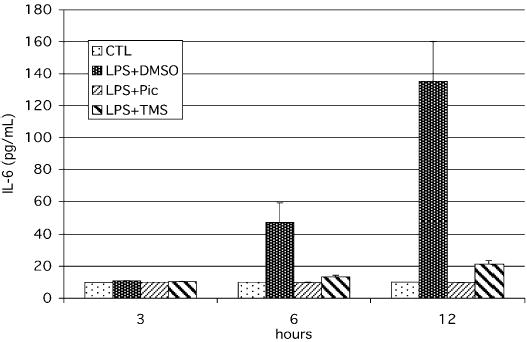
RAW macrophages were treated with 100 μM piceatannol, 100 μM TMS, or solvent (DMSO) for 1 h before stimulation with 1 μg/mL LPS. Aliquots of the supernatants were removed from the wells at the indicated time points, and IL-6 levels were analyzed by ELISA.
Piceatannol possesses antioxidative properties because of its hydroxylated phenol structure (19). Antioxidants had previously been shown to exert a protective effect against septic shock in various animal model (25), so we decided to investigate whether the inhibitory effects of piceatannol required the presence of the phenolic hydroxyl groups. A methylated form of piceatannol, trans-3,3′,4,5′-tetramethoxystilbene (TMS), in which the four hydroxyl groups were replaced by methoxy groups, was still able to inhibit LPS-induced IL-6 up-regulation, illustrating that the antioxidative properties of piceatannol are dispensable for its ability to inhibit LPS-induced gene expression and that its capability to inhibit IRF3 activation might be related to its role as a tyrosine kinase inhibitor.
In addition to IL-6, LPS activation of macrophages also leads to the induction of other proinflammatory cytokines such as TNF-α, which is one of the first cytokines to be expressed after contact of cells with endotoxin. Because TNF-α is a major contributor to the development of sepsis (24), we examined the effects of piceatannol on the LPS-mediated induction of TNF-α. As shown in Figure 4A, piceatannol, and to a lesser extent TMS, significantly inhibited the LPS-induced stimulation of TNF-α.
Fig. 4. Inhibition of LPS-induced TNF-α, ICAM-1, and MCP-1 production by piceatannol.
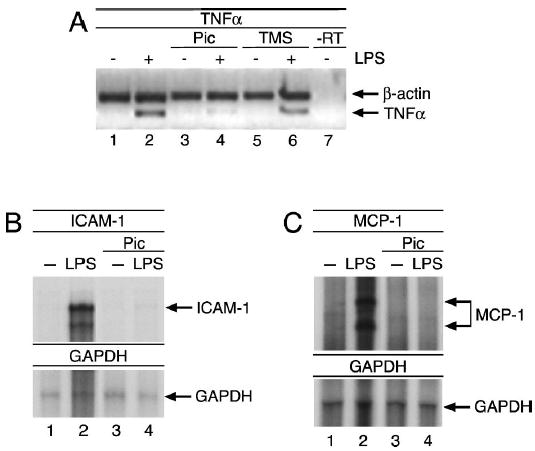
(A) U373 cells were treated as in Figure 3, and TNF-α mRNA levels were analyzed by RT-PCR. (Band C) RAW cells were treated with 100 μM piceatannol or solvent in the presence of 50 μg/mL cycloheximide for 1 h before stimulation with 1 μg/mL LPS for 6 h. Total RNA was prepared, and ICAM-1 (B) and MCP-1 (C) mRNA levels were determined by RNase protection assay. GAPDH was used as an internal control.
Besides the induction of proinflammatory cytokines, LPS activation of macrophages also triggers the release of a variety of other proinflammatory mediators that attract leukocytes to the site of infection (14, 24). For example, macrophage chemoattractant protein 1 (MCP-1) and intracellular adhesion molecule 1 (ICAM-1) are among the LPS-induced gene products that promote leukocyte infiltration into the infected tissue (26). In the course of progression to sepsis, these factors contribute to the continuous and autoamplifying immune response that ultimately leads to multiorgan failure. Because both MCP-1 and ICAM-1 harbor an ISRE in their respective promoters and are therefore dependent on IRF3 for their induction in response to LPS, we tested piceatannol’s abilities to prevent their transcriptional up-regulation after LPS stimulation. Similar to its inhibitory effects on IL-6 and TNF-α induction, piceatannol was also effective in blocking LPS-induced MCP-1 and ICAM-1 expression (Fig. 4, B and C).
Sepsis represents a combination of excessive inflammatory response and inappropriate coagulation reaction (27). Tissue factor (TF) is a key mediator between the immune system and coagulation (28) and is the principal trigger of coagulation by forming the activating VIIa-TF complex. Subsequent amplification of coagulation involves processing of prothrombin to thrombin and the generation of fibrin. The consequent deposition of fibrin clots in the microvasculature hinders tissue oxygenation and causes multiorgan failure. As anticipated, administration of either piceatannol or TMS was able to inhibit the LPS-induced expression of TF (Fig. 5), suggesting that these stilbene derivatives might be able to inhibit the coagulation cascade in vivo.
Fig. 5. Piceatannol inhibits LPS induction of tissue factor.
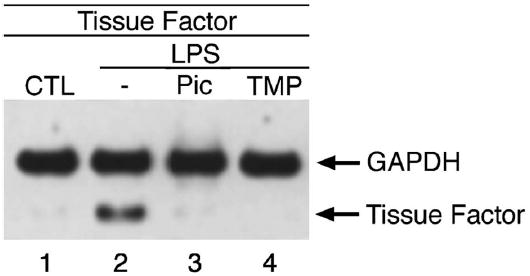
RAW cells were treated with 100 μM piceatannol or 100 μM TMS in the presence of 50 μg/mL cyclohexamide for 1 h before incubation with 1 μg/mL LPS for an additional 6 h. Total RNA was isolated and analyzed by RT-PCR for tissue factor and GAPDH expression levels.
Based on the ability of piceatannol or TMS to abrogate LPS-induced gene transcription, we reasoned that these stilbenes might exert a protective affect against endotoxic shock. Intraperitoneal injection of 1000 μg LPS into NIH Swiss mice results in a >70% mortality after 3 days (Fig. 6A) However, when piceatannol or TMS was administered before the LPS challenge, the mortality associated with septic shock syndrome was reduced to < 35% (Dunnett’s test: P < 0.05). Importantly, TMS displayed the same efficacy as piceatannol, thereby supporting the notion that the protective effect is independent of the antioxidative properties of piceatannol.
Fig. 6. Piceatannol prevents LPS-induced septic shock and tissue damage.
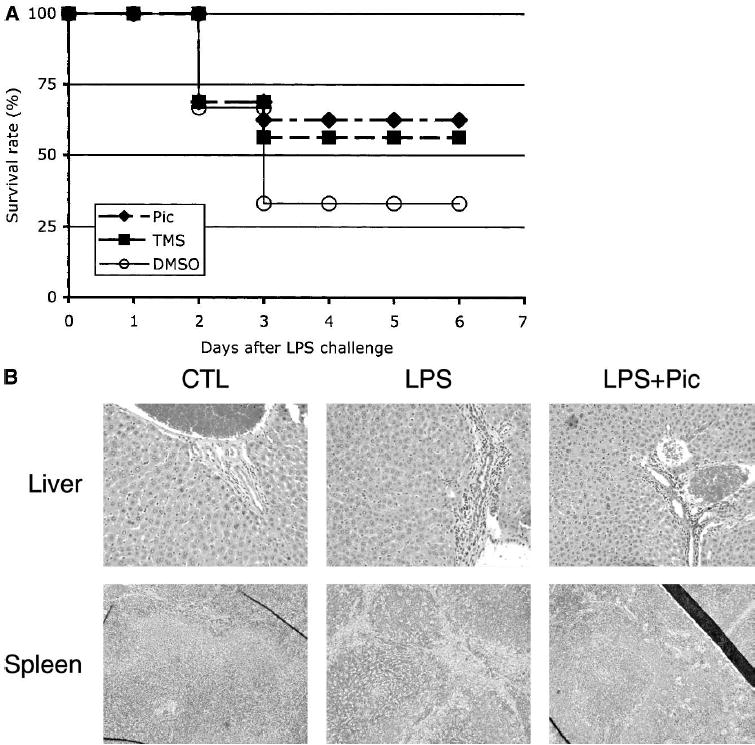
(A) Mice (16 animals/treatment group) were injected i.p. with Piceatannol, TMS, or DMSO (as solvent control) 1 h before challenge with 1000 μg LPS as described under Materials and Methods. (B) Mice were treated as described in A, and livers and spleens were sectioned and stained with H&E. Liver sections are shown at 200× magnification, and spleen sections at 100× magnification.
The liver and the spleen are the first organs to display signs of inflammation after LPS administration, as evidenced by the migration of granulocytes and leukocytes from the blood into the connective tissues. Therefore, we evaluated the pathohistologic consequences of 36 h LPS challenge without or with concomitant piceatannol administration on these organs (Fig. 6B). Tissue sections of livers derived from LPS-challenged animals showed a clear infiltration of leukocytes into the parenchyma at the portal triads. Similarly, analysis of the spleen sections revealed the presence of severe inflammation as documented by the “starry sky” appearance that is indicative of reactive follicles. In contrast, sections obtained from the organs of animals that had received piceatannol before the LPS challenge showed greatly reduced symptoms of inflammation. Not only was a diminished number of infiltrating leukocytes visible at the portal areas, but importantly, these leukocytes were retained in the collagen layer and did not enter the parenchyma. Likewise, the spleens of piceatannol- and LPS-treated animals displayed clearly organized follicles resembling those found in the spleens of the untreated animals.
We also examined the livers and spleens of animals that had received TMS instead of piceatannol. In accordance with our previous findings on the effectiveness of TMS to reproduce the in vitro effects of piceatannol, and the observed ability of TMS to prevent endotoxic shock, the compound proved as effective as piceatannol in inhibiting leukocyte infiltration and tissue inflammation (data not shown). These data provide further evidence that the inhibition of the in vivo inflammatory response by piceatannol and TMS provides a protective effect against endotoxic shock-associated multiorgan injury.
DISCUSSION
Activation of IRF3 is an important step in the cellular immune response and is essential for the cytokine and chemokine production of myeloid cells after they encounter viral or bacterial pathogens (29, 30). Although the production of these circulating inflammatory mediators is a critical element of the innate immune response, their uncontrolled and excessive generation leads to severe pathologic processes such as disseminated intravascular coagulation (DIC) and septic shock.
Numerous attempts to intercept the development and progression of septic shock were aimed at individual mediators of the inflammation or coagulation system (25, 27). However, because the occurrence of septic shock syndrome is based on a highly complex network of often functionally redundant pathways (14), the pharmacologic intervention at the level of an individual component failed to provide a satisfying therapeutic strategy (31).
In this report we demonstrate that piceatannol and TMS inhibit LPS-induced activation of IRF3 and consequently abrogate the induction of its downstream target genes. Indeed, piceatannol was able to preclude the IRF3-mediated increase in the expression levels of several prominent LPS-induced genes that play a crucial role in the development and progression of septic shock syndrome. Intriguingly, during the preparation of this manuscript, Sakaguchi et al. reported a significantly increased resistance to LPS-induced endotoxic shock in IRF3-deficient mice (17).
The ability of piceatannol and related stilbene derivatives to confer protection against septic shock in a widely used animal model is likely related to their ability to target multiple elements in the inflammatory response as well as the coagulation cascade that are under the control of IRF3. Importantly, stilbene derivatives are found in many plants that have been subject to human consumption for hundreds of years, suggesting a low toxicity of these compounds (19, 32, 33). Indeed, when mice were given 1000 μg piceatannol per diem for 30 days in pilot studies, no adverse physiologic or histologic side effects could be detected.
In summary, our results support the concept that the pharmacologic modulation of IRF3 activation by small molecule inhibitors such as stilbene derivatives offers a novel and promising target for the development of new therapeutic strategies directed against endotoxic shock syndrome.
Acknowledgments
The authors would like to thank Dr. Nissi Varki for assistance in the analysis and interpretation of the histologic material and Syngen Inc. (India) for providing TMS. The authors are also grateful to Drs. Dennis Otero and Robert Rickert for technical assistance and helpful discussions.
References
- 1.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 2.Shuai K, Ziemiecki A, Wilks AF, Harpur AG, Sadowski HB, Gilman MZ, Darnell JEJ. Polypeptide signalling to the nucleus through tyrosine phosphorylation of JAK and Stat proteins. Nature. 1993;366:580–583. doi: 10.1038/366580a0. [DOI] [PubMed] [Google Scholar]
- 3.Imada K, Leonard WJ. The Jak-STAT pathway. Mol Immunol. 2000;37:1–11. doi: 10.1016/s0161-5890(00)00018-3. [DOI] [PubMed] [Google Scholar]
- 4.Schindler C, Darnell JE., Jr Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu Rev Biochem. 1995;64:621–651. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 5.David M. Signal transduction by type I interferons. Biotechniques. 2002;33(Suppl) [PubMed] [Google Scholar]
- 6.Miyamoto M, Fujita T, Kimura Y, Maruyama M, Harada H, Sudo Y, Miyata T, Taniguchi T. Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements. Cell. 1988;54:903–913. doi: 10.1016/s0092-8674(88)91307-4. [DOI] [PubMed] [Google Scholar]
- 7.Mamane Y, Heylbroeck C, Genin P, Algarte M, Servant MJ, LePage C, DeLuca C, Kwon H, Lin R, Hiscott J. Interferon regulatory factors: the next generation. Gene. 1999;237:1–14. doi: 10.1016/s0378-1119(99)00262-0. [DOI] [PubMed] [Google Scholar]
- 8.Taniguchi T. IRF-1 and IRF-2 as regulators of the interferon system and cell growth. Indian J Biochem Biophys. 1995;32:235–239. [PubMed] [Google Scholar]
- 9.Navarro L, Mowen K, Rodems S, Weaver B, Reich N, Spector D, David M. Cytomegalovirus activates interferon immediate-early response gene expression and an interferon regulatory factor 3-containing interferon-stimulated response element-binding complex. Mol Cell Biol. 1998;18:3796–3802. doi: 10.1128/mcb.18.7.3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol. 1998;18:2986–2996. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Navarro L, David M. p38-dependent activation of interferon regulatory factor 3 by lipopolysaccharide. J Biol Chem. 1999;274:35535–35538. doi: 10.1074/jbc.274.50.35535. [DOI] [PubMed] [Google Scholar]
- 13.Lin R, Mamane Y, Hiscott J. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol. 1999;19:2465–2474. doi: 10.1128/mcb.19.4.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 15.Shelley O, Murphy T, Paterson H, Mannick JA, Lederer JA. Interaction between the innate and adaptive immune systems is required to survive sepsis and control inflammation after injury. Shock. 2003;20:123–129. doi: 10.1097/01.shk.0000079426.52617.00. [DOI] [PubMed] [Google Scholar]
- 16.Karaghiosoff M, Steinborn R, Kovarik P, Kriegshauser G, Baccarini M, Donabauer B, Reichart U, Kolbe T, Bogdan C, Leanderson T, Levy D, Decker T, Muller M. Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nature Immunol. 2003;4:471–477. doi: 10.1038/ni910. [DOI] [PubMed] [Google Scholar]
- 17.Sakaguchi S, Negishi H, Asagiri M, Nakajima C, Mizutani T, Takaoka A, Honda K, Taniguchi T. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem Biophys Res Commun. 2003;306:860–866. doi: 10.1016/s0006-291x(03)01049-0. [DOI] [PubMed] [Google Scholar]
- 18.Su L, David M. Distinct mechanisms of STAT phosphorylation via the interferon-alpha/beta receptor. Selective inhibition of STAT3 and STAT5 by piceatannol. J Biol Chem. 2000;275:12661–12666. doi: 10.1074/jbc.275.17.12661. [DOI] [PubMed] [Google Scholar]
- 19.Waffo Teguo P, Fauconneau B, Deffieux G, Huguet F, Vercauteren J, Merillon JM. Isolation, identification, and antioxidant activity of three stilbene glucosides newly extracted from Vitis vinifera cell cultures. J Nat Prod. 1998;61:655–657. doi: 10.1021/np9704819. [DOI] [PubMed] [Google Scholar]
- 20.Kageura T, Matsuda H, Morikawa T, Toguchida I, Harima S, Oda M, Yoshikawa M. Inhibitors from rhubarb on lipopolysaccharide-induced nitric oxide production in macrophages: structural requirements of stilbenes for the activity. Bioorg Med Chem. 2001;9:1887–1893. doi: 10.1016/s0968-0896(01)00093-1. [DOI] [PubMed] [Google Scholar]
- 21.Lin R, Heylbroeck C, Genin P, Pitha PM, Hiscott J. Essential role of Interferon Regulatory Factor 3 in Direct Activation of RANTES Chemokine Transcription. MolCell Biol. 1999;19:959–966. doi: 10.1128/mcb.19.2.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol. 2002;76:5532–5539. doi: 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lang CH, Silvis C, Deshpande N, Nystrom G, Frost RA. Endotoxin stimulates in vivo expression of inflammatory cytokines tumor necrosis factor alpha, interleukin-1beta, -6, and high-mobility-group protein-1 in skeletal muscle. Shock. 2003;19:538–546. doi: 10.1097/01.shk.0000055237.25446.80. [DOI] [PubMed] [Google Scholar]
- 24.Hack CE, Aarden LA, Thijs LG. Role of cytokines in sepsis. Adv Immunol. 1997;66:101–195. doi: 10.1016/s0065-2776(08)60597-0. [DOI] [PubMed] [Google Scholar]
- 25.Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nature Med. 2003;9:517–524. doi: 10.1038/nm0503-517. [DOI] [PubMed] [Google Scholar]
- 26.Yan W, Zhao K, Jiang Y, Huang Q, Wang J, Kan W, Wang S. Role of p38 MAPK in ICAM-1 expression of vascular endothelial cells induced by lipopolysaccharide. Shock. 2002;17:433–438. doi: 10.1097/00024382-200205000-00016. [DOI] [PubMed] [Google Scholar]
- 27.Freeman BD, Zehnbauer BA, Buchman TG. A meta-analysis of controlled trials of anticoagulant therapies in patients with sepsis. Shock. 2003;20:5–9. doi: 10.1097/01.shk.0000068327.26733.10. [DOI] [PubMed] [Google Scholar]
- 28.Esmon C. The protein C pathway. Crit Care Med. 2000;28:S44–S48. doi: 10.1097/00003246-200009001-00010. [DOI] [PubMed] [Google Scholar]
- 29.Hiscott J, Pitha P, Genin P, Nguyen H, Heylbroeck C, Mamane Y, Algarte M, Lin R. Triggering the interferon response: The role of IRF3 Transcription Factor. J Int Cytol Res. 1999;19:1–13. doi: 10.1089/107999099314360. [DOI] [PubMed] [Google Scholar]
- 30.Doyle S, Vaidya S, O’Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 31.Opal SM. Clinical trial design and outcomes in patients with severe sepsis. Shock. 2003;20:295–302. doi: 10.1097/01.shk.0000084343.58020.57. [DOI] [PubMed] [Google Scholar]
- 32.Ko SK, Lee SM, Whang WK. Anti-platelet aggregation activity of stilbene derivatives from Rheum undulatum. Arch Pharm Res. 1999;22:401–403. doi: 10.1007/BF02979065. [DOI] [PubMed] [Google Scholar]
- 33.Palmieri L, Mameli M, Ronca G. Effect of resveratrol and some other natural compounds on tyrosine kinase activity and on cytolysis. Drugs Exp Clin Res. 1999;25:79–85. [PubMed] [Google Scholar]