

Abstract
Photolyase uses light energy to split UV-induced cyclobutane dimers in damaged DNA, but its molecular mechanism has never been directly revealed. Here, we report the direct mapping of catalytic processes through femtosecond synchronization of the enzymatic dynamics with the repair function. We observed direct electron transfer from the excited flavin cofactor to the dimer in 170 ps and back electron transfer from the repaired thymines in 560 ps. Both reactions are strongly modulated by active-site solvation to achieve maximum repair efficiency. These results show that the photocycle of DNA repair by photolyase is through a radical mechanism and completed on subnanosecond time scale at the dynamic active site, with no net change in the redox state of the flavin cofactor.
Keywords: photocycle, radical mechanism, ultrafast kinetics
One of the detrimental effects of UV radiation on the biosphere is the formation of cyclobutane pyrimidine dimers (Pyr<>Pyr) between two adjacent thymine bases in DNA (1). Pyr dimers bring both DNA and RNA polymerases to a standstill and may result in mutation or cell death. Photolyase (EC 4.1.99.3), which is a photoenzyme that exists in all three branches of life, harnesses blue or near-UV light energy to cleave the cyclobutane ring of the Pyr<>Pyr and, thus, prevents the harmful effects of UV radiation (2, 3).
Photolyase is a flavoprotein and contains two noncovalently bound chromophores. One chromophore is the fully reduced flavin–adenine dinucleotide (FADH–), the catalytic cofactor that carries out the repair function upon excitation by either direct photon absorption or resonance energy transfer from the second chromophore, which is an antenna pigment (methenyltetrahydrofolate or deazaflavin) that harvests sunlight and enhances repair efficiency. The model for the catalytic reaction (3, 4) proposes that the excited flavin cofactor transfers an electron to the Pyr<>Pyr to generate a charge-separated radical pair (FADH· + Pyr<>Pyr·–). The anionic ring of the dimer is split by a [2 + 2] cycloreversion, and the excess electron returns to the flavin radical to restore the catalytically competent FADH– form and close the catalytic photocycle (Fig. 1A). This hypothetical radical mechanism has not been directly proven [although it was proposed ≈20 years ago (5) and supported by extensive biochemical data (6, 7), spectroscopic studies (8–10), and computer modeling (11, 12), as well as recent structural determination (13, 14)], and the radical intermediates have not yet been captured. Here, we report our direct mapping of the repair processes by following the temporal evolution of reactants and intermediate states, and we uncover the complete dynamics of the catalytic photocycle.
Fig. 1.

Repair of damaged DNA by photolyase. (A) Schematic representation of DNA-repair processes by photolyase through an electron-transfer radical mechanism. The key catalytic reactions, charge separation (k1) and ring splitting (k2), are given at the bottom. Our study follows the evolution of the flavin cofactor. (B) The steady-state repair of dinucleotide thymine dimer (200 μM) by photolyase (10 μM) by illumination of the reaction mixture under 360-nm light. The thymine monomer formation was detected by increase in absorption at 260 nm.
Experimental Methods
Photolyase and Dimer Substrates. We used Escherichia coli photolyase depleted of the antenna cofactor (EPL-FADH–) as the repair enzyme and cyclobutane thymine dimer (T<>T) in a dinucleotide, oligonucleotide, or polynucleotide as the substrate. E. coli photolyase was prepared as described in ref. 15. The photoantenna molecule methenyltetrahydrofolate was removed during purification by photodecomposition (16). For all femtosecond-resolved experiments, a concentration of 400 μM was used in reaction buffer, containing 50 mM Tris (pH 7.4), 50 mM NaCl, 1 mM EDTA, 10 mM DTT, and 50% (vol/vol) glycerol. The flavin cofactor in all purified samples contains the neutral radical FADH· form. To reduce the flavin cofactor to the catalytic state FADH–, the sample was purged with high-purity nitrogen to remove oxygen and then illuminated with a high-intensity lamp (150 W) under anaerobic conditions, with a cutoff filter at 550 nm to ensure that the sample was exposed at wavelengths of >550 nm. The resulting fully reduced enzyme does not absorb significantly at wavelengths of >500 nm. Substrates of dinucleotide cyclobutane thymine dimer and dimer-containing oligo(dT)12–18 and poly(dT) were prepared by acetone-sensitized irradiation with a UVB lamp (8 W) according to standard procedures (17). The mixture of the fully reduced enzyme with substrates was prepared under yellow light and anaerobic conditions, and it had no measurable absorption at >500 nm. The equilibrium binding constants of E. coli photolyase for T<>T in DNA or polymer substrates are typically 107 to 108 and 103 to 104 M–1 for dinucleotide dimers (18). The steady-state studies readily detected the repaired thymine formation after exposure of the enzyme complexes to visible light (Fig. 1B).
Femtosecond Methods. All experimental measurements were carried out by using the femtosecond-resolved fluorescence up-conversion and transient-absorption techniques, as described in ref. 19. Briefly, the femtosecond pulse after the two-stage amplifier (Spitfire, Spectra-Physics) has a temporal width of 110 fs, with an energy of >2 mJ and a repetition rate of 1 kHz. The laser beam is then split into two equal parts to pump two optical parametric amplifiers (OPA-800C, Spectra-Physics). We used the pump wavelength at 400 nm by direct doubling of the fundamental 800 nm from the first optical parametric amplifier through a 0.2-mm-thick β barium borate (BBO) crystal. The pulse energy was attenuated to 140 nJ before entering the sample cell. For fluorescence up-conversion experiments, the fluorescence emission was collected by a pair of parabolic focus mirrors and mixed with another fundamental pulse in a 0.2-mm BBO crystal through a noncollinear configuration. The up-converted signal was detected by a photomultiplier after passing through a double-grating monochromator. The response time in this noncollinear geometry is 350–450 fs, as determined from the up-conversion signal of Raman scattering by water in the range of 450–460 nm. For transient-absorption measurements, the various probe wavelengths of 500–700 nm were generated by mixing the idler or signal with the fundamental from the second optical parametric amplifier. The sensitivity of the transient-absorption method can reach 10–4 to 10–5 of the absorbance change. The pump-beam polarization for fluorescence up-conversion experiments was set at a magic angle (54.7°) with respect to the acceptance axis of the up-conversion crystal (vertical), whereas the gating-beam polarization was set parallel to this axis by using a half-wave plate. For the transient-absorption measurements, the pump-beam polarization was set at a magic angle directly with respect to the probe beam, which was vertical. Samples were kept in various stirring quartz cells during irradiation to avoid heating and photobleaching under anaerobic conditions.
To map the catalytic processes, we used a femtosecond laser pulse (λpump = 400 nm; pulse width, Δt = 70 fs) to initiate excitation of the cofactor FADH– and synchronize the dynamics with repair function of the photolyase. Another femtosecond pulse, delayed in time, probes the dynamic changes of the flavin cofactor. Specifically, we characterized initial decay dynamics (k1 in Fig. 1 A) of the reactant FADH–* by using a femtosecond-resolved fluorescence up-conversion technique. Subsequently, we monitored the dynamics of the radical intermediate FADH·, its formation at the rate of k1, and its decay at the rate of k2 (Fig. 1 A) by using a sensitive transient-absorption method (19).
Results and Discussion
Active-Site Solvation. We first characterized the photophysics of FADH– in the active site without substrate. By gating a series of fluorescence wavelengths of the weak FADH–* emission from the blue side to the red side (Fig. 2A), we obtained the wavelength-resolved fluorescence dynamics shown in Fig. 2B. The observed transients, corresponding to systematic decays from 475 to 620 nm, show significant solvation dynamics at the active site, after the sudden change of the cofactor dipole upon excitation (20, 21). All blue-side transients (λfl <510 nm) have an initial decay within 2 ps (Fig. 2B Inset), showing ultrafast liberation of local protein residues and trapped water molecules (22). Subsequently, slow solvation occurs from 66 ps until merging with the cofactor lifetime of 1.3 ns in the entire emission, reflecting long-time relaxation of the local environment. Table 1 shows the time constants and relative amplitudes of the transients with a series of exponential decays. The observation of continuous solvation processes, consistent with the highly polar active site from the x-ray structure (4), reveals a dynamic active site upon function initiation. Although the substrate recognition may push certain water out of the binding pocket, we observed similar solvation dynamics with repaired thymine monomers in the active site. The observed active-site solvation directly controls the catalytic reactions of DNA repair, as described below.
Fig. 2.

Ultrafast fluorescence spectroscopy of a photolyase in the absence and presence of substrate. (A) Absorption and emission spectra of photolyase containing FADH– cofactor (EPL-FADH–) and no second chromophore in the absence of substrate. The pump wavelength was fixed at 400 nm for all experiments. Nine fluorescence wavelengths were gated from the blue side to the red side. (B) Four typical gated fluorescence transients, with systematic decays from 475 to 550 nm, reflecting significant solvation at the active site. Inset shows fluorescent transients at early time points. (C) The fluorescence transients at 550 nm with and without the substrate thymine dimer. The enzyme concentration was 0.4 mM, and the substrate concentration was 8 mM. Inset shows the fluorescence transients at 480 and 550 nm in the presence of substrate. The initial ultrafast solvation at 480 nm is still present.
Table 1.
Fitting results of the fluorescence transients shown in Fig. 2B
| λ | 475 nm | 480 nm | 490 nm | 550 nm |
|---|---|---|---|---|
| α1 | 0.38 | 0.29 | 0.21 | -0.99 |
| α2 | 0.36 | 0.34 | 0.21 | 0.20 |
| α3 | 0.22 | 0.28 | 0.36 | — |
| α4 | 0.04 | 0.09 | 0.22 | 0.80 |
| τ1 | 0.98 | 1.10 | 1.21 | 0.31 |
| τ2 | 66 | 77 | 87 | 212 |
| τ3 | 277 | 308 | 690 | — |
| τ4 | 1,300 | 1,300 | 1,300 | 1,300 |
All femtosecond-resolved transients are fitted by the following equation: 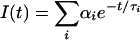 , where i = 1 - 4. The time constants are given in picoseconds. The lifetime of FADH-* is 1.3 ns in the enzyme, and all other time constants represent solvation dynamics. The negative coefficient corresponds to formation dynamics at the red-side emission.
, where i = 1 - 4. The time constants are given in picoseconds. The lifetime of FADH-* is 1.3 ns in the enzyme, and all other time constants represent solvation dynamics. The negative coefficient corresponds to formation dynamics at the red-side emission.
Capture of the FADH· Intermediate. In the presence of substrate, the fluorescence transients drastically changed and became much faster (Fig. 2C). At the blue side, the ultrafast solvation is still clear (Fig. 2C Inset). For example, the transient gated at 480-nm emission shows initial solvation in 2.7 ps. At the red side (λfl ≥ 510 nm), all fluorescence transients become identical. At 550-nm emission, the transient can be represented either by a stretched-single-exponential decay, Ae–(t/τ)β, with τ = 170 ps and β = 0.71, or a double-exponential decay of 60- and 335-ps lifetimes, with 45% and 55% of the total amplitude, respectively. The dynamics does not follow a single-exponential decay because of its strong coupling with the slow active-site solvation. Thus, the observed 170 ps (β = 0.71) represents the dynamics (k1) of excited-state quenching directly by the substrate in the highly solvated active site.
To determine whether the quenching occurs by an electron-transfer process, we searched for the proposed intermediate species, FADH·. At >500 nm, only FADH· and the excited-state FADH–* have absorption (Fig. 3A). Thus, the dynamics of FADH· intermediate can be followed by probing at appropriate wavelengths. At 690 nm, FADH–* absorption dominates, and, at this wavelength, we observed drastically different dynamics in the absence and presence of substrate (Fig. 3B Inset), consistent with the fluorescence decay dynamics at 550-nm emission (Fig. 2C). In the presence of substrate, the transient is dominantly represented by FADH–* with the same stretched-single-exponential or double-exponential decay (Fig. 3B). When the probe was tuned to 625 nm, we observed very different dynamics (Fig. 3 C), and the transient decayed much slower. This result is significant because it shows the capture of the hypothesized FADH· intermediate, proving that the excited-state quenching of FADH–* by the substrate is through an electron-transfer process and the catalytic reaction follows a radical mechanism, which has been a longstanding unresolved issue. The transient at 625 nm is the sum of two dynamic processes of FADH–* and FADH·. With the same stretched β value of 0.71, we obtain a time constant of 560 ps (1/k2) for the decay of FADH·, which represents the electron-return process from the repaired thymine to FADH· to restore the catalytically active FADH–. By using double-exponential decay for the charge-separation process, we also obtained a double-exponential decay of FADH· in 460 and 1,240 ps. When the probe is tuned at wavelengths of 625–500 nm, all resulting transients gave the same decay dynamics of FADH·, as shown also with the probe of 510 nm (Fig. 3C Inset). The observed non-single-exponential decay of FADH· presumably reflects the strong coupling with the slow solvation at the active site. After charge separation, the local environment relaxes again, and the active site is in a continuous dynamic motion.
Fig. 3.

Determination of forward and back electron transfer in photolyase photocycle by ultrafast absorption spectroscopy. (A) Absorption spectra of EPL-FADH· (red) and EPL-FADH–* (blue), as well as EPL-FADH– before (black) and after (green) the repair experiment. The absorption profile of EPL-FADH–* was obtained from ref. 8 and calibrated by our four transient-absorption data at 510, 580, 620, and 690 nm, relative to FADH·. (B) The absorption transient probed at 690 nm, showing a dominant contribution of FADH–* decay (95%) with a minor signal from FADH·. The enzyme concentration is 0.4 mM, and the substrate concentration is 8 mM. Inset shows the drastically different dynamics with and without the substrate. (C) Absorption transients probed at 625 and 510 nm (Inset) show both FADH–* and the intermediate FADH· dynamics. (D) Absorption transients probed at 690 nm for a series of substrates, dinucleotide thymine dimer, and oligo(dT)12–18 and poly(dT) with dimer. The oligomer concentration is 1 mM, and the total dimer concentration in poly(dT) is ≈5 mM. Inset shows the early time points of the reaction.
Electron Return and Catalytic Photocycle. The restoration of FADH– by electron return from the repaired thymine monomers in 560 ps (β = 0.71) is supported by a number of observations. With 170 ps for the charge separation and 1.3 ns of the FADH–* lifetime in the active site, we obtained a quantum yield of 0.87 for the k1 process, consistent with the thymine-formation quantum yield of 0.89 given in ref. 18, indicating that the efficiency of the ring cleavage is nearly 100% and the charge recombination before dimer splitting (which would result in a futile cycle and, hence, lower quantum yield) must occur in a much longer time than 560 ps. The observed complete repair process in 560 ps is also consistent with the recent observation (10) of thymine formation in 589 ps probed at 260 nm for Anacystis nidulans photolyase. Also, the FADH· signal decays to zero (Fig. 3C), and no detectable steady-state FADH· absorption was observed after the repair reaction (Fig. 3A), excluding the possibility that FADH· stays in the neutral radical form after repair and is subsequently reduced photochemically by an electron from a neighboring Trp residue in the enzyme (19, 22, 23) or by a chemical donor in the active site. Thus, the excess electron in the repaired thymine bases does return to FADH· to close the catalytic photocycle and lead to net-zero electron changes. The entire DNA repair is completed in 560 ps through a radical mechanism.
van der Waals Contacts and Adenine Mediation. We further examined the repair dynamics by using substrates of oligo(dT)12–18 and poly(dT) containing T<>Ts. The results of these experiments with the 690-nm probe are shown in Fig. 3D. It was conceivable that the complex binding might be different for various substrates resulting in different reaction dynamics, but our repair results surprisingly showed nearly identical temporal behaviors, indicating that the distances between FADH–* and T<>T in complexes with dinucleotide, oligonucleotide, and polynucleotide substrates are very similar. This observation is consistent with the recent x-ray photolyase-product complex structure of A. nidulans photolyase with repaired DNA, showing a direct van der Waals contact in 3–4 Å between the cofactor and the dimer thymines inside the active-site hole (14).
Crystal structures of photolyases from three different species (4, 13, 24) show a bent configuration of the flavin cofactor in the active site. The adenine moiety was proposed to mediate the electron transfer between the cofactor and the substrate (12). In A. nidulans photolyase-product structure (14), the adenine has two hydrogen bonds with the thymine residues of the dimer at 3.1 and 3.2 Å, and the isoalloxazine ring is 4.3 Å away from the thymine residues. However, our results suggest a direct electron jump from the cofactor to the substrate, not following the hopping mechanism bridged by the adenine moiety. Otherwise, without substrate, we would observe intramolecular electron transfer from the isoalloxazine ring to the adenine moiety in both fluorescence and absorption measurements. In the absence of substrate, we observed only solvation dynamics and lifetime emission of FADH–* (Figs. 2B and 3B Inset), and we did not observe evidence for alternate decay pathways. Also, the reduction potentials [–(1.9–1.4) V vs. normal hydrogen electrode (NHE) for T<>T (25) and –2.52 V vs. NHE for adenine (26)] strongly favor electron transfer to the dimer. However, it is possible that the adenine moiety of FADH–, because of its proximity to both the isoalloxazine ring and the T<>T, may mediate the repair reaction by anchoring the dimer through hydrogen bonding and modulating the electron jump between the cofactor and the substrate through a superexchange mechanism.
Dynamic Control and Repair Efficiency. Fig. 4 shows a reaction scheme based on data from this study and previous studies on DNA repair by photolyase. Upon function initiation by a blue-light photon, the active site starts a continuous dynamical motion, which is strongly coupled with catalytic electron transfer reactions. To achieve high repair efficiency, the quantum yields of both charge-separation and ring-splitting reactions must be optimized to minimize the nonproductive pathway of charge recombination before ring cleavage. Unlike other sensitizer systems that have fast charge recombination leading to a low-repair quantum yield (27), the active-site solvation and proximate adenine in photolyase appear to be critical to strategically slowing down the charge separation (170 ps) and recombination by dynamically tuning the redox potentials of reaction species and stabilizing the charge-separated radical intermediates, leaving enough time to cleave the cyclobutane ring (560 ps) to reach a maximum-repair quantum yield (0.87).
Fig. 4.

Evolution of catalytic reactions of DNA repair by photolyase along the coordinate. Active-site solvation strongly modulates the charge-separation, ring-splitting, and electron-return processes, resulting in slow charge separation (170 ps) and a stretched-single-exponential-decay dynamics (β = 0.71). The charge recombination must be slower than the complete ring splitting (560 ps) to eliminate possible ring reclosure and achieve a maximum-repair quantum yield (0.87).
One subtle issue that remains unresolved is whether the splitting of the cyclobutane ring in 560 ps is asynchronously concerted or sequential. Recent quantum chemical studies (28, 29) predicted stepwise splitting in which the first-step cleavage is a downhill reaction and is expected to be ultrafast. Thus, for effective repair, the charge recombination needs to take longer time than the complete ring cleavage, as described here, to prevent cyclobutane ring reclosure. With the fully resolved dynamic behavior of the flavin cofactor reported here, it should become possible to probe the dimer radical and the thymine product in the UV region and understand their dynamics.
Conclusions
Here, we report our direct observation of thymine dimer repair in DNA by photolyase. With femtosecond resolution, we followed the photocycle and mapped out the temporal evolution of catalytic reactions. We captured the catalytic intermediate of flavin radical (FADH·) and directly proved the electron-transfer radical mechanism of the photocycle that was proposed approximately two decades ago (5). Active-site solvation was observed to occur on picosecond-to-nanosecond time scales and to have a critical role in the continuous modulation of catalytic reactions. These synergistic motions in the active site of the damaged DNA–enzyme complex, optimized by evolution, reveal its perfect correlation of structural integrity and dynamical locality to ensure maximum repair efficiency (0.87) on the ultrafast time scale of 560 ps.
Acknowledgments
We thank Dr. Sang-Tae Kim (Applied Biosystems) for help with substrate preparation and advice on enzyme assay. This work was supported by National Institutes of Health Grant GM31082 (to A.S.) and Selective Investment in the Ohio State University Physics Department (D.Z.).
Author contributions: A.S. and D.Z. designed research and wrote the paper; and Y.-T.K., C.S., L.W., A.S., and D.Z. performed research and analyzed data.
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on May 3, 2005.
Abbreviations: Pyr<>Pyr, cyclobutane pyrimidine dimer; T<>T, cyclobutane thymine dimer.
See accompanying Profile on page 16125.
References
- 1.Brash, D. E. (1997) Trends Genet. 13, 410–414. [DOI] [PubMed] [Google Scholar]
- 2.Rupert, C. S., Goodgal, S. H. & Herriott, R. M. (1958) J. Gen. Physiol. 41, 451–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sancar, A. (2003) Chem. Rev. 103, 2203–2237. [DOI] [PubMed] [Google Scholar]
- 4.Park, H. W., Kim, S. T., Sancar, A. & Deisenhofer, J. (1995) Science 268, 1866–1872. [DOI] [PubMed] [Google Scholar]
- 5.Sancar, G. B., Jorns, M. S., Payne, G., Fluke, D. J., Rupert, C. S. & Sancar, A. (1987) J. Biol. Chem. 262, 492–498. [PubMed] [Google Scholar]
- 6.Payne, G. & Sancar, A. (1990) Biochemistry 29, 7715–7727. [DOI] [PubMed] [Google Scholar]
- 7.Ramsey, A. J., Alderfer, J. L. & Jorns, M. S. (1992) Biochemistry 31, 7134–7142. [DOI] [PubMed] [Google Scholar]
- 8.Kim, S. T., Heelis, P. F., Okamura, T., Hirata, Y., Mataga, N. & Sancar, A. (1991) Biochemistry 30, 11262–11270. [DOI] [PubMed] [Google Scholar]
- 9.Langenbacher, T., Zhao, X., Bieser, G., Heelis, P. F., Scancar, A. & Michel-Beyerle, M. E. (1997) J. Am. Chem. Soc. 119, 10532–10536. [Google Scholar]
- 10.MacFarlane, A. W., IV, & Stanley, R. J. (2003) Biochemistry 42, 8558–8568. [DOI] [PubMed] [Google Scholar]
- 11.Sanders, D. B. & Wiest, O. (1999) J. Am. Chem. Soc. 121, 5127–5134. [Google Scholar]
- 12.Antony, J., Medvedev, D. M. & Stuchebrukhov, A. A. (2000) J. Am. Chem. Soc. 122, 1057–1065. [Google Scholar]
- 13.Komori, H., Masui, R., Kuramitsu, S., Yokoyama, S., Shibata, T., Inoue, Y. & Miki, K. (2001) Proc. Natl. Acad. Sci. USA 98, 13560–13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mees, A., Klar, T., Gnau, P., Hennecke, U., Eker, A. P. M., Carell, T. & Essen, L. O. (2004) Science 306, 1789–1793. [DOI] [PubMed] [Google Scholar]
- 15.Sancar, A., Smith, F. W. & Sancar, G. B. (1984) J. Biol. Chem. 259, 6028–6032. [PubMed] [Google Scholar]
- 16.Heelis, P. F., Payne, G. & Sancar, A. (1987) Biochemistry 27, 4634–4640. [DOI] [PubMed] [Google Scholar]
- 17.Wulff, D. L. & Fraenkel, G. (1961) Biochim. Biophys. Acta 51, 332–339. [DOI] [PubMed] [Google Scholar]
- 18.Kim, S. T. & Sancar, A. (1991) Biochemistry 30, 8623–8630. [DOI] [PubMed] [Google Scholar]
- 19.Saxena, C., Sancar, A. & Zhong, D. (2004) J. Phys. Chem. B 108, 18026–18033. [Google Scholar]
- 20.Pal, S. K. & Zewail, A. H. (2004) Chem. Rev. 104, 2099–2124. [DOI] [PubMed] [Google Scholar]
- 21.Lu, W., Kim, J., Qiu, W. & Zhong, D. (2004) Chem. Phys. Lett. 388, 120–126. [Google Scholar]
- 22.Wang, H., Saxena, C., Quan, D., Sancar, A. & Zhong, D. (2005) J. Phys. Chem. B 109, 1329–1333. [DOI] [PubMed] [Google Scholar]
- 23.Li, Y. F., Heelis, P. F. & Sancar, A. (1991) Biochemistry 30, 6322–6329. [DOI] [PubMed] [Google Scholar]
- 24.Tamada, T., Kitadokoro, K., Higuchi, Y., Inaka, K., Yasui, A., de Ruijter, P. E., Eker, A. P. M. & Miki, K. (1997) Nat. Struct. Biol. 4, 887–891. [DOI] [PubMed] [Google Scholar]
- 25.Heelis, P. F., Deeble, D. J., Kim, S. T. & Sancar, A. (1992) Int. J. Radiat. Biol. 62, 137–143. [DOI] [PubMed] [Google Scholar]
- 26.Seidel, C. A. M., Schulz, A. & Sauer, M. H. M. (1996) J. Phys. Chem. 100, 5541–5553. [Google Scholar]
- 27.Song, Q. H., Tang, W. J., Hei, X. M., Wang, H. B., Guo, Q. X. & Yu, S. Q. (2005) Eur. J. Org. Chem. 6, 1097–1106. [Google Scholar]
- 28.Voityuk, A. A. & Rösch, N. (1997) J. Phys. Chem. A 101, 8335–8338. [Google Scholar]
- 29.Durbeej, B. & Eriksson, L. A. (2000) J. Am. Chem. Soc. 122, 10126–10132. [Google Scholar]