

Abstract
Keratocytes in the corneal stroma express keratan sulfate-containing proteoglycans including cornea-specific keratocan. On plastic dishes, human, bovine and rabbit keratocytes lose their characteristic dendritic morphology and keratocan expression when cultured in serum-containing media. Herein, we demonstrated that murine keratocytes also acquired a fibroblastic shape and lost keratocan expression after first passage when cultured on plastic in the presence of serum. In contrast, cells expanded on human amniotic membrane (AM) stromal matrix maintained a 3-dimensional dendritic morphology and expressed keratocan mRNA and protein for at least 8 passages before senescence. When keratocytes were cultured on AM, the promoter activity of TGF-β2 and TGF-βRII was downregulated as compared to that on plastic. Furthermore, cells on AM continuously retained Smad 2 and Smad 4 in the cytoplasm and did not express α-SMA even when 10 ng/ml TGF-β1 was added in a serum-free medium for up to 5 days. In parallel to such downregulation of TGF-β signaling, keratocan promoter-driven ECFP expression was observed in cells cultured either on AM in the presence of serum, or on plastic containing serum treated with a neutralizing antibody to TGF-β. Collectively, these results indicate that downregulation of Smad-mediated TGF-β signaling is an important mechanism for cultured keratocytes to maintain a normal phenotype while continuously expanded in a serum-containing medium. This strategy of suppressing TGF-β signaling, achieved by AM stromal matrix in part via suppression of TGF-β gene transcription, can be used to expand keratocytes in culture without the use of AM in the future.
Keratocytes, a unique population of neural crest-derived cells embedded in the corneal stroma, play a major role in maintaining corneal transparency. Different culturing methods have been explored to study the mechanism whereby normal keratocytes are regulated in vitro. Bovine (1), rabbit (2) and human (3;4) keratocytes cultured on plastic rapidly lose their characteristic dendritic morphology, and acquire a fibroblastic morphology when exposed to a fetal bovine serum (FBS)-containing medium. Furthermore, these exposed cells downregulate the expression of keratan sulfate-containing proteoglycans (5–8), keratocan (3;9), and CD34 (4), and upregulate that of chondroitin-dermatan sulfate-containing proteoglycans (9;10) and α-SMA (2;4;9;11). Similarly, murine corneal fibroblasts cultured on plastic in a FBS-containing medium lose expression of corneal stroma-specific keratocan mRNA, but continue to express mimecan and lumican mRNAs (12;13). To overcome such a detrimental effect of FBS, a serum-free culture system has to be used to maintain a normal dendritic morphology and expression of keratan sulfate-containing proteoglycans at the expense of cell expansion (1).
TGF-β signaling is essential for corneal development and plays a pivotal role in normal and abnormal wound healing (14;15). One important mediator of TGF-β signaling is the Smad pathway. The binding of TGF-β• • ligands • • to TGF-β • • receptors triggers phosphorylation of Smad 2 and Smad 3. Smad 4 binds with phosphorylated Smad 3 in a complex and is translocated to the nucleus to transactivate target genes (16). Therefore, the translocation of these Smads into the nucleus is a common effector in the Smad-mediated pathway (17). In 1% serum or a serum-free medium, addition of TGF-β1 differentiates keratocytes into myofibroblasts with prominent focal adhesions, and upregulates expression of α-SMA, integrin α5β1, cadherins, type I and III collagens, biglycan and the EDA (EIIIA) form of fibronectin (9–11;18). It remains unclear whether suppression of TGF-β signaling is not only important to prevent myofibroblast differentiation but also essential for maintaining the normal keratocyte phenotype.
Recently, we reported a culture system that can achieve effective expansion of human keratocytes by growing them on the stromal surface of the AM in an FBS-containing medium (3;4). They maintained a dendritic morphology, continued to express corneal stroma-specific keratocan for at least 5 passages (at 1:2 split), and did not express α-SMA under serum-containing conditions or addition of TGF-β1 (4). Previously, we have reported that transcript expression of TGF-β2, TGF-β3, and TGF-βRII is suppressed in both serum-containing and serum-free media, of which the latter is challenged by exogenous TGF-β1, in cultured human corneal and limbal fibroblasts (19) as well as human conjunctival and pterygium fibroblasts (20). Suppression of TGF-β signaling is coupled with downregulation of α-SMA, integrin α5 and EDA form of fibronectin transcripts. Herein, we report that murine keratocytes can be similarly expanded on AM for at least 8 passages without losing their normal phenotype in a serum-containing medium and that suppression of Smad-mediated TGF-β signaling pathway is pivotal in maintaining keratocan-expressing phenotype.
MATERIALS AND METHODS
Isolation and Culture of Keratocytes on Plastic or AM
Six to ten week-old CD-1 albino mice of were obtained from Charles Rivers (Boston, MA) and handled according to ARVO guidelines for care of animals in ophthalmic research. After euthanasia, the eyes were enucleated by forceps, washed profusely in PBS, and incubated in DMEM containing 20 mM HEPES, 15 mg/ml dispase II (Roche, Indianapolis, IN) and 100 mM sorbitol at 4 oC for 18 h as previously reported (21;22). The entire corneal epithelium loosened by this treatment was subsequently removed by vigorous shaking. Under a dissecting microscope, the corneal stroma was separated from the sclera at the corneoscleral limbus by pressing down the limbus with a 27 G needle while the eye was held with a forcep. Isolated corneal stromas were incubated overnight at 37 oC in DMEM containing 1.25 mg/ml collagenase A (Roche, Indianapolis, IN), 50 μg/ml gentamicin and 20 mM HEPES in a non-coated plastic dish until the tissue became “smeared” onto the dish bottom. Digested corneal stromas in collagenase A were centrifuged at 800 x g for 5 min. Keratocytes were resuspended in DMEM containing 20 mM HEPES, ITS (5 μg/ml insulin, 5 μg/ml transferrin and 5 ng/ml sodium selenite), 50 μg/ml gentamicin and 1.25 μg/ml amphotericin B with or without 10% FBS. This keratocyte-containing cell suspension was then seeded on plastic dishes or on the stromal side of the AM (Bio-Tissue, Miami, FL) fastened to a culture insert as previously described (23).
The suspension of keratocytes prepared from 3 – 4 murine corneal buttons was seeded on a 35 mm plastic dish or on the stromal surface of one 32 mm AM insert as reported (3). Cells were cultured in DMEM supplemented with 10% FBS (DMEM/10% FBS), and the medium was changed every 2 – 3 days. When cells reached 80–90% confluence, they were dissociated into single cells by incubation in 0.05% trypsin and 0.53 mM EDTA in HBSS at 37 oC for 5 min in plastic dishes or for 20 min in AM inserts, followed by vigorous pipetting. After centrifuging at 800 x g for 5 min, cells were resuspended in DMEM/10% FBS and seeded on a plastic dish or AM stroma. They were cultured in DMEM containing 10% FBS, 20 mM HEPES, 50 μg/ml gentamicin and 1.25 μg/ml amphotericin B.
TGF-β 1 Challenge and Neutralizing Antibody
To assess whether TGF-β1 affected the cell phenotype, triggered Smad 2 and Smad 4 nuclear translocation, and differentiated keratocytes into myofibroblasts, 10 ng/ml human recombinant TGF- β1 (Sigma, St Louis, MO) was added to serum-free DMEM/ITS cells for 3 h or 5 days when cells expanded on AM were passed to 24 well plastic dishes and AM inserts, respectively. In addition, primary keratocytes were seeded and cultured on AM or plastic for 3 days in DMEM/ITS or DMEM/10% FBS for 24 h, of which the latter was treated with or without 10 μg of a monoclonal antibody neutralizing TGF-β1, -β2, and -β3 (R&D Systems, Minneapolis, MN) per ml of DMEM medium for 48 h before adenoviral transfection.
Morphological Analysis
The corneal stroma after immediate surgical isolation was assessed for cell morphology by phase contrast microscopy and for viability by Live/Dead Assay® (Molecular Probes, Eugene, OR) according to a published method (24) by incubating them for 30 min with 2.5 ml of 2 mM calcein-AM and 4 mM ethidium homodimer in PBS. Morphology of primary and subcultured cells on plastic and AM were assessed by phase-contrast microscopy. Images were photographed with a NikonTe-2000u Eclipse epi-fluorescent microscope (Nikon, Tokyo, Japan).
Assays of Cell Proliferation
To verify that cells indeed proliferated on AM, the passage 2 (P2) cells that were continuously cultured on either AM or plastic were subcultured at a density of 10,000 cells per 24-well plastic dish in DMEM/ITS or DMEM/10% FBS or on AM in DMEM/10% FBS. Cells were terminated at day 3 and day 7 for MTT assay (Roche) according to the manufacturer’s instruction. This assay measured by absorbance at 550 nm yielded a linear correlation for cell numbers above 2,500 cells using P2 murine corneal fibroblasts (not shown). Cells at day 7 were also immunostained using an anti-Ki67 antibody. The number of Ki67 positive nuclei was randomly measured in 10 fields under high magnification (400 x) for each culture. Experiments were performed in triplicate.
Transient Transfection and TGF-β Promoter Assays
Freshly isolated cells expanded on AM were subcultured on plastic and AM inserts. Upon reaching 60–80% confluence, cells in each 24 well or AM insert were co-transfected with 1.0 μg/ml plasmid DNA containing TGF-β2 or TGF- β RII promoter-luciferase and 1.0 μg/ml pCMV/Sport/Bgal (Invitrogen) using GeneJammer® (Stratagene) according to manufacturer’s protocol. The plasmid containing TGF-β2 or TGF-β RII promoter-luciferase was constructed by inserting either human TGF-β2 promoter (−1729 to +63) or TGF-β RII promoter (−1883 to +50) upstream of Luc+ in pGL3-basic (Promega, Madison, WI). TGF-β2 promoter (25) was kindly provided by Dr Kim (NIH, Maryland) and was inserted into Kpn I and Hind III of pGL3-basic. TGF-β RII promoter (26) was amplified by PCR using genomic DNA of human corneal fibroblast as the template, the forward primer of 5’-GTACGGTACCCATCAAAGAAGTTATGG TTC-3’, and the reverse primer of 5’-GTACAAGCTTACTCAACTTCAACTCAG CGC-3’. PCR program used was 95°C, 30 seconds; 55°C, 30 seconds; 72°C, 2 minutes; for 30 cycles. The amplified TGF-β RII promoter fragment was then digested with Kpn I and Hind III, gel purified (Qiagen, Valencia, CA), and inserted at the same sites on pGL3-basic. TGF-β2 and TGF-β RII promoter activities were measured by the Luciferase Assay System® (Promega) and normalized with the β-galactosidase activity.
Adenoviral Transfection
A pKerapr3.2-intron-ECFP/BpA plasmid DNA was constructed by insertion of an ECFP fragment generated by PCR using pECFP-N1 (Clonetech Palo Alto, CA) as template and two restriction enzyme sites-tagged primers (ECFP-RI, 5’- GATCGAATTCCCACCGGTCGCCACCAT GGTG-3’ and ECFP-Sal I, 5’-: GTTACTCGACTTACTTGTACATCTCGTC CATG-3’). The resulting PCR fragment was digested with EcoR I and Sal I simultaneously and the ligated to the EcoR I and Sal I sites of the pKera3.2-int-MCS-BPA plasmid vector (12). The fidelity of PCR amplified ECFP was confirmed by DNA sequencing. Next, the Kerapr3.2-intron-ECFP/BpA DNA fragment (6.0 kb) was excised from the pKerapr3.2-intron-ECFP/BpA plasmid with Not I and Kpn I digestion and ligated into pAd-Track plasmid vector, which was kindly provided by Dr. Wei Li (Bascom Palmer Eye Institute, Miami, FL) and contains a CMV-EGFP expression cassette (27). The final construct was designated as pAd-Kerapr3.2-intron-ECFP/BpA and used to generate recombinant adenoviral plasmid by homologous recombination in E. coli according to a previously published method (27), and replication-deficient recombinant adenoviruses in the 293 cells according to previously published method (28). Large scale adenovirus preparation was prepared as previously described (12). Purified viruses were aliquoted in 50% glycerol and stored at −80°C. The viral titer (PFU per milliliter) for adenovirus preparation was determined in 293 cells using 96 well plates and a series of diluted virus for transfection. After 7 days checked the GFP expression under an inverted fluorescence microscope and estimated titer. The Aden-track-Kerapr3.2-intron-ECFP/BpA adenovirus had a titer of 3x1011 infectious particles per ml (PFU per ml). Cells were then transfected by aden-track-Kerapr3.2-intron-ECFP/BpA adenovirus (50 pfu) for 24 h. Transfection efficiency was judged by expression of EGFP, and expression of keratocan by expression of ECFP in the same cell using a NikonTe-2000u Eclipse epi-fluorescent microscope equipped with appropriate filters.
Immunostaining
To assess protein expression of α-SMA, keratocan, CD34, fibronectin, Smad 2 and Smad 4, culture dishes or frozen sections were fixed in cold methanol for 10 min at –20 oC, blocked and permeabilized as previously described (29). After blocking with 1% BSA and 1% goat serum for 30 min, cells were incubated overnight with the following antibodies to α-SMA (1:100 dilution, DAKO, Carpintera, CA), CD34 (1:100, Santa Cruz), fibronectin (1:100, Sigma), Smad 2 (1:50, Santa Cruz, Temecula, CA) Smad 4 (1:50, Santa Cruz), and keratocan (1:50, rabbit antiserum against mouse keratocan N-terminal peptide) (VRQAYEIQDPEDWDVHDDFYC, Invitrogen) (27). This peptide was conjugated to a sulfolink® column (Pierce, Rockford, IL), which was then used to purify anti-mouse keratocan antibody according to the manufacturer’s instruction. The secondary antibodies (Sigma) used at 1:100 dilution were RITC-anti-rabbit, FITC-anti-mouse and FITC-anti-goat for immunofluorescence. ABC method (Vector Laboratories, Burlingame, CA) with DAB was applied to CD34 and fibronectin staining. Cell nuclei were counterstained with 10 μg/ml Hoechst 33342, propidium iodine or hematoxylin (all from Sigma).
Reverse Transcription-Polymerase Chain Reaction
Total RNAs were extracted by Trizol® reagent from cells cultured on plastic or AM and two murine corneas as a positive control. Total RNAs equivalent to 1 x 105 cultured cells or 2 corneas were subjected to RT-PCR as recommended by Promega. The final concentration of RT reaction was 10 mM Tris-HCl [pH 9.0], 5 mM MgCl2, 50 mM KCl, 0.1 % Triton X-100, 1 mM each dNTP, 1 unit/μl recombinant RNase in ribonucleases inhibitor, 15 units AMV reverse transcriptase, 0.5 μg Oligo(dT)15 primer and total RNA in a total volume of 20 μl. The reaction was kept at 42 °C for 60 min. One tenth sample from RT was used for subsequent PCR with the final concentration of PCR reaction being 10 mM Tris-HCl [pH 8.3], 50 mM KCl, 1.5 mM Mg(OAc)2, 1.25 units of Taq DNA polymerase in a total volume of 50 μl using primers shown in Table 1. The PCR mixture was first denatured at 94 °C for 5 min then amplified for 30 cycles (94 °C, 1 min; 60 °C, 1min; 72 °C, 1 min) using PTC-100 Programmable thermal Controller (MJ Research Inc, USA). After amplification, 15 μl of each PCR product and 3 μl of 6 X loading buffer were mixed and electrophoresed on a 1.5 % agarose gel in 0.5 X Tris-boric acid-EDTA (TBE) containing 0.5 μg/ml ethidium bromide. Gels were photographed and scanned.
Western Blot Analysis
Two normal murine corneas were minced with a blade as a positive control. Conditioned media were collected from passage 2 plastic and AM cultures. These samples and cells cultured on either plastic or AM were extracted from 2 dishes at 4 °C overnight with 1 ml of 4 M guanidine-HCl containing 10 mM sodium acetate, 10 mM sodium EDTA, 5 mM aminobenzamidine, and 0.1 M ɛ-amino-n-caproic acid. The extracts were dialyzed exhaustively overnight against distilled water and the water insoluble fraction was dissolved in 0.1 M Tris-acetate solution (pH 6.0) containing 6 M urea. The protein concentration was measured by spectrophotometer at 280 nm. One hundred μg protein aliquots were incubated with 0.1 U/ml endo-β-galactosidase (Seikagaku, Tokyo, Japan), or 0.1 U/ml keratanase II (Seikagaku, Tokyo, Japan) in PBS at 37 °C overnight. Each sample was added with an equal volume of 2 x SDS sample buffer, boiled for 5 min, electrophoresed on a 4 – 15% gradient SDS-PAGE gel, and transferred to a nitrocellulose membrane. These membranes were pre-incubated with the blocking buffer, probed with affinity-purified antiserum against mouse keratocan and alkaline phosphatase-conjugated goat anti-rabbit IgG (Pierce) as a secondary antibody, and visualized with Western Blue® (Promega).
RESULTS
In Vivo Morphology and Keratocan Expression of Murine Keratocytes
In CD-1 albino murine globes, there is a visible boundary that demarcates the limbus between the cornea and the sclera (Fig. 1A). Such a demarcation facilitated the surgical isolation of the corneal stroma. Following the removal of an intact sheet of the ocular surface epithelium from the globe by dispase II (22), the corneal stroma was dissected from the adjacent sclera (Fig. 1B). Live/Dead Assay® revealed that an overwhelming majority of keratocytes were viable (stained green) and exhibited a 3–dimensional dendritic morphology in the corneal stroma (Fig. 1C). Some dead cells (stained red) were found in the cut edge of the excised stroma (Fig. 1C, inset, marked by arrowheads). Keratocytes in the stroma expressed keratocan as evidenced by positive staining with an affinity-purified antibody against mouse keratocan peptide (Fig. 1D). In contrast, corneal epithelial cells or endothelial cells were not stained (Fig. 1D, inset). These results indicated that in vivo murine keratocytes also exhibited a characteristic dendritic morphology and specifically expressed keratocan.
Fig. 1.
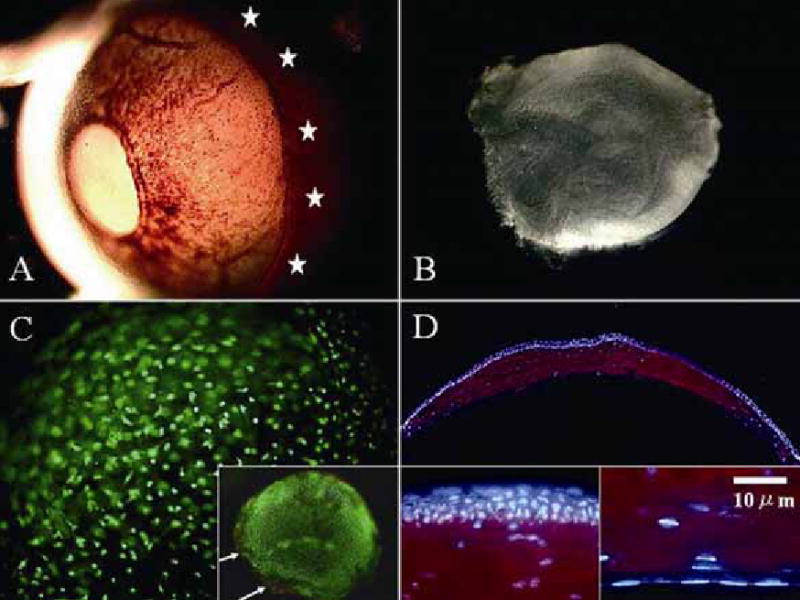
In vivo keratocyte morphology and keratocan expression. An enucleated CD-1 albino mouse eye shows a distinguishable limbal region under transillumination (A, marked by stars). The corneal stroma is dissected from the adjacent sclera (see Methods) (B). Under high magnification, Live/Dead assay® shows a dendritic morphology of all viable (stained green) keratocytes distributed in a three dimension manner (more visible on the plane in focus) (C). Dead cells (stained red) are found only in the excised stromal edge (C, inset, white arrows). Immunostaining demonstrates keratocan expression exclusively in the corneal stroma (D). Corneal epithelial cells and endothelial cells do not express keratocan (D, inset).
In Vitro Morphology and Proliferation of Keratocytes Cultured on AM
Keratocytes were then isolated from the corneal stroma by collagenase digestion. An average of 5,000 cells was obtained per mouse cornea. Cells at a density of 3,000 per cm2 were seeded on either plastic or AM in DMEM/10% FBS. Within 12 h after seeding, cells attached on either substrate, and exhibited a distinctly different morphology. Cells on plastic dishes were evenly distributed on the flat surface and adopted a spindle-shaped morphology with a broad stellate cytoplasm (Fig. 2A and 2B). They became confluent in 4 to 5 days. In contrast, cells on AM were dendritic or satellite in shape and had a triangular cell body and a scanty cytoplasm which formed extensive intercellular networks, and projected their dendritic processes in a 3 -dimensional manner (Fig. 2D and 2E). They became confluent in 10 days.
Fig. 2.
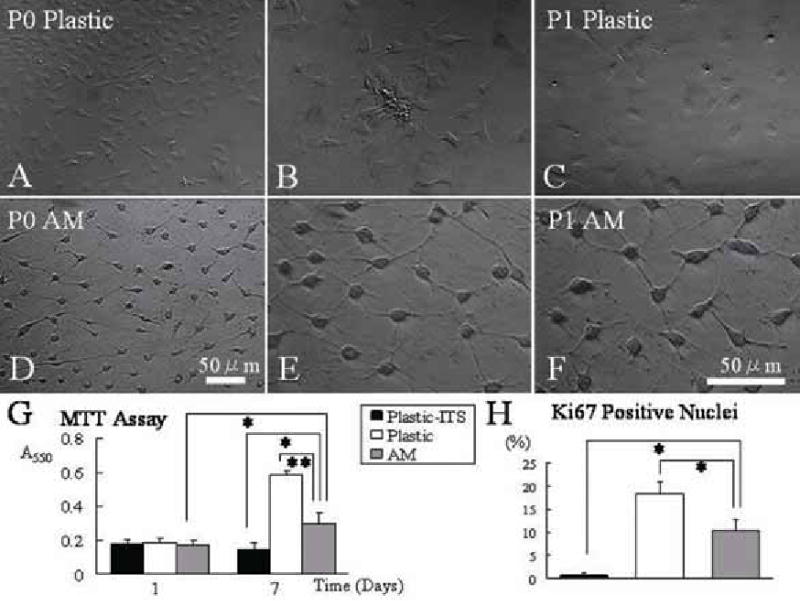
Cellular morphology and proliferation on plastic and AM cultures. Passage 0 (P0) cells expanded on plastic are spindle-shaped with a broad satellite cytoplasm (A and B), and become more flattened after subcultured to P1 (C). In contrast, cells on AM maintain a triangular shaped cell body and a scanty cytoplasm with dendritic processes (D and E) even after subcultured to P1 (F). Extensive intercellular cell-cell contacts are evident in cells expanded on AM. Micrograph A and D are taken at same magnification and B to F at higher magnification. Both MTT assay (G) and quantitation of Ki67 nuclear staining (H) show that cells on AM in DMEM/10% FBS had a higher proliferative activity than cells on plastic in DMEM/ITS, but less than cells on plastic in DMEM/10% FBS (*p<0.05 and **p<0.01, for both G and H).
Upon reaching 80–90% confluence, cells expanded on AM or plastic were trypsinized and continuously passaged onto the same type of substrate as used in the primary culture (P0). Cells subcultured on plastic at P1 became more flattened (Fig. 2C). In contrast, cells expanded on AM subcultures still maintained a dendritic morphology with pronounced intercellular contacts (Fig. 2F). They continuously preserved such a dendritic morphology until passage 8, when cells became senescent (not shown). Using MTT assay, cells of P2 plastic cultures in DMEM/ITS did not show an increase of cell number during the one week of culturing, while cells on plastic in DMEM/10% FBS rapidly expanded in number (Fig. 2G). Cells cultured on AM in DMEM/10% FBS were intermediate between that of the above two conditions (Fig. 2G, p<0.05 cf. DMEM/ITS and p<0.01 cf. DMEM/10% FBS). At day 7, the number of Ki67 positive nuclei in cells cultured on AM in DMEM/10% FBS was significantly more than that of cells on plastic in DMEM/ITS, but less than cells cultured on plastic in DMEM/10% FBS (Fig. 2H, both p<0.01). Collectively, these results confirmed that cells cultured on AM continued to proliferate and maintained a dendritic morphology in a FBS-containing medium.
Phenotypic Characterization of Cells Expanded on AM
To confirm that dendritic cells expanded on AM were indeed keratocytes and not myofibroblasts, immunostaining was performed for the expression of keratocan and α-SMA, respectively. For primary cultures (P0), a majority of dendritic cells cultured on plastic in DMEM/ITS for 5 days (Fig. 3A) expressed keratocan (Fig. 3B) but not α-SMA (Fig. 3C). In contrast, cells cultured on plastic in DMEM/10% FBS were not dendritic (Fig. 3D) and did not express keratocan (Fig. 3E); instead some cells expressed α-SMA (Fig. 3F). However, dendritic cells expanded on AM in DMEM/10% FBS (Fig. 3G) maintained keratocan expression (Fig. 3H), and did not express α-SMA (Fig. 3I). In DMEM/10% FBS, CD34 was not expressed by cells cultured on plastic (Fig 4A), but expressed by cells cultured on AM (Fig. 4C). In contrast, fibronectin was expressed extracellularly and intracellularly by cells cultured on plastic (Fig. 4B), but not expressed by cells cultured on AM (Fig. 4D). These results collectively indicated that the keratocyte phenotype was maintained by AM.
Fig. 3.
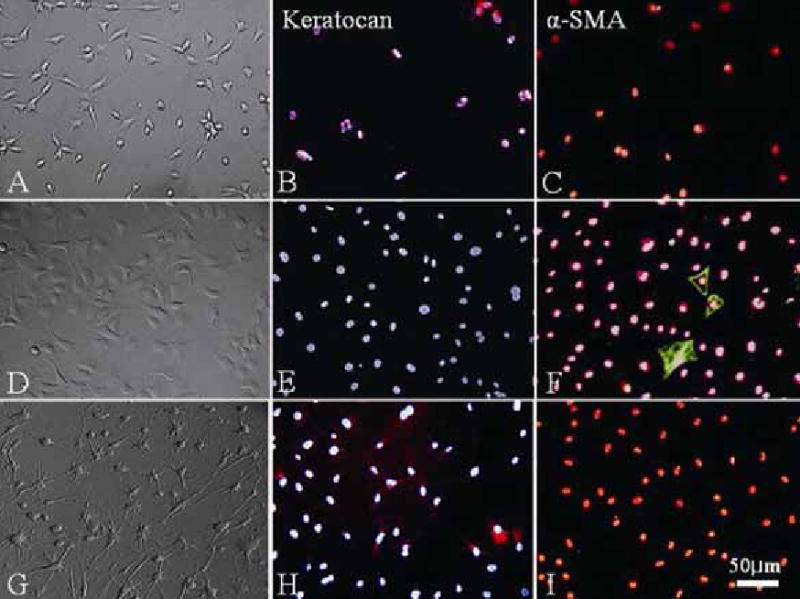
Keratocan and α-SMA expression. On plastic, cells of primary cultures (P0) in serum-free DMEM/ITS have a dendritic morphology (A) and expressed keratocan (B), but not α-SMA (C). Cells cultured in DMEM/10% FBS expand in number (D) but completely lose keratocan expression (E), while some cells express α-SMA (F). In contrast, P2 cells expanded on AM in DMEM/10% FBS continue to be dendritic, form cell-cell networks (G) similar to P0 cultures, and express keratocan (H), but not α-SMA (I).
Fig. 4.
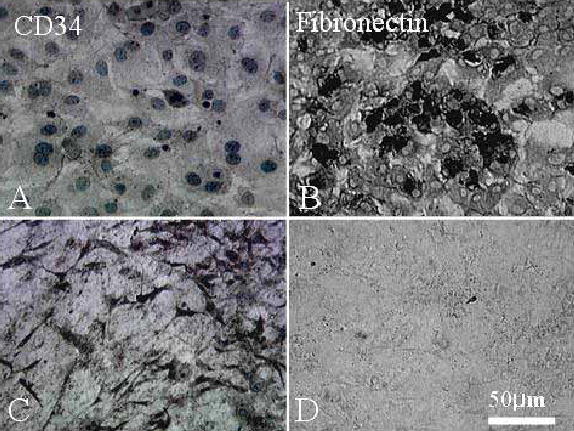
CD34 and fibronectin expression. CD34 is not expressed (A), but fibronectin is expressed intracellularly and extracellularly (B) by P2 cells cultured on plastic. In contrast, CD34 is maintained (C) while fibronectin is not expressed (D) by P2 cells cultured on AM.
To confirm transcript expression of keratocan, total RNAs were extracted from cells on plastic and AM, and subjected to RT-PCR. The result showed that keratocan transcript (of the size of 1065 bp) was expressed by cells cultured on plastic at passage 0, but lost at passage 1 (Fig. 5A) and thereafter (not shown). In contrast, the keratocan transcript was continuously expressed in an abundant amount from passage 0 to passage 3 (Fig. 5A) and up to passage 8 (not shown) when cultured on AM.
Fig. 5.
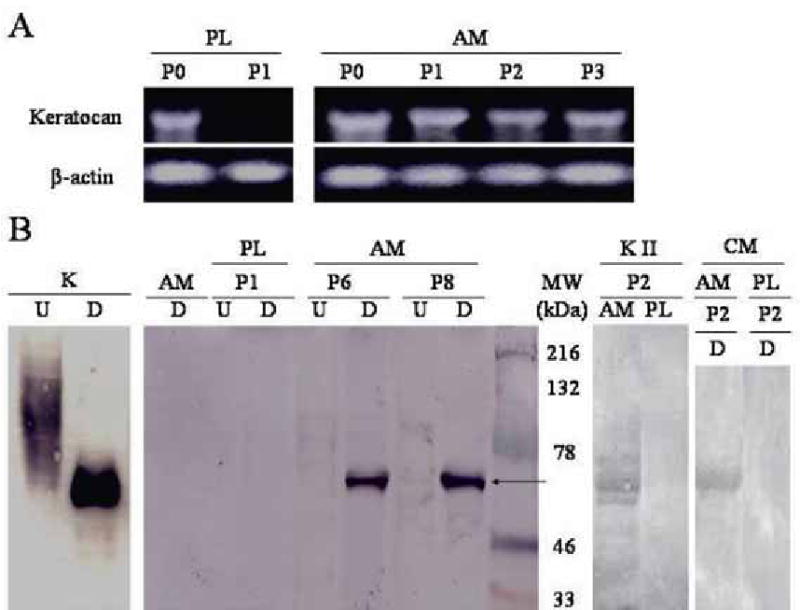
Expression of keratocan transcript and proteins. (A) Expression of keratocan transcript (size of 1065bp) is maintained in AM cultures from passage 0 (P0) to passage 3 (P3). In contrast, such expression is maintained on plastic at P0, but lost at P1. β -actin was used as a loading control. (B) Cornea stroma extracted with 4 M guanidine-HCl from the normal corneal stroma (K), cells cultured on AM at P6 and P8 (designated as A6 and A8, respectively), and cell cultured on plastic at P1 (designated as P1) are treated with (D) or without (U) endo-β-galactosidase to remove keratan sulfate, and subjected to Western blot using an antibody to murine keratocan. The undigested samples of the normal cornea and A6 and A8 show high MW smearing, while the digested counterparts reveal a 50 kD band of keratocan (arrow). A similar 50 kDa band was obtained from P2 cultures on AM, but not from P2 cultures on plastic by keratanase II digestion (KII, P2, AM and PL). AM alone without cells (AM) does not express keratocan. The digested sample of the conditioned media of P2 AM cultures, but not those of P2 plastic cultures, also shows a 50 kD band.
To verify the keratocan protein expression, insoluble matrix proteins were extracted by 4 M guanidine HCl and subjected to Western blot analysis using an antibody against the core protein of keratocan. The sample from the normal murine corneal stroma, which was used as the positive control, showed a dense smearing in the high molecular weight region (Fig. 5B, K-U). Nevertheless, the same sample after digestion with endo-β-galactosidase showed a positive protein band of ~50 kDa (Fig. 5B, K–D). The undigested sample of AM cultures at passages 6 and 8 showed a similar faint smearing in the same high molecular weight region (AM P6 and P8-U, respectively). Both samples after digestion with endo-β-galactosidase showed a strong positive protein band of 50 kDa (AM P6 and P8, D, AM, respectively). A similar 50 kDa band was obtained from P2 cultures on AM, but not from P2 cultures on plastic using digestion by keratanase II (KII, P2, AM and PL). In contrast, there was no smearing in the undigested sample, nor the protein band detected after digestion in plastic cultures at passage 1 (PL P1-U and D) and thereafter (not shown). The negative control of pure AM extract alone without any cultured cells did not contain any keratocan without (not shown) or with endo-β-galactosidase digestion (AM, D). Because keratocan expression was strongly observed in the extracellular matrix of in vivo murine corneas, conditioned media from P2 murine keratocyte cultures was also examined for keratocan expression. The result showed that the digested samples of the conditioned medium from AM culture, but not plastic cultures, showed a 50 kD band (Fig. 4B). Collectively, these data indicated that in DMEM/10% FBS cells expanded on AM, but not plastic, expressed keratocan in the matrix and the conditioned medium.
Transient and Sustained Suppression of Smad-dependent TGF-β Signaling in Keratocytes Cultured on AM
Because FBS contains TGF-β1 (30) and TGF-β signaling is involved in promoting myofibroblast differentiation (9–11;18), we speculated that suppression of TGF-β signaling might also be involved in the maintenance of the normal keratocyte phenotype. We added 10 ng/ml TGF-β1 to both plastic and AM cultures containing DMEM/ITS and examined the Smad-mediated TGF-β signaling by immunolocalization of Smad 2 and Smad 4. As shown in Figure 6, cells on AM or plastic responded differently to exogenous addition of TGF-β1. Nuclear localization of Smad 4 was found 16% of cells on plastic without TGF-β1, but increased to 67% and 85% of cells cultured on plastic after 3 h and 5 days of TGF-β1 challenge, respectively (Fig. 6A). A similar trend was noted for nuclear localization of Smad 2 (Fig. 6A). In contrast, no cell cultured on AM showed nuclear localization of Smad 2 and Smad 4 at 3 h and 5 days of TGF-β1 challenge (Fig. 6A). These results indicated that TGF-β signaling mediated by Smad was continuously suppressed in cells cultured on AM even in the presence of 10% FBS.
Fig. 6.
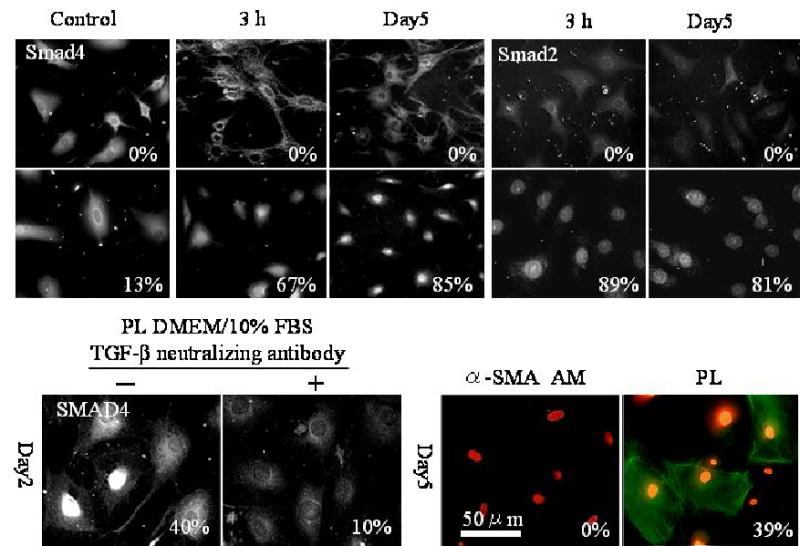
Suppression of Smad-mediated TGF-β signaling by AM. (A) Immunostaining shows that Smad 4 nuclear translocation takes place only in a small percentage of cells (13%) cultured on plastic in DMEM/ITS, but increased to 67% and 85% after 10 ng/ml TGF-β1 is added for 3 h. and 5 days, respectively. A similar trend is noted for Smad 2 nuclear localization. (B) Nuclear localization of Smad 4 is noted in 40% of cells on plastic in DMEM/10% FBS, but in 10% of cells when TGF-β neutralizing antibody is added for two days. (C) Expression of α-SMA is noted in 39% of cells cultured on plastic after 10 ng/ml TGF-β1 for 5 days, but is negative in cells cultured on AM.
To confirm that TGF-β was indeed responsible for Smad signaling in DMEM/10%FBS, we added a neutralizing antibody to three TGF-β isoforms in the plastic cultures, and noted that nuclear translocation of Smad 4was indeed prevented (Fig. 6B). To demonstrate whether nuclear localization of Smad 4 also correlated with downstream of TGF-β signaling, we quantified α-SMA expression in parallel. Thirty-nine percent of cells cultured on plastic differentiated into α-SMA-expressing myofibroblasts, but no cell on AM expressed α-SMA even after 5 days of continuous stimulation with TGF-β1 (Fig. 6C). Taken together, these results demonstrated that Smad-mediated TGF-β signaling was inhibited in cells cultured on AM and suppression of Smad-mediated TGF-β signaling correlated with prevention of cells from differentiating into myofibroblasts.
Inhibition of TGF-β2 and TGF-βRII Transcriptional Activity in Keratocytes Cultured on AM
To determine whether the aforementioned downregulation of TGF-β signaling was mediated by suppressing TGF-β genes at the transcriptional level, TGF-β2 and TGFβ-RII promoter activities were evaluated by transient transfection. As compared to cells cultured on plastic and adjusted by background transfection with CMV-β Gal, the promoter activity of TGF-β2 and TGF-β RII was decreased 4.1 folds and 2.6 folds, respectively, in cells cultured on AM (Fig. 7, both p<0.001). These data supported the notion that downregulation of TGF-β signaling was indeed mediated by suppressing TGF-β2 and TGFβ-RII genes at the transcriptional level in cells expanded on AM.
Fig. 7.
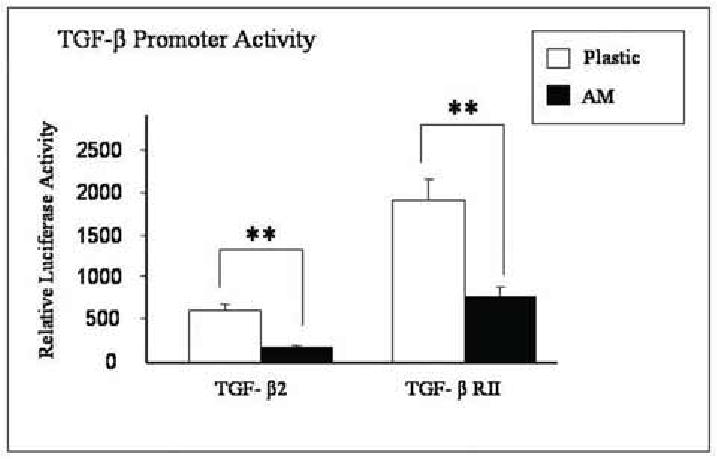
Suppression of TGF-β promoter activity by AM. Mouse corneal fibroblasts (P1) cultured on plastic and AM are cotransfected with TGF-β2 promoter-luc plus CMV-β-galactosidase or TGF-βRII promoter–luc plus CMV-β-galactosidase for 48 hours. Cell extracts are assayed for both activities of luciferase and β-galactosidase. The relative luciferase unit of the promoter activity of TGF-β2 and TGF-βRII is suppressed in cells cultured on AM (** p < 0.01).
Suppression of TGF-β Signaling Maintained Keratocan Expression
To demonstrate a direct link between downregulation of TGF-β signaling and keratocan expression, 50 multiplicity of infectivity (M.O.I.) of Aden-track-Kerapr3.2-intron-ECFP/BpA was added to cells cultured on plastic in either DMEM/ITS or DMEM/10% FBS, of which the latter was further treated with or without an antibody to neutralize all three TGF-β isoforms. Transfection efficiency was revealed by EGFP (green fluorescence) driven by CMV in the same construct, while expression of keratocan promoter was revealed by ECFP (blue fluorescence) in the same cell. Cells retained the dendritic morphology after transfection in the positive control cultured on plastic in DMEM/ITS (Fig. 8A) or on AM in DMEM/10% FBS (Fig. 8B). Cells also maintained a flattened bipolar morphology in the negative control cultured on plastic in DMEM/10% FBS (Fig. 8C). These results indicated that transfection itself did not alter their respective characteristic cell morphology. Interestingly, the fibroblastic morphology did not revert to a dendritic morphology after the addition of TGF-β neutralizing antibody for 2 days (Fig. 8D). The overall transfection efficiency was more than 80% in these experiments (Fig. 8I). Under such a high transfection rate, keratocan promoter-driven ECFP expression was observed in 30 – 40% of cells cultured on plastic in DMEM/ITS (Fig. 8E), 15 – 20% of cells cultured on AM in DMEM/10% FBS (Fig. 8F), but less than 2% of cells cultured on plastic in DMEM/10% FBS (Fig. 8G). These results corroborated with the aforementioned pattern of keratocan transcript and protein expression in these three cultures (Fig. 5). ECFP expression was restored to 10 –15% of cells cultured on plastic in DMEM/10% FBS when TGF-β neutralizing antibody was added (Fig. 8H). Collectively, these results indicated that TGF-β in 10% FBS was indeed responsible for the suppression of keratocan expression for cells cultured on plastic, and that keratocan expression by cells cultures on AM in 10% FBS was correlated with suppression of Smad-mediated TGF-β signaling.
Fig. 8.
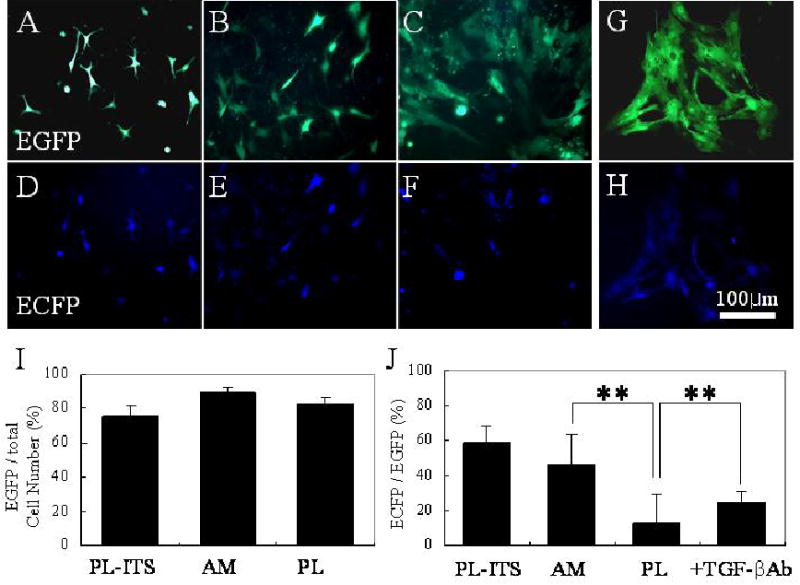
Keratocan promoter activity. Aden-track-Kerapr3.2-intron-ECFP/BpA adenovirus (50 M.O.I.) is used to transfect primary cultures in DMEM/ITS on plastic (A), DMEM/10% FBS on AM (B), and DMEM/10% FBS on plastic without (C) or with TGF-β neutralizing antibody (D). Transfection efficiency determined by EGFP expression (green fluorescence) is more than 80 % in these experiements (I). On the same field, cells expressing EGFP was then analyzed for keratocan promoter activity by monitoring ECFP expression (blue fluorescence). Keratocan expression is noted in 59 ± 9.5 % cells cultured on plastic in DMEM/ITS (E) and 46 ± 6.2 % of cells on AM cultures in DMEM/10% FBS (F), and is significantly reduced in cells cultured on plastic in DMEM/10% FBS (13 ± 2.6%, G), (**p<0.01, J), but is partially restored to 25 ± 5.6 % (**p<0.01, J) when added with a TGF-β neutralizing antibody (H).
DISCUSSION
In this report, we have demonstrated that murine keratocytes expanded in an FBS-containing medium preserved a native 3-dimensional dendritic morphology with extensive intercellular contacts, and continuously expressed keratocan and CD34, but not fibronectin and α-SMA, for up to 8 passages so long as they were cultured on AM stromal matrix (Fig. 2–4). These findings are in agreement with what we have reported in human corneal keratocytes (3;4). Previously, it has been noted that other small leucine rich proteoglycans are maintained or upregulated in plastic cultures of murine fibroblasts at late passages (12;13). We noted herein that, expression of keratocan transcript and protein was lost by cells cultured on plastic in the presence of 10% FBS as early as passage 1, but continuously maintained by cells on AM for up to passage 8 (at 1:2 split) before senescence (Fig. 5). Furthermore, based on the digestability with endo-β-galactosidase and keratanase II, keratocan expressed by these keratocytes expanded on AM was glycosylated with keratan sulfate, secreted into the conditioned medium, and deposited in the extracellular matrix (Fig. 5). This feature is known to be unique to keratocytes in the corneal stroma (31). In the presence of 10% FBS, cells cultured on AM were indeed proliferating to an extent less than their counterparts cultured on plastic (Fig. 2). Therefore, we conclude that the phenotype of murine keratocytes, defined by a dendritic morphology, positive expression of keratan sulfate-containing keratocan, and CD34, and negative expression of fibronectin and α-SMA, is maintained on AM even when cells are stimulated to proliferate by 10% FBS.
We speculate that TGF-β is a factor in FBS that is responsible for the loss of the normal keratocyte phenotype when cells are cultured on plastic dishes. TGF-β is well known for its action in stimulating fibroblasts to differentiate into myofibroblasts (32;33). Nevertheless, it remains unknown whether suppression of TGF-β signaling, which can prevent myofibroblast differentiation, is also essential for maintaining the normal keratocyte phenotype. Although an earlier study showed that addition of TGF-β1 in DMEM supplemented with 1% platelet-poor horse serum (deficient in TGF-β1) decreases the expression of keratan sulfate proteoglycans at the transcript and protein levels in bovine cells (9;10), expression of keratocan was not defined in these two studies. In the present study, we noted that the keratocyte phenotype correlated not only with negative expression of α-SMA and fibronectin expression, i.e., myofibroblast differentiation (Fig. 3 and 4), but also with suppression of Smad-mediated TGF-β signaling (Fig. 6). It is known that Smad-mediated TGF-β signaling, judged by nuclear translocation of Smad 2 and Smad 4, takes place in less than an hour, and maximizes in 3–4 hours.(34) However, subsequent interplay and crosstalk between Smad-mediated signaling and other signaling pathways can modify the final outcome of transactivation of targeted genes.[for review see (32;33)] That was why we examined nuclear localization of Smad 2 and Smad 4 not only at 3 h but also at 5 days to make sure that crosstalks and interplays with other signaling pathways would not deter Smad signaling. Our results showed that following exognenous addition of 10 ng/ml TGF-β 1 in DMEM/IT’S, the proportion of cells exhibiting nuclear translocation of Smad 2 and Smad 4 on plastic cultures continuously increased from 3 h to 5 days presumably due to TGF-β 1 autoinduction(35), eventually resulting in expression of α-SMA in 5 days (Fig. 6). Chronic nuclear localization of Smads ensures expression of Smad-targeted genes including α-SMA(36;37). Indeed, such chronic nuclear location of Smad 2 and Smad 4 correlated with eventual expression of α-SMA gene in 5 days (Fig. 6) and upregulation of TGF-β 2 and TGF-β RII promoter activities (Fig. 7). On the contrary, if TGF-β signaling is intercepted or modified by other signaling pathways, the nuclear translocation of Smad will be transient. [for review see (32;33)] In this study, however, we showed that nuclear exclusion of Smad 2 and Smad 4 occurred at both 3 h and 5 days in cells cultured on AM, eventually leading to suppression of α-SMA (Fig. 6). Furthermore, by interceding Smad-mediated signaling to the nucleus, subsequent transcription of TGF-β 2 and TGF-β RII was also aborted as evidenced by their promoter activities (Fig. 7). These results strongly support the notion that suppression of Smad-mediated TGF-β signaling by AM is potent, complete and sustained for a prolonged period of time despite exogenous challenge of TGF-β 1. They also explain why clinical transplantation of AM is effective in reducing scar formation during ocular surface reconstruction in ophthalmology [for reviews see (38–40)].
To draw a causative link between suppression of Smad-mediated TGF-β signaling and keratocan expression, we added TGF-β neutralizing antibody to the plastic cultures in DMEM containing 10% FBS. Such a maneuver effectively blocked Smad 4 nuclear translocation (Fig. 6B), and at the same time upregulated keratocan promoter activity (Fig. 8). Collectively, these findings further support that suppression of Smad-mediated TGF-β signaling is pivotal not only in preventing myofibroblast differentiation, but also in sustaining keratocan-expressing keratocyte phenotype. Deriving from such a strategy of suppressing TGF-β signaling, we have developed a serum-free medium to promote keratocyte proliferation while maintaining their normal phenotype (manuscript in preparation). Furthermore, our preliminary studies showed that such suppression of TGF-β signaling was retained in soluble AM extracts (data not shown), suggesting that this action does not require cell-matrix contact. Recently we reported that soluble non-glycosylated lumican isolated from AM promotes mouse corneal wound healing{Yeh L-K, 2005 5386 /id. Investigation is in progress to identify the exact molecular component(s) in AM soluble extracts that is responsible for downregulating TGF-β signaling so that we might develop new therapeutics in combating scar formation in the future.
Footnotes
This work were Supported by a research grant from TissueTech, Inc. and in part by an unrestricted grant from Ocular Surface Research & Education Foundation, Miami, FL and National Institutes of Health Grant EY06819 (to SCGT) and EY12486 (to C.-Y. Liu).
The abbreviations used are: α-SMA, α-smooth muscle actin; FBS, fetal bovine serum; AM, human amniotic membrane; TGF-β, transforming growth factor beta.
References
- 1.Beals MP, Funderburgh JL, Jester JV, Hassell JR. Invest Ophthalmol Vis Sci. 1999;40:1658–1663. [PubMed] [Google Scholar]
- 2.Jester JV, Barry-Lane PA, Cavanagh HD, Petroll WM. Cornea. 1996;15:505–516. [PubMed] [Google Scholar]
- 3.Espana EM, He H, Kawakita T, Di Pascuale MA, Raju VK, Liu CY, Tseng SC. Invest Ophthalmol Vis Sci. 2003;44:5136–5141. doi: 10.1167/iovs.03-0484. [DOI] [PubMed] [Google Scholar]
- 4.Espana EM, Kawakita T, Liu CY, Tseng SCG. Invest Ophthalmol Vis Sci. 2004;45:2985–2991. doi: 10.1167/iovs.04-0201. [DOI] [PubMed] [Google Scholar]
- 5.Dahl IM, Johnsen W, Anseth A, Prydz H. Exp Cell Res. 1974;88:193–197. doi: 10.1016/0014-4827(74)90634-x. [DOI] [PubMed] [Google Scholar]
- 6.Dahl IM, Coster L. Exp Eye Res. 1978;27:175–190. doi: 10.1016/0014-4835(78)90087-8. [DOI] [PubMed] [Google Scholar]
- 7.Dahl IM. Exp Eye Res. 1981;32:419–433. doi: 10.1016/s0014-4835(81)80021-8. [DOI] [PubMed] [Google Scholar]
- 8.Hassell JR, Schrecengost PK, Rada JA, SundarRaj N, Sossi G, Thoft RA. Invest Ophthalmol Vis Sci. 1992;33:547–557. [PubMed] [Google Scholar]
- 9.Funderburgh JL, Mann MM, Funderburgh ML. J Biol Chem. 2003;278:45629–45637. doi: 10.1074/jbc.M303292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Funderburgh JL, Funderburgh ML, Mann MM, Corpuz L, Roth MR. J Biol Chem. 2001;276:44173–44178. doi: 10.1074/jbc.M107596200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petridou S, Masur SK. Invest Ophthalmol Vis Sci. 1996;37:1740–1748. [PubMed] [Google Scholar]
- 12.Liu C, Arar H, Kao C, Kao WW. Gene. 2000;250:85–96. doi: 10.1016/s0378-1119(00)00165-7. [DOI] [PubMed] [Google Scholar]
- 13.Gendron RL, Liu CY, Paradis H, Adams LC, Kao WW. Mol Vis. 2001;7:107–113. [PubMed] [Google Scholar]
- 14.Jester JV, Petroll WM, Cavanagh HD. Prog Retin Eye Res. 1999;18:311–356. doi: 10.1016/s1350-9462(98)00021-4. [DOI] [PubMed] [Google Scholar]
- 15.Saika S, Saika S, Liu CY, Azhar M, Sanford LP, Doetschman T, Gendron RL, Kao CW, Kao WW. Dev Biol. 2001;240:419–432. doi: 10.1006/dbio.2001.0480. [DOI] [PubMed] [Google Scholar]
- 16.Attisano L, Wrana JL. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 17.Shi Y, Massague J. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 18.Jester JV, Ho-Chang J. Exp Eye Res. 2003;77:581–592. doi: 10.1016/s0014-4835(03)00188-x. [DOI] [PubMed] [Google Scholar]
- 19.Tseng SC, Li DQ, Ma X. J Cell Physiol. 1999;179:325–335. doi: 10.1002/(SICI)1097-4652(199906)179:3<325::AID-JCP10>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 20.Lee SB, Li DQ, Tan DTH, Meller D, Tseng SCG. Curr Eye Res. 2000;20:325–334. [PubMed] [Google Scholar]
- 21.Espana EM, Romano AC, Kawakita T, Di Pascuale M, Smiddy R, Tseng SC. Invest Ophthalmol Vis Sci. 2003;44:4275–4281. doi: 10.1167/iovs.03-0089. [DOI] [PubMed] [Google Scholar]
- 22.Kawakita T, Espana EM, He H, Yeh LK, Liu CY, Tseng SC. Invest Ophthalmol Vis Sci. 2004;45:3507–3512. doi: 10.1167/iovs.04-0266. [DOI] [PubMed] [Google Scholar]
- 23.Meller D, Pires RTF, Tseng SCG. Br J Ophthalmol. 2002;86:463–471. doi: 10.1136/bjo.86.4.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poole CA, Brookes NH, Clover GM. J Cell Sci. 1993;106:685–692. doi: 10.1242/jcs.106.2.685. [DOI] [PubMed] [Google Scholar]
- 25.Noma T, Glick AB, Geiser AG, O'Reilly MA, Miller J, Roberts AB, Sporn MB. Growth Factors. 1991;4:247–255. doi: 10.3109/08977199109043910. [DOI] [PubMed] [Google Scholar]
- 26.Bae HW, Geiser AG, Kim DH, Chung MT, Burmester JK, Sporn MB, Roberts AB, Kim SJ. J Biol Chem. 1995;270:29460–29468. doi: 10.1074/jbc.270.49.29460. [DOI] [PubMed] [Google Scholar]
- 27.Chen J, Huber BT, Grand RJ, Li W. J Immunol. 2001;167:1297–1305. doi: 10.4049/jimmunol.167.3.1297. [DOI] [PubMed] [Google Scholar]
- 28.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grueterich M, Espana E, Tseng SC. Invest Ophthalmol Vis Sci. 2002;43:63–71. [PubMed] [Google Scholar]
- 30.Kropf J, Schurek JO, Wollner A, Gressner AM. Clin Chem. 1997;43:1965–1974. [PubMed] [Google Scholar]
- 31.Liu CY, Siraishi A, Kao CWC, Converse RL, Funderburgh JL, Corpuz LM, Conrad GW, Kao WWY. J Biol Chem. 1998;273:22584–22588. doi: 10.1074/jbc.273.35.22584. [DOI] [PubMed] [Google Scholar]
- 32.Massague J, Wotton D. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Derynck R, Zhang YE. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 34.Inman GJ, Nicolas FJ, Hill CS. Mol Cell. 2002;10:283–294. doi: 10.1016/s1097-2765(02)00585-3. [DOI] [PubMed] [Google Scholar]
- 35.Ghahary A, Shen Q, Shen YJ, Scott PG, Tredget EE. J Cell Physiol. 1998;174:301–309. doi: 10.1002/(SICI)1097-4652(199803)174:3<301::AID-JCP4>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 36.Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A. Mol Biol Cell. 2005;16:1987–2002. doi: 10.1091/mbc.E04-08-0658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lan HY, Mu W, Tomita N, Huang XR, Li JH, Zhu HJ, Morishita R, Johnson RJ. J Am Soc Nephrol. 2003;14:1535–1548. doi: 10.1097/01.asn.0000067632.04658.b8. [DOI] [PubMed] [Google Scholar]
- 38.Tseng SCG. Bioscience Rep. 2002;21:481–489. doi: 10.1023/a:1017995810755. [DOI] [PubMed] [Google Scholar]
- 39.Sippel KC, Ma JJK, Foster CS. Curr Opin Ophthalmol. 2001;12:269–281. doi: 10.1097/00055735-200108000-00006. [DOI] [PubMed] [Google Scholar]
- 40.Dua HS, Gomes JA, King AJ, Maharajan VS. Surv Ophthalmol. 2004;49:51–77. doi: 10.1016/j.survophthal.2003.10.004. [DOI] [PubMed] [Google Scholar]