

Abstract
The human type 1 (placenta, breast tumors, prostate tumors) and type 2 (adrenals, gonads) isoforms of 3β-hydroxysteroid dehydrogenase/isomerase (3β-HSD1 and 3β-HSD2) are encoded by two distinct genes that are expressed in a tissue-specific pattern. Our recent studies have shown that His156 contributes to the 14-fold higher affinity that 3β-HSD1 exhibits for substrate and inhibitor steroids compared to human 3β-HSD2 containing Tyr156 in the otherwise identical catalytic domain. Our structural model of human 3β-HSD localizes His156 or Tyr156 in the subunit interface of the enzyme homodimer. The model predicts that Gln105 on one enzyme subunit has a higher probability of interacting with His156 on the other subunit in 3β-HSD1 than with Tyr156 in 3β-HSD2. The Q105M mutant of 3β-HSD1 (Q105M1) shifts the Michaelis-Menten constant (Km) for 3β-HSD substrate and inhibition constants (Ki) for epostane and trilostane to the much lower affinity profiles measured for wild-type 3β-HSD2 and H156Y1. However, the Q105M2 mutant retains substrate and inhibitor kinetic profiles similar to those of 3β-HSD2. Our model also predicts that Gln240 in 3β-HSD1 and Arg240 in 3β-HSD2 may be responsible for the 3-fold higher affinity of the type 1 isomerase activity for substrate steroid and cofactors. The Q240R1 mutation increases the isomerase substrate Km by 2.2-fold to a value similar to that of 3β-HSD2 isomerase and abolishes the allosteric activation of isomerase by NADH. The R240Q2 mutation converts the isomerase substrate, cofactor and inhibitor kinetic profiles to the 4- to 14-fold higher affinity profiles of 3β-HSD1. Thus, key structural reasons for the substantially higher affinities of 3β-HSD1 for substrates, coenzymes and inhibitors have been identified. These structure and function relationships can be used in future docking studies to design better inhibitors of the 3β-HSD1 that may be useful in the treatment hormone-sensitive cancers and preterm labor.
The human type 1 (placenta, mammary gland, prostate) and type 2 (adrenals, ovary, testis) isoforms of 3ß-hydroxysteroid dehydrogenase (EC 1.1.1.145)/steroid Δ5 -Δ4-isomerase (EC 5.3.3.1) (3β-HSD11 and 3β-HSD2) are encoded by two distinct genes which are expressed in a tissue-specific pattern (1). As shown in Figure 1, human 3β-HSD1 catalyzes the conversion of 3ß-hydroxy-5-ene-steroids (dehydroepiandrosterone or DHEA, pregnenolone) to 3-oxo-4-ene-steroids (androstenedione, progesterone), and human 3β-HSD2 converts 17α-hydroxypregnenolone and pregnenolone to ultimately produce cortisol and aldosterone in the human adrenal, respectively (2). 17α-Hydroxylase/17–20 lyase (CYP17) in the human adrenal gland converts pregnenolone to DHEA, which is the major circulating steroid in humans as DHEA-sulfate (2). In placenta, androstenedione is converted by aromatase and 17β-hydroxysteroid dehydrogenase (17β-HSD) to estradiol, which participates in the cascade of events that initiates labor in humans (2,3). Placental 3β-HSD1 also converts pregnenolone to progesterone to help maintain the uterus in a quiescent state throughout human pregnancy (3). In addition to placenta and other human peripheral tissues, the 3β-HSD1 is selectively expressed in breast tumors (4) and prostate tumors (5,6), where it catalyzes the first step in the conversion of circulating DHEA to estradiol or testosterone to promote tumor growth. Determination of the structure/function relationships of human 3β-HSD1 and 3β-HSD2 may lead to the development of highly specific inhibitors of 3β-HSD1 that can help control the timing of labor and slow the growth of hormone-sensitive tumors without inhibiting 3β-HSD2, so that steroidogenesis in the adrenal gland to produce cortisol and aldosterone is not compromised.
Figure 1.

Human 3β-hydroxysteroid dehydrogenase is expressed as two-tissue specific isoforms (3β-HSD1 and 3β-HSD2) as a key, rate-limiting enzyme in the steroid biosynthetic pathways that produce estradiol, testosterone, cortisol and aldosterone.
The two-step reaction of 3β-HSD/isomerase using dehydroepiandrosterone (DHEA) as substrate is shown in Figure 2. This reaction scheme shows the reduction of NAD+ to NADH by the rate-limiting 3β-HSD activity and the requirement of this NADH for the activation of isomerase on the same enzyme protein. Because the isomerase reaction is irreversible, the 3β-HSD/isomerase cannot convert androstenedione to DHEA (7,8). According to our stopped-flow fluorescence spectroscopy study, NADH induces a time-dependent conformational change in the enzyme structure as the isomerase activity reaches a maximum over 1 minute after the addition of the coenzyme (9). The intermediate steroid, 5-androstene-3,17-dione, remains bound during the reaction sequence (7,9). Our homology model (10) of human 3β-HSD structure (Figure 3) predicts that a difference in the amino acid sequence at position 240 (Gln in 3β-HSD1 and Arg in 3β-HSD2) may be responsible for the 3-fold higher affinities of 3β-HSD1 for isomerase substrate, NAD+ and NADH compared to 3β-HSD2. The current study tests this prediction using site-directed mutagenesis.
Figure 2.

3β-HSD/isomerase catalyzes two sequential reactions on a single enzyme protein. The human 3β-HSD and isomerase activities are represented using dehydroepiandrosterone (DHEA) as substrate.
Figure 3.

(A) Homology model of the catalytic domain of human 3β-HSD1 showing Gln240, NAD+, DHEA and the catalytic residues, Tyr154 and Lys158. (B) Homology model of the catalytic domain of human 3β-HSD2 showing Arg240+, NAD+, DHEA and the catalytic residues, Tyr154 and Lys158.
In addition, we recently reported (11) that His156 in 3β-HSD1 is responsible for the 14-fold higher affinities that 3β-HSD1 exhibits for substrate (DHEA) and inhibitor (epostane) steroids compared to 3β-HSD2 with Tyr156 in the otherwise identical catalytic domains (Tyr154-Pro-His156/Tyr156-Ser-Lys158). Because our structural model localizes His156/Tyr156 in the subunit interface (Figure 4), the structural basis for the differences in 3β-HSD1 and 3β-HSD2 are investigated in this report using site-directed mutagenesis to determine if subunit interactions between Gln105 and His156 or Tyr156 are involved.
Figure 4.
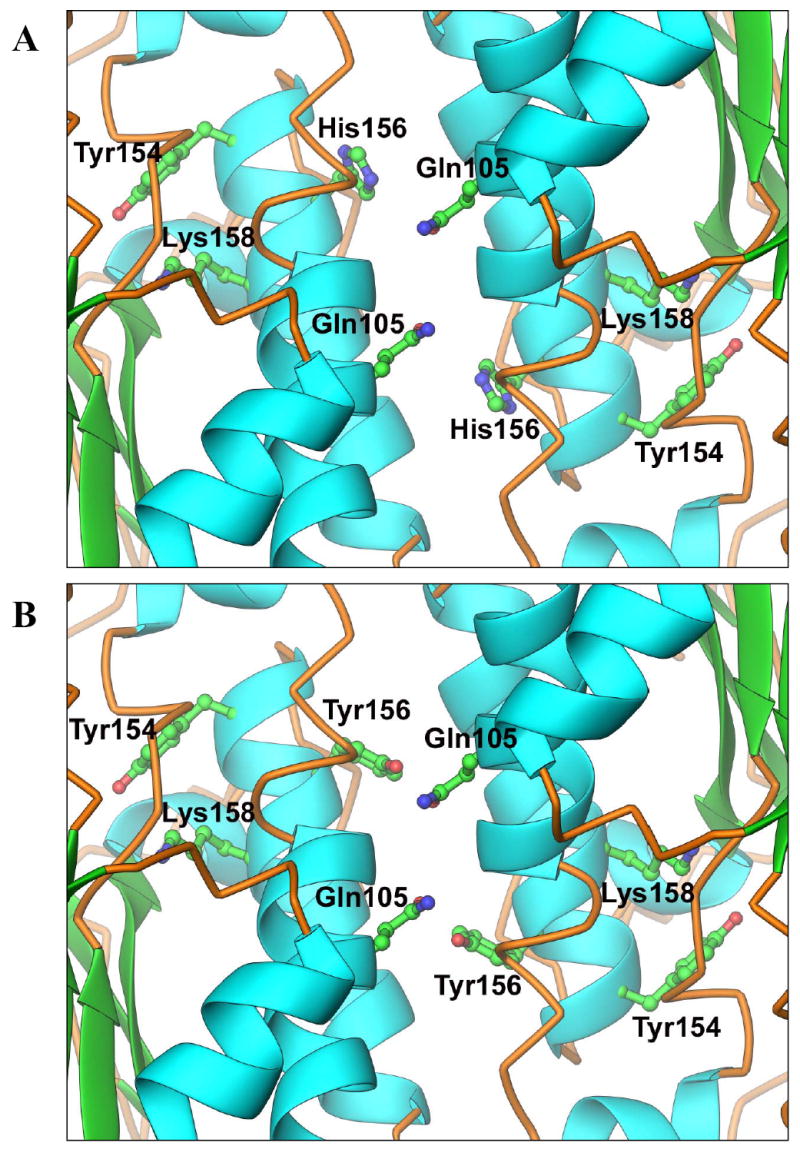
(A) Homology model of the human 3β-HSD1 dimer interface showing the critical hydrogen-binding residues, Gln105 and His156, as well as the catalytic residues, Tyr154 and Lys158. (B) Homology model of the human 3β-HSD2 dimer interface in which Tyr156 replaces His156.
Experimental Procedures
Materials
Dehydroepiandrosterone and pyridine nucleotides were purchased from Sigma Chemical Co. (St. Louis, MO); 5-androstene-3,17-dione from Steraloids Inc. (Newport, RI); reagent grade salts, chemicals and analytical grade solvents from Fisher Scientific Co. (Pittsburgh, PA). Glass distilled, deionized water was used for all aqueous solutions.
Site-directed mutagenesis
Using the Advantage cDNA PCR kit (BD Biosciences Clontech, Palo Alto, CA) and pGEM-3βHSD1 or pGEM-3βHSD2 as template (12), double-stranded PCR-based mutagenesis was performed with the primers in Table 1 to create the cDNA encoding the Q105M1, Q105M2, Q240R1 and R240Q2 mutant enzymes. The presence of the mutated codon and integrity of the entire mutant 3β-HSD cDNA were verified by automated dideoxynucleotide DNA sequencing using the Big Dye Terminator Cycle Sequencing Ready Reaction kit (PE Applied Biosystems, Foster City, CA). Chou-Fasman and Garnier-Osguthorpe-Robson analysis of each mutant enzyme was used to choose amino acid substitutions that produced no apparent changes in the secondary structure of the protein (Protylze program, Scientific and Educational Software, State Line, PA)
Table 1.
Oligonucleotide primers used for site-directed mutagenesis.
| Mutation | Direction | Nucleotide sequence of primer1 |
|---|---|---|
| Q105M1 | Forward | 5′-GGTACCATGCTCCTGTTAGAGGC –3′ |
| Reverse | 5′-CAGGAGCATGGTACCTTTCACATTG-3′ | |
| Q105M2 | Forward | 5′-AAGGTACCATGCTACTGTTGGAGGC-3′ |
| Reverse | 5′-ACAGTAGCATGGTACCTTTCACATTG-3′ | |
| Q240R1 | Forward | 5′-GCCCTGCGGGACCCCAAGAAGGC -3′ |
| Reverse | 5′-GGTCCCGCAGGGCCCTCAAGGC -3′ | |
| R240Q2 | Forward | 5′-GCTCTGCAGGACCCCAAGAAGGC -3′ |
| Reverse | 5′-GGTCCTGCAGAGCCCTCAAGGC -3′ |
The mutated codons are in the bold, italic font.
Expression and purification of the mutant and wild-type enzymes
The mutant 3ß-HSD cDNA was introduced into baculovirus as previously described (12). Recombinant baculovirus was added to 1.5 x 109 Sf9 cells (1L) at a multiplicity of infection of 10 for expression of each mutant enzyme. The expressed mutant and wild-type enzymes were separated by SDS-polyacrylamide (12%) gel electrophoresis, probed with our anti-3β-HSD polyclonal antibody and detected using the ECL western blotting system with antirabbit, peroxidase-linked secondary antibody (Amersham Pharmacia Biotech, Piscataway, NJ). Each expressed enzyme was purified from the 100,000 g pellet of the Sf9 cells (2 L) by our published method (7,8) using Igepal CO 720 (Rhodia, Inc., Cranbury, NJ) instead of the discontinued Emulgen 913 detergent (Kao Corp, Tokyo). Each expressed, purified mutant and wild-type enzyme produced a single major protein band (42.0 kDa) on SDS-polyacrylamide (12%) gel electrophoresis that co-migrated with the human wild-type 1 control enzyme. Protein concentrations were determined by the Bradford method using bovine serum albumin as the standard (13).
Kinetic studies
Michaelis-Menten kinetic constants for the 3ß-HSD substrate were determined for the purified mutant and wild-type enzymes in incubations containing dehydroepiandrosterone (3–100 μM) plus NAD+ (0.2 mM) and purified enzyme (0.04 mg) at 27 °C in 0.02 M potassium phosphate, pH 7.4. The slope of the initial linear increase in absorbance at 340 nm per min (due to NADH production) was used to determine 3β-HSD activity. Kinetic constants for the isomerase substrate were determined at 27 °C in incubations of 5-androstene-3,17-dione (20-100 μM), with or without NADH (0.05 mM) and purified enzyme (0.01-0.04 mg) in 0.02 M potassium phosphate buffer, pH 7.4. Isomerase activity was measured by the initial absorbance increase at 241 nm (due to androstenedione formation) as a function of time. Blank assays (zero-enzyme, zero-substrate) assured that specific isomerase activity was measured as opposed to non-enzymatic, "spontaneous" isomerization (8). In addition, isomerase incubations without added coenzyme (NADH) were used to measure any basal (zero-coenzyme) isomerase activity in the mutants, and this basal activity was subtracted as a blank and reported in the kinetic table legends. Changes in absorbance were measured with a Varian (Sugar Land, TX) Cary 219 recording spectrophotometer. The Michaelis-Menten constants (Km, Vmax) were calculated from Lineweaver-Burke (1/S vs. 1/V) plots and verified by Hanes-Woolf (S vs. S/V) plots (14). The kcat values (min −1) were calculated from the Vmax values (nmol/min/mg) and represent the maximal turnover rate (nmol product formed/min/nmol enzyme dimer).
Kinetic constants for the 3ß-HSD cofactor were determined for the purified mutant and wild-type enzymes in incubations containing NAD+ (10-100 μM), dehydroepiandrosterone (100 μM) and purified enzyme (0.04 mg) in 0.02 M potassium phosphate, pH 7.4, at 27 °C using the spectrophotometric assay at 340 nm. Kinetic constants for the isomerase cofactor were determined in incubations of NADH (0-50 μM), 5-androstene-3,17-dione (100 μM) and purified enzyme (0.01–0.04 mg) in 0.02 M potassium phosphate buffer, pH 7.4 at 27 °C using the spectrophotometric assay at 241 nm. Zero-coenzyme blanks were used as described above for the substrate kinetics.
Inhibition constants (Ki) were determined for the inhibition of the wild-type 1, wild-type 2, Q240R1 and R240Q2 3β-HSD activities by epostane and trilostane using conditions that were appropriate for each enzyme species. For Q240R1 and 3β-HSD1, the incubations at 27 °C contained sub-saturating concentrations of dehydroepiandrosterone (4.0 and 8.0 μM, relative to substrate Km values of 3.7–6.1 μM), epostane or trilostane (0–1.0 μM), NAD+ (0.1 mM) and purified human type 1 enzyme (0.03–0.04 mg) in 0.02 M potassium phosphate buffer, pH 7.4. For Q105M1, Q105M2, R240Q2 and 3β-HSD2, similar incubations contained 10.0 μM or 20.0 μM dehydroepiandrosterone and the concentration ranges of epostane and trilostane were 0–10.0 μM. Dixon analysis (I versus 1/V) was used to determine the type of inhibition and calculate the Ki values (14).
Modeling and sequence alignment
Amino acid and nucleotide sequences were retrieved from the SWISS-PROT protein sequence database (15). Crystallographic coordinates were retrieved from the protein data bank (PDB) (16). The three dimensional structure of human type 1 3β-HSD/isomerase was modeled using the crystal structure (17) of UDP-galactose 4-epimerase from E. coli (Protein Data Bank Accession Code 1A9Z) as we previously described (10). The modeled images were produced using the Ribbons 2.0 program (18).
RESULTS
Site-Directed Mutagenesis, Expression and Purification of the Mutant Enzymes
The cDNA encoding the Q105M1 mutant of 3β-HSD1, the Q105M2 mutant of 3β-HSD2, the Q240R1 mutant of 3β-HSD1 and the R240Q2 mutant of 3β-HSD2 were produced by double-stranded, PCR-based mutagenesis and inserted into baculovirus. These amino acids were predicted to be critical residues by our modeled tertiary/quaternary structure for human 3β-HSD (Figures 3 and 4). The locations of these targeted residues plus other key amino acids are indicated in the primary structures of 3β-HSD1 and 3β-HSD2 (Figure 5). As shown by the immunoblots in Figure 6A, the baculovirus system successfully expressed the mutant enzyme proteins in Sf9 cells. Each expressed enzyme was highly purified according to SDS-PAGE (Figure 6B) using our published method (7,8).
Figure 5.

Amino acid sequence of the human type 1 3β-HSD/isomerase. Single letter abbreviations for amino acids in the 42.0 kDa monomer of the homodimer are shown. The key amino acids discussed in this report are labeled with position numbers. The amino acids that form the two α-helices in the subunit interface are underlined.
Figure 6.

Western immunoblots and SDS-Polyacrylamide gel electrophoresis of the mutant and wild-type enzymes by baculovirus. (A) For the western blots, the Sf9 cell homogenate (3.0 μg) containing the Q105M1, Q105M2, Q240R1, or R240Q2 plus the purified control wild-type 3β-HSD (0.05 μg) were separated by SDS-polyacrylamide (12%) gel electrophoresis. The 42.0 kDa band of the enzyme monomer was detected using our anti-3β-HSD antibody. (B) For SDS-PAGE, each lane was over-loaded with 4.0 μg of purified protein, and the bands were visualized by Coomassie Blue staining.
Substrate Kinetic Analyses of the Mutant Enzymes
The Michaelis-Menten kinetic values measured for substrate utilization by purified Q105M1, Q105M2, 3β-HSD1, Q240R1, R240Q2 and 3β-HSD2 are summarized in Table 2. For the dehydrogenase activity, the Q105M1 mutant demonstrates an 11-fold higher Km for substrate and 2-fold higher kcat than 3β-HSD1, which are similar to those of 3β-HSD2 and to those previously reported for the H156Y1 mutant enzyme (DHEA Km= 42.4 μM, kcat= 7.2 min−1, 11). The Q105M2 mutant exhibits DHEA kinetic constants that are similar to those of 3β-HSD2 with almost identical utilization efficiencies (kcat/Km). The Q105M1 and Q105M2 mutant enzymes retain isomerase substrate Km values similar to the appropriate 3β-HSD1 or 3β-HSD2 isoform. Both Q105M mutant enzymes exhibit reduced isomerase activity (kcat) compared to their wild-type enzyme counterpart. The Q240R1 mutant has a slightly elevated Km and diminished kcat for DHEA to yield a substrate utilization efficiency (kcat/Km) that is similar to wild-type 3β-HSD2. The R240Q2 mutant exhibits a decreased Km and elevated kcat for DHEA to yield a utilization efficiency (kcat/Km) that lies mid-way between wild-type 3β-HSD1 and 3β-HSD2. The Q240R1 mutation shifts the isomerase substrate Km value to a 2.2-fold higher level that is comparable to the isomerase substrate Km of wild-type 3β-HSD2. Similarly, the R240Q2 mutation shifts the isomerase substrate Km and kcat values from the wild-type 2 to the wild-type 1 profile.
Table 2.
Substrate kinetics for the 3β-HSD and isomerase activities of the purified mutant and wild-type enzymes.
| 3β-HSD1 | Isomerase2 | |||||
|---|---|---|---|---|---|---|
| Purified Enzyme | Km μM | kcat min−1 | kcat/Km min−1 uM−1 | Km μM | kcat min−1 | kcat/Km min−1 uM−1 |
| Q105M1 | 40.5 | 6.8 | 0.17 | 33.0 | 34.8 | 1.06 |
| Q240R1 | 6.1 | 1.4 | 0.23 | 62.3 | 33.3 | 0.53 |
| 3β-HSD1 | 3.7 | 3.3 | 0.89 | 27.9 | 50.2 | 1.25 |
| Q105M2 | 25.0 | 4.5 | 0.18 | 80.8 | 66.7 | 0.82 |
| R240Q2 | 32.7 | 13.0 | 0.40 | 17.5 | 34.4 | 1.97 |
| 3β-HSD2 | 47.3 | 7.1 | 0.15 | 88.4 | 81.5 | 0.92 |
Kinetic constants for the 3β-HSD substrate (DHEA) were determined in incubations at 27°C containing NAD+ (200 μM), dehydroepiandrosterone (3–100 μM) and purified enzyme (0.04 mg) in 0.02 M potassium phosphate, pH 7.4. Each Km and kcat value represents the mean of triplicate measurements with a standard deviation ≤7% of single mutant enzyme preparations.
Kinetic constants for the isomerase substrate (5-androstene-3,17-dione) were determined in incubations at 27°C of NADH (50 μM), 5-androstene-3,17-dione (20–100 μM) and purified enzyme (0.01–0.04 mg) in 0.02 M potassium phosphate buffer, pH 7.4.
Coenzyme Kinetic Analyses of the Mutant Enzymes
In agreement with our previously reported results obtained with the H156Y1 mutant of human 3β-HSD1 (11), the Q105M1 mutant enzyme exhibits the shift in 3β-HSD substrate kinetics to the 3β-HSD2 profile (Table 2), but the mutation does not affect the Km values of NAD+ as the cofactor for the 3β-HSD1 reaction or of NADH as the allosteric activator of type 1 isomerase (Table 3). The Q105M2 mutant enzyme retains Km and kcat values for NAD+ utilization and for the NADH-activation of isomerase that are similar to those measured for wild-type 3β-HSD2 (Table 3). The Q240R1 mutation has little effect on the Km value of 3β-HSD1 for NAD+ but disrupts the concentration-dependent activation of type 1 isomerase by NADH (Table 3, Figure 7). The basal isomerase activity of Q240R1 in the absence of NADH is 22% of the wild-type 3β-HSD1 isomerase after maximal activation by NADH. In contrast, the basal isomerase activities of 3β-HSD1, 3β-HSD2, R240Q2, Q105M1 and Q105M2 are only 5–10% of the kcat of NADH-activated isomerase, and NADH stimulates that basal activity in a concentration dependent-manner (Figure 7). The R240Q2 mutant of 3β-HSD2 shifts the Km of NAD+ utilization to a 2.3-fold lower value that is similar to the value measured for 3β-HSD1 and to a 3.9-fold lower Km for the NADH-activation of isomerase, which is comparable to that of type 1 isomerase (Table 3).
Table 3.
Cofactor kinetics for the 3β-HSD and isomerase activities of the purified mutant and wild-type enzymes.
| 3β-HSD NAD+1 | Isomerase NADH2 | |||||
|---|---|---|---|---|---|---|
| Purified Enzyme | Km μM | kcat min−1 | kcat/Km min−1 uM−1 | Km μM | kcat min−1 | kcat/Km min−1 uM−1 |
| Q105M1 | 38.8 | 2.3 | 0.06 | 4.9 | 28.8 | 5.88 |
| Q240R1 | 25.6 | 1.3 | 0.05 | N.D.3 | N.D. | N.D. |
| 3β-HSD1 | 34.1 | 3.5 | 0.10 | 4.6 | 45.0 | 9.78 |
| Q105M2 | 61.4 | 8.7 | 0.14 | 7.2 | 69.8 | 9.69 |
| R240Q2 | 37.2 | 11.2 | 0.30 | 3.2 | 35.5 | 11.09 |
| 3β-HSD2 | 86.3 | 7.1 | 0.08 | 12.6 | 99.1 | 7.87 |
Kinetic constants for the 3β-HSD cofactor were determined in incubations at 27°C containing NAD+ (13–100 μM), dehydroepiandrosterone (100 μM) and purified enzyme (0.03 mg) in 0.02 M potassium phosphate, pH 7.4. Each Km and kcat value represents the mean of triplicate measurements with a standard deviation ≤6% of single mutant enzyme preparations.
Kinetic constants for the isomerase cofactor were determined in incubations at 27°C of NADH (3–50 μM), 5-androstene-3,17-dione (100 μM) and purified enzyme (0.01 mg) in 0.02 M potassium phosphate buffer, pH 7.4.
N.D., not determined. The specific isomerase activity of Q240R1 in the absence of NADH was increased only slightly at each NADH concentration tested (2–50 μM). In contrast, NADH dramatically stimulates the 3β-HSD1, 3β-HSD2, Q105M1, Q105M2 and R240Q2 isomerase activities in a concentration-dependent manner to allow the calculation of kinetic constants.
Figure 7.
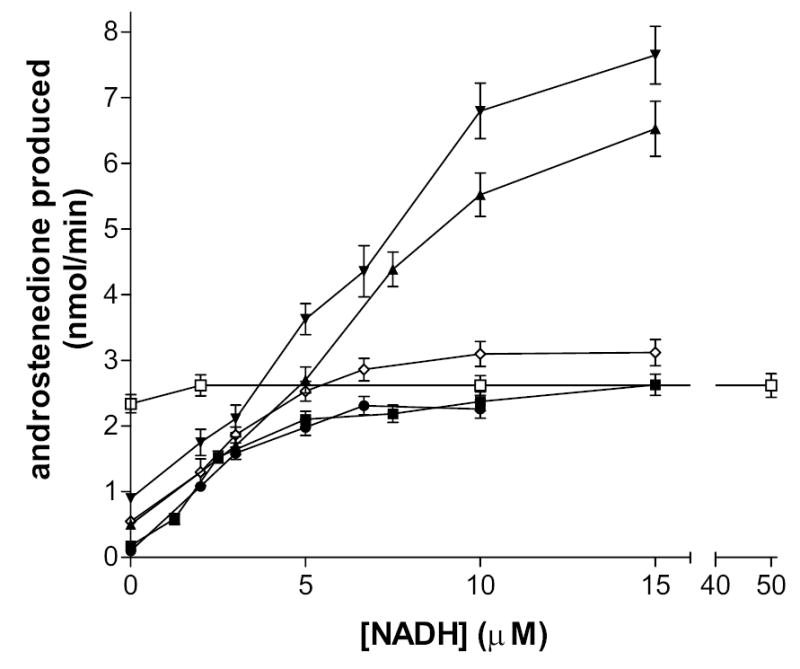
Stimulation of the isomerase activity of the wild-type and mutant enzymes by NADH. Isomerase activity (nmol androstenedione produced/min) was determined spectrophotometrically at 240 nm in incubations of NADH (3-50 μM), 5-androstene-3,17-dione (100 μM) and purified enzyme (0.01 mg) in 0.02 M potassium phosphate buffer, pH 7.4, at 27oC. The NADH vs velocity curves of the human mutant 3β-HSD enzymes- Q240R1(□), R240Q2 (⋄), Q105M1 (•), Q105M2 (▾)- were compared to those measured for the human wild-type enzymes- 3β-HSD1 (▪) and 3β-HSD2 (▴). Data points are the means of triplicate determinations, and the cross bars represent standard deviations.
Kinetic Analyses of the Inhibition of the Mutant Enzymes
The inhibition constant values (Ki) derived from Dixon analyses (I vs 1/v) show that epostane and trilostane inhibit the 3β-HSD activity of Q105M1 with 21- to 26-fold lesser affinities compared to the Ki values measured using the wild-type 3β-HSD1 enzyme. However, Q105M2 retains the lower affinity profiles of inhibition by epostane and trilostane that are observed with wild-type 3β-HSD2 (Table 4). While the Q240R1 mutant enzyme is inhibited by epostane and trilostane with the same high-affinity profiles as measured for 3β-HSD1, the R240Q2 mutation of 3β-HSD2 shifts the inhibition constants (Ki) to values that are 5- to 14-fold lower for epostane and trilostane, respectively (Table 4). According to the Dixon plots (not shown), epostane and trilostane inhibited the 3β-HSD activities of Q105M1, Q105M2, Q240R1, R240Q2, 3β-HSD1 and 3β-HSD2 in a competitive manner.
Table 4.
Comparison of inhibition constants for the purified Q105M1, Q105M2, Q240R1, R240Q2, 3β-HSD1 and 3β-HSD2 activities.
| Inhibitor steroid Ki (μM)1 | ||
|---|---|---|
| Enzyme | Epostane | Trilostane |
| Q105M1 | 1.84 | 2.08 |
| Q240R1 | 0.07 | 0.13 |
| 3β-HSD1 | 0.07 | 0.10 |
| Q105M2 | 1.64 | 2.07 |
| R240Q2 | 0.21 | 0.12 |
| 3β-HSD2 | 0.98 | 1.66 |
For 3β-HSD1, Q240R1 and R240Q2, the epostane and trilostane concentration ranges were 0–0.75 μM. For Q105M1, Q105M2 and 3β-HSD2, the epostane and trilostane concentration ranges were 0–7.5 μM. Inhibition constants (Ki from Dixon plots) were determined in incubations at 27°C containing the inhibitor plus dehydroepiandrosterone (4.0 μM or 8.0 μM for Q240R1 and 3β-HSD1; 10.0 μM or 20.0 μM for Q105M1, Q105M2, R240Q2 or 3β-HSD2), NAD+ (0.2 mM) and purified enzyme (0.03–0.04 mg) in 0.02 M potassium phosphate, pH 7.4.
DISCUSSION
Our studies of the structure/function of human 3β-HSD took a dramatic turn in 2002 when we reported that it is possible to selectively inhibit human 3β-HSD1 without affecting the activity of human 3β-HSD2 (11). Since that discovery, our primary focus has been on determining the structural basis for the 14- to 16-fold higher affinity of purified human 3β-HSD1 for substrate and inhibitor steroids compared to human 3β-HSD2. There is a strict tissue-specific distribution of the two human 3β-HSD isoforms. 3β-HSD1 is expressed in mammary gland, breast tumors, prostate, prostate tumors and placenta, and 3β-HSD2 is expressed in the adrenals, testes and ovaries. We also reported that 3β-HSD1 transfected into human breast tumor MCF-7 tet-off cells have a 12-fold greater affinity for substrate and inhibitor steroids compared to the 3β-HSD2 that we transfected into the MCF-7 tet-off cells (19). These recent discoveries identify human 3β-HSD1 as a target enzyme for inhibition in hormone-sensitive tumors.
The primary significance of human 3β-HSD1 in breast cancer is its role in the conversion of circulating DHEA to androstenedione, which is then converted by aromatase to estrone and to estradiol by 17β-HSD in breast tumors. Selective inhibition of 3β-HSD1 in breast tumors blocks the intracrine production estradiol but does not block 3β-HSD2 in adrenals to interfere with the steroidogenesis that produces cortisol and aldosterone (Figure 1). Inhibition of 3β-HSD1 in breast tumors is most useful in postmenopausal women, when ovarian 3β-HSD2 and aromatase no longer function in the biosynthesis of estradiol (20). Although aromatase inhibitors effectively block estradiol production in breast tumors, aromatase is also present at high levels in human osteoblasts (21). While the 3β-HSD isoform in bone has not been identified, the incubation of human osteoblast cells with trilostane does not diminish osteoblastic activity (22). Thus, the inhibition of 3β-HSD1 in breast tumors may not produce a high incidence of osteoporosis that has been observed using the aromatase inhibitors (21,23). In terms of the hormonal treatment of prostate cancer, inhibitors of 5α-reductase like finasteride have not proven to be effective as a therapy for prostate malignancies (24). Unlike the benign hyperplastic prostate, the growth of prostate tumors is stimulated by testosterone as well as by its 5α-reductase product steroid, 5α-dihydrotestosterone (24). The selective inhibition human 3β-HSD1 activity to block testosterone production in prostate tissue and its tumors may yield a new treatment for prostate cancer that will avoid the initial flare of tumor growth seen with LHRH agonists like leuprolide (25). Other enzyme targets for inhibition in the hormonal treatment of breast or prostate cancer include human sulfatase to block the conversion of circulating estrone-sulfate or DHEA-sulfate to the free steroids in tissues and isoforms of human 17β-hydroxysteroid dehydrogenase (17β-HSD1 and 17β-HSD5) that function as reductases to block the conversion of estrone to estradiol (26,27). Thus, human 3β-HSD1 is one of several steroid metabolizing enzymes currently under study as potential therapeutic targets for the treatment of hormone-sensitive cancers. Although our data obtained with epostane and trilostane demonstate that it is possible to selectively inhibit 3β-HSD1 with them, we are not suggesting that these compounds be used to treat breast or prostate cancer. After the complete structural basis for the kinetic differences between human 3β-HSD2 and 3β-HSD2 has been elucidated by studies like this one, docking routines can be performed to design better inhibitors with greater specificity for 3β-HSD1 than epostane and trilostane.
The selective inhibition of steroid biosynthesis via 3β-HSD1 in human placenta has another clinical implication. The onset of labor in human pregnancy could be delayed by selectively inhibiting the activity of placental 3β-HSD1 near term to decrease estradiol production from DHEA of fetal origin (2,3) without interfering with cortisol or aldosterone production by 3β-HSD2 in the maternal adrenal gland. Estradiol participates in the cascade of events leading to parturition in humans by stimulating the biosynthesis of oxytocin receptors in the myometrium (28). In addition, inhibition of placental 3β-HSD1 would inhibit progesterone production to diminish uterine quiescence (3).
In our recent structure/function study (11), the catalytic residues for human type 1 3β-HSD activity were identified as Tyr154 and Lys158 (Figure 5). The presence of His156 in human 3β-HSD1 versus Tyr156 in 3β-HSD2 was also shown to be responsible for the 14- to17-fold greater affinity of the type 1 enzyme for 3β-HSD substrate steroids and inhibitors compared to the type 2 enzyme (11). In this report, we have localized the critical His156/Tyr156 residue in the subunit interface of the enzyme dimer by homology modeling (Figure 4) and tested this prediction by mutagenesis of a potential key hydrogen-binding partner, Gln105, on the other subunit of 3β-HSD1 and 3β-HSD2 to produce Q105M1 and Q105M2, respectively. The His156/Tyr156 difference between the two isoenzymes is responsible only for the higher affinity of 3β-HSD1 for dehydrogenase substrate and inhibitor steroids but is not involved in the higher affinity of 3β-HSD1 for coenzymes and for the isomerase substrate steroid (11).
The homology model predicts that Gln105 is positioned to hydrogen-bond to the imidazole ring of His156 across the subunit interface of 3β-HSD1, and to hydrogen-bond to the phenolic group of Tyr156 on the adjacent subunit of 3β-HSD2. In this model, the subunits are anti-parallel relative to each other at a helical interface, as has been reported for human 17β-HSD1 (29) and E. coli UDP-galactose-4-epimerase (17). Many members of the short-chain dehydrogenase-reductase family are anti-parallel homodimers, which create a four-helix bundle subunit interface that is critical to the function of the adjacent active sites in each monomer (29). Based on a cataloging of interactions between Gln-His and Gln-Tyr residues in high resolution protein crystal structures, the Gln-Tyr side chain interactions are tightly clustered in only a few geometries compared to the extensive range of geometries observed for side chain interactions between Gln and His (30). Thus, the Q105M1 mutation may disrupt key hydrogen-bonding interactions between Gln105 and His156 in the subunit interface of 3β-HSD1. However, the Q105M2 mutation has little effect because the Gln105-Tyr156 hydrogen-bond may either not exist or is relatively weak between the subunits of 3β-HSD2 due to the restrictive geometry of this interaction. Sub-optimal hydrogen bonds are commonly observed in protein-protein interfaces, to the extent that over 17% of buried donors or acceptors do not participate in hydrogen bonds in high resolution structures (31). The 11-fold higher Km for the dehydrogenase substrate, 2-fold lower dehydogenase kcat and the 21- to 26-fold lower Ki values of trilostane and epostane measured for Q105M1 compared 3β-HSD1 in this study are almost identical to the kinetic shifts previously reported for the H156Y mutant of 3β-HSD1 (11). As seen previously with the H156Y mutant, the Km values of coenzyme and isomerase substrate utilization for 3β-HSD1 are not modified by the Q105M1 mutation. In sharp contrast, the Q105M2 mutant exhibits substrate, coenzyme or inhibitor kinetics of both the dehydrogenase and isomerase activities that are similar to those of 3β-HSD2, which emphasizes the different interactions between the subunits of the two isoforms. This interaction may be critical to the binding orientation of the substrate steroid in the adjacent catalytic sites. Thus, the interactions between Gln105 and His156 in the subunit interface of 3β-HSD1 appear to be key structural reasons for its 11- to 26-fold higher affinity for the DHEA dehydrogenase substrate and inhibitor steroids relative to 3β-HSD2. In addition, the Q105M1 mutant data supports our model of the anti-parallel orientation of the two 3β-HSD1 monomers.
Human 3β-HSD1 utilizes NAD+ as the cofactor for the dehydrogenase reaction and NADH as the allosteric activator of isomerase with 2.5- to 2.7-fold lower Km values than 3β-HSD2 (11). We recently showed that the 3β-HSD and isomerase activities share the same coenzyme domain on a single enzyme protein by shifting cofactor-specificity from NAD(H) to NADP(H) for both activities with a two-point mutation, D36AK37R (10). In addition, 3β-HSD1 utilizes the isomerase substrate steroid with a 3.2-fold lower Km value compared to 3β-HSD2 (11). In our current study, homology modeling has predicted that a non-identical amino acid in the two isoenzyme structures may be responsible for these differences: Gln240 in 3β-HSD1 and Arg240 in 3β-HSD2 (Figure 3). Site-directed mutagenesis produced Q240R1 in 3β-HSD1 and R240Q2 in 3β-HSD2 to test this prediction.
The Q240R1 mutation shifts the isomerase substrate kinetic values from the 3β-HSD1 to the 3β-HSD2 profile. The Q240R1 mutation does not affect the NAD+ Km value but decreases the NAD+ kcat by 2.7-fold and abolishes the concentration-dependent activation of type 1 isomerase by NADH. Most significantly, the Q240R1 mutant greatly diminishes the allosteric activation of the isomerase activity of 3β-HSD1 by NADH. The NADH vs velocity plot for Q240R1 in Figure 7 shows that the isomerase is almost fully active at the 3β-HSD1 level without NADH. This suggests that the substitution of positively charged Arg240 for uncharged Gln240 shifts most of the enzyme into the isomerase conformation. Less enzyme in the 3β-HSD conformation explains the 2.4- to 2.8-fold lower Kcat and unchanged Km values measured for the utilization of DHEA and NAD+ by the 3β-HSD activity of Q240R1.
The R240Q2 mutation of 3β-HSD2 shifts the Km value of the isomerase substrate steroid to a 2.4-fold lower value, shifts the Km value for NADH to a 4-fold lower value, shifts the Km of NAD+ utilization to a 2.3-fold lower value and lowers the inhibition constants (Ki) of epostane by 5-fold and of trilostane by 14-fold. These changes create a 3β-HSD2 mutant enzyme that is similar to 3β-HSD1 in these kinetic properties but that retains the high DHEA substrate Km value of 3β-HSD2. Because the Ki values measured for epostane and trilostane of R240Q2 are shifted to the higher-affinity kinetic profile of 3β-HSD1, these data show that the isomerase substrate and coenzyme binding properties are related to the competitive binding of 3β-HSD inhibitor steroids. The H156Y1 and Q105M1 mutations that affect only 3β-HSD DHEA substrate kinetics but not isomerase substrate or coenzyme kinetics shift the inhibition kinetics of 3β-HSD1 to the much lower-affinity profile of 3β-HSD2. These opposing effects of R240Q2 and Q105M1 on the Ki values of epostane and trilostane support our model of the two-step, sequential 3β-HSD and isomerase activities. In this model, the NADH produced by the 3β-HSD reaction induces a change in enzyme conformation around a single steroid binding site, and this conformational change activates the isomerase reaction on the bifunctional enzyme protein (9).
In summary, three of the most important observations of this study are: 1) the Q105M1 mutant decreases the affinity of 3β-HSD1 for DHEA substrate and inhibitor steroids so that it mimics 3β-HSD2; 2) Q240R1 abolishes the allosteric activation of the isomerase activity by NADH; and 3) the R240Q2 mutant increases the affinity of 3β-HSD2 for isomerase substrate, NADH and inhibitor steroids so that it mimics the 3β-HSD1 enzyme. Determining the roles of Gln105, His156 and Gln240 in human 3β-HSD1 in comparison those of Gln105, Tyr156 and Arg240 in 3β-HSD2 provides key structural data for use in future docking studies that may produce highly specific inhibitors of 3β-HSD1. Efforts are underway using our genetically-engineered, soluble form of human type 1 3β-HSD/isomerase (32) to grow enzyme crystals that will provide a complete enzyme structure using X-ray diffraction for the future docking studies.
Footnotes
This work was supported by NIH grants HD20055, CA114717 (JLT) and DK26546 (VP,TCU).
The abbreviations used are: HSD, hydroxysteroid dehydrogenase; DHEA, dehydroepiandrosterone; LHRH, luteinizing hormone releasing hormone.
References
- 1.Rheaume E, Lachance Y, Zhao HF, Breton N, Dumont M, de Launoit Y, Trudel C, Luu-The V, Simard J, Labrie F. Mol Endocrinol. 1991;5:1147–1157. doi: 10.1210/mend-5-8-1147. [DOI] [PubMed] [Google Scholar]
- 2.Rainey WE, Carr BR, Sasano H, Suzuki T, Mason JI. Trends Endocrinol Metab. 2002;13:234–239. doi: 10.1016/s1043-2760(02)00609-4. [DOI] [PubMed] [Google Scholar]
- 3.Kacsoh, B. (2000) Endocrine Physiology, pp. 566–567, McGraw-Hill Companies, Inc., New York.
- 4.Gingras S, Moriggi R, Groner B, Simard J. Molecular Endocrinology. 1999;13:66–81. doi: 10.1210/mend.13.1.0221. [DOI] [PubMed] [Google Scholar]
- 5.Geldof AA, Dijkstra I, Newling DW, Rao BR. Anticancer Res. 1995;15:1349–1354. [PubMed] [Google Scholar]
- 6.Gingras S, Simard J. Endocrinology. 1999;140:4573–4584. doi: 10.1210/endo.140.10.7038. [DOI] [PubMed] [Google Scholar]
- 7.Thomas JL, Myers RP, Strickler RC. J Steroid Biochem. 1989;33:209–217. doi: 10.1016/0022-4731(89)90296-3. [DOI] [PubMed] [Google Scholar]
- 8.Thomas JL, Berko EA, Faustino A, Myers RP, Strickler RC. J Steroid Biochem. 1988;31:785–793. doi: 10.1016/0022-4731(88)90287-7. [DOI] [PubMed] [Google Scholar]
- 9.Thomas JL, Frieden C, Nash WE, Strickler RC. J Biol Chem. 1995;270:21003–21008. doi: 10.1074/jbc.270.36.21003. [DOI] [PubMed] [Google Scholar]
- 10.Thomas JL, Duax WL, Addlagatta A, Brandt S, Fuller RR, Norris W. J Biol Chem. 2003;37:5483–35490. doi: 10.1074/jbc.M304752200. [DOI] [PubMed] [Google Scholar]
- 11.Thomas JL, Mason JI, Brandt S, Spencer BR, Norris W. J Biol Chem. 2002;277:42795–42801. doi: 10.1074/jbc.M208537200. [DOI] [PubMed] [Google Scholar]
- 12.Thomas JL, Evans BW, Blanco G, Mercer RW, Mason JI, Adler S, Nash WE, Isenberg KE, Strickler RC. J Steroid Biochem Mol Biol. 1998;66:327–334. doi: 10.1016/s0960-0760(98)00058-2. [DOI] [PubMed] [Google Scholar]
- 13.Bradford MM. Analyt Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 14.Segel, I. H. (1975) Enzyme Kinetics, pp. 109–111, John Wiley & Sons, New York.
- 15.Bairoch A, Apweiler R. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thoden JB, Frey PA, Holden HM. Biochemistry. 1996;35:2557–66. doi: 10.1021/bi952715y. [DOI] [PubMed] [Google Scholar]
- 18.Carson, M. (1997) Methods Enzymol.277, pp. 493–505, Carter, C. W., and Sweet, R. M., eds, Academic Press, New York.
- 19.Thomas JL, Umland TC, Scaccia LA, Boswell EL, Kacsoh B. Endocrine Res. 2004;30:935–941. doi: 10.1081/erc-200044164. [DOI] [PubMed] [Google Scholar]
- 20.Simpson ER. Fertil Steril. 2002;77:S6–10. doi: 10.1016/s0015-0282(02)02984-9. [DOI] [PubMed] [Google Scholar]
- 21.Dowsett M. Semin Oncol. 2003;30:58–69. doi: 10.1016/s0093-7754(03)00300-2. [DOI] [PubMed] [Google Scholar]
- 22.Kasperk CH, Wakley GK, Hierl T, Ziegler R. Bone J Miner Res. 1997;12:464–471. doi: 10.1359/jbmr.1997.12.3.464. [DOI] [PubMed] [Google Scholar]
- 23.Nawata H, Tanaka S, Tanaka S, Takayanagi R, Sakai Y, Yanase T, Ikuyama S, Haji M. J Steroid Biochem Mol Biol. 1995;53:265–174. doi: 10.1016/0960-0760(95)00031-t. [DOI] [PubMed] [Google Scholar]
- 24.Reddy GK. Clin Prostate Cancer. 2004;2:206–208. doi: 10.1016/s1540-0352(11)70045-2. [DOI] [PubMed] [Google Scholar]
- 25.Mcleod DG. Urology. 2003;61:3–7. doi: 10.1016/s0090-4295(02)02393-2. [DOI] [PubMed] [Google Scholar]
- 26.Pasqualini JR. Biochim Biophys Acta. 2004;1654:123–43. doi: 10.1016/j.bbcan.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Penning TM, Jin Y, Steckelbroeck S, Lanisnik Rizner T, Lewis M. Mol Cell Endocrinol. 2004;215:63–72. doi: 10.1016/j.mce.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 28.Richter ON, Kubler K, Schmolling J, Kupka M, Reinsberg J, Ulrich U, van der Ven H, Wardelmann E, van der Ven K. Mol Hum Reprod. 2004;10:339–346. doi: 10.1093/molehr/gah039. [DOI] [PubMed] [Google Scholar]
- 29.Ghosh D, Sawicki M, Pletnev V, Erman M, Ohno S, Nakajin S, Duax WL. J Biol Chem. 2001;276:18457–18463. doi: 10.1074/jbc.M100538200. [DOI] [PubMed] [Google Scholar]
- 30.Singh , J., and Thornton, J. M. (1992) Atlas of Protein Side-Chain Interactions, Vol. 1, pp. 244–245, 264–265, IRL press, Oxford.
- 31.Xu D, Tsai CJ, Nussinov R. Protein Engineering. 1997;10:999–1012. doi: 10.1093/protein/10.9.999. [DOI] [PubMed] [Google Scholar]
- 32.Thomas JL, Mason JI, Blanco G, Veisaga ML. J Mol Endocrinol. 2001;27:77–83. doi: 10.1677/jme.0.0270077. [DOI] [PubMed] [Google Scholar]