

Abstract
Eukaryotic translation initiation factor (eIF) 5 is crucial for the assembly of the eukaryotic preinitiation complex. This activity is mediated by the ability of its C-terminal HEAT domain to interact with eIF1, eIF2, and eIF3 in the multifactor complex and with eIF4G in the 48S complex. However, the binding sites for these factors on eIF5–C-terminal domain (CTD) have not been known. Here we present a homology model for eIF5-CTD based on the HEAT domain of eIF2Bε. We show that the binding site for eIF2β is located in a surface area containing aromatic and acidic residues (aromatic/acidic boxes), that the binding sites for eIF1 and eIF3c are located in a conserved surface region of basic residues, and that eIF4G binds eIF5-CTD at an interface overlapping with the acidic area. Mutations in these distinct eIF5 surface areas impair GCN4 translational control by disrupting preinitiation complex interactions. These results indicate that the eIF5 HEAT domain is a critical nucleation core for preinitiation complex assembly and function.
Keywords: general amino acid control, ribosome preinitiation complex, translation initiation, translational control
In eukaryotic translation initiation, the 40S ribosomal subunit binds 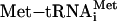 , 5′-capped mRNA, and the 60S subunit in a coordinated manner, setting up the 80S initiation complex with the anticodon of
, 5′-capped mRNA, and the 60S subunit in a coordinated manner, setting up the 80S initiation complex with the anticodon of 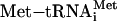 base-paired at the ribosomal P site to the first start codon of the mRNA (for review, see ref. 1). At least 11 eukaryotic initiation factors (eIFs) mediate this process.
base-paired at the ribosomal P site to the first start codon of the mRNA (for review, see ref. 1). At least 11 eukaryotic initiation factors (eIFs) mediate this process. 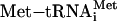 binds the 40S subunit in a ternary complex (TC) with eIF2 and GTP to form the 43S preinitiation complex. Subsequent joining of the 43S particle to the mRNA/eIF4F assembly produces the 48S preinitiation complex, which then scans for the first AUG codon. Correct AUG pairing with the
binds the 40S subunit in a ternary complex (TC) with eIF2 and GTP to form the 43S preinitiation complex. Subsequent joining of the 43S particle to the mRNA/eIF4F assembly produces the 48S preinitiation complex, which then scans for the first AUG codon. Correct AUG pairing with the 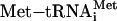 anticodon triggers eIF5-dependent GTP hydrolysis for eIF2, leading to dissociation of the eIFs and formation of the 40S initiation complex. The GDP-bound eIF2 that is released after GTP hydrolysis is recycled to eIF2-GTP by the pentameric guanylate exchange factor eIF2B.
anticodon triggers eIF5-dependent GTP hydrolysis for eIF2, leading to dissociation of the eIFs and formation of the 40S initiation complex. The GDP-bound eIF2 that is released after GTP hydrolysis is recycled to eIF2-GTP by the pentameric guanylate exchange factor eIF2B.
The C-terminal domain (CTD) of eIF5 is an important nucleation core of the preinitiation complex assembly and mediates formation of the multifactor complex (MFC) with eIF1, eIF2 TC, and eIF3 (2, 3). It contains unique aromatic/acidic boxes (AA boxes) 1 and 2. These are also found in the CTDs of eIF2Bε (the catalytic subunit of eIF2B) and mammalian eIF4G (4). The AA boxes in eIF5 and eIF2Bε are required for binding to the lysine-rich segment [lysine box (K box)] present in the N-terminal domain of the common substrate, the β subunit of eIF2 (4). The ability of eIF5-CTD to bind eIF3c is strongly enhanced by its interaction with the eIF2β K box, then leading to rapid and tight MFC assembly (5).
The integrity of the translation initiation machinery is critical for proper cellular response to different stress stimuli (6). In yeast, amino acid starvation activates Gcn2p kinase to phosphorylate eIF2, rendering eIF2 a competitive inhibitor of eIF2B and thereby reducing the level of functional TC. Although this is inhibitory for general protein synthesis, it selectively promotes translation of GCN4 by a mechanism involving four upstream ORFs (uORFs) in the GCN4 5′ leader, which, in turn, activates transcription of hundreds of genes, including ≈70 amino acid-biosynthetic genes (7, 8) (general control response). When TC is plentiful (nonstarvation conditions), the 40S subunit tethered to GCN4 mRNA after uORF1 translation quickly reacquires TC to translate uORF2, -3, or -4, inhibiting GCN4 translation. When the TC level is reduced by a starvation signal, the reaquisition of TC by the 40S subunit is delayed, and the 40S subunit bypasses uORF2–4 to set up the initiation complex at the GCN4 start codon and to start translation of GCN4. As expected, mutations altering eIF2 or eIF2B subunits that reduce the functional TC level (8), or MFC mutations altering eIF5-CTD and eIF3c that delay TC binding to the 40S subunit (9–11), constitutively derepress GCN4 translation by allowing the ribosome in the GCN4 mRNA leader to bypass inhibitory uORF2–4 (general control derepressed, or Gcd phenotype).
Mutations in eIF5-CTD also cause the Gcn– phenotypes (general control nonderepressible). In this phenotype, the restriction of the mutant eIF5-CTD function at a higher temperature (36°C) increases the frequency of scanning past uORF1, thereby impairing GCN4 translation derepression (11, 12). This underlines the role of eIF5-CTD in postassembly processes during the scanning or AUG recognition. In support of this role, eIF5-CTD can bind simultaneously to eIF4G and eIF3c-N-terminal domain, promoting mRNA binding to the 43S complex (11–14).
In this report, we identify binding sites for initiation factors on eIF5-CTD. To achieve this, we homology-modeled the 3D structure of eIF5-CTD, based on the structure of the eIF2Bε C-terminal HEAT domain (15). We identified two highly conserved charged areas on the surface of the structure: an acidic surface, composed of AA-box amino acids and an adjacent basic surface that is composed mainly of lysine residues and specifically conserved in eIF5 homologues. Site-directed mutagenesis identified the acidic surface as the interface to eIF2β and the basic surface as the interface to eIF3c and eIF1. Yeast strains carrying eIF5-CTD mutations predicted to alter these surfaces displayed pronounced general control phenotypes. Thus, the basic and acidic surfaces of eIF5-HEAT domain are critical not only for translation initiation but also for accurate control of GCN4 translation upon amino acid starvation.
Methods
Plasmids and Yeast Strains. Plasmids, oligonucleotides, and yeast strains used in this study are listed in Tables 1, 2, and 3, respectively, which are published as supporting information on the PNAS web site. Plasmid and yeast strain constructions are described in Supporting Text, which is published as supporting information on the PNAS web site.
Biochemical Assays. GST pull-down assays with 35S-labeled proteins, synthesized with the TnT/T7 system (Promega) in a rabbit reticulocyte lysate, were conducted as described (2, 5, 11, 16). The amounts of bound 35S-labeled protein were quantitated with STORM (Molecular Dynamics). Coimmunoprecipitation of FLAG (FL)-tagged eIF2 and hemagglutinin (HA)-tagged eIF3 was done as described (2, 11).
Results
Homology Modeling of Yeast eIF5 HEAT Domain. To perform site-directed mutagenesis of eIF5-CTD without altering its structure (“surface” mutagenesis), we modeled the eIF5-CTD structure based on the eIF2Bε C-terminal HEAT domain structure, which was solved recently (15). Alignment of 22 eIF5 and 10 eIF2Bε sequences from diverse eukaryotes suggests that the eIF5-CTD has eight α-helices similar to those of the eIF2Bε-CTD (Fig. 5, which is published as supporting information on the PNAS web site) (15). The predicted eIF5-CTD α-helices align very well with the eIF2Bε-CTD α-helices except for helix 2, consistent with the idea that this helix of eIF2Bε-CTD, together with the first one, is involved specifically in guanine–nucleotide exchange (15). Fig. 6A, which is published as supporting information on the PNAS web site, shows the homology-modeled structure of yeast eIF5-CTD calculated with the program swiss-model (17), based on the alignment in Fig. 5. Nine point mutations of hydrophobic residues in yeast eIF5-CTD have been reported to cause a temperature-sensitive (Ts–) phenotype (11). The homology model indicates that most of these residues are buried in the hydrophobic core of the domain.
The modeled structure of eIF5-CTD has extensive negatively charged surfaces, similar to the “acidic belt” described for eIF2Bε-CTD (15) (Fig. 6 B and C), but only the residues in one of the negatively charged surface areas are highly conserved, designated here as area I (Fig. 1A and Fig. 6Ac). Compared with eIF2Bε-CTD, eIF5-CTD has additional acidic surfaces, in place of the “basic patch” near the N-terminal helical hairpin of eIF2Bε-CTD (15) (Fig. 6 B and C). These additional acidic surfaces of eIF5-CTD are also found in human eIF5-CTD (data not shown). A positively charged surface designated area II in Figs. 1A and 6A is conserved in all eIF5 proteins examined. Surface area I is contiguous with the negatively charged C terminus (amino acids 396–405), which is disordered in the crystal structure of eIF2Bε-CTD (15) and thus not present in our model.
Fig. 1.

Identification of MFC partner-binding surfaces on eIF5-CTD. (A) Positions of mutations are located on the surface of the eIF5-CTD structure model (Center); and after rotating 60° to the left (Left) and to the right (Right). Areas I and II, defined in the text, are circled. Residues changed in the AN1, BN1, and BN2 mutants are colored in red and dark and light blue, respectively, and labeled in Left and Right, respectively. Residues deleted by W391Δ and E396Δ are colored in orange and yellow, respectively, with the first deleted residues labeled in Center. The last nine residues of eIF5 (residues 397–405) are not present in this model. (B) Yeast eIF5 amino acids from position 339 to 400 are arranged to show the predicted secondary structure. Residues predicted to participate in α-helices 5–8 (defined in Fig. 5) are boxed with the helix numbers in boldface. Highlighted with orange are AA-box amino acids (AA box 1 in helices 6 and 7; AA box 2 in helix 8), whereas highlighted with green in blue letters are basic residues highly conserved in all eIF5. Conserved acidic residues are shown in red. Arrows indicate the site of mutations. Thick arrows indicate regions deleted by W391Δ and E396Δ. Circled in red are the residues altered by Ts– point mutations (11). (C) GST pull-down assays. GST alone (lanes 2) or GST-eIF5 (lanes 3 and 10) and its mutants (lanes 4–8 and 11) were allowed to bind the 35S partners indicated in the middle. The complex was analyzed by SDS/PAGE and autoradiography. Five or 3 μg of GST or its fusion protein was used in lanes 1–8 or 9–11, respectively. Percent of 35S protein pulled down is shown below each gel. Lanes 1 and 7, 20% in-put amounts; N. T., not tested. (D) Summary of effects of eIF5 mutations, indicated to the right, on GST-eIF5 binding to eIF2β-N (a), eIF3c-N (b), and eIF1 (c). Binding assays as in C were performed several times, and the fractions of 35S partners bound to GST-eIF5 mutants were compared with the fraction of the same partners bound to wild-type GST-eIF5. Average values (boxes) and SDs (bars) are presented. (E) Hypothetical model of MFC assembly. eIF5-CTD is depicted as the same orientation as in A Center, with areas I and II in red and blue, respectively. Parts of eIF1, eIF2β-K-box, eIF3c, and eIF4G are drawn as differently colored objects.
Identification of the eIF2β-Binding Face. We showed previously that the 15-aa-deletion of eIF5 C terminus designated W391Δ, isolated in Ts– mutant screening, abolishes the interaction with eIF2 without affecting binding to eIF3 (5, 11). This deletion removes a part of helix 8 (see Fig. 1 A and B) and the following highly acidic C-terminal tail of 10 amino acids. To test the effect of removing the C-terminal tail alone, we constructed the mutant E396Δ, which lacks E396 and the following nine amino acids (E396 is colored yellow in Fig.1A and is the last amino acid of the model). As shown in Fig. 1C, at the top, a GST-eIF5 fusion protein carrying this mutation reduced binding to 35S-eIF2β by 2-fold, without affecting binding to 35S-eIF1, 35S-eIF3c, and 35S-eIF4G (lane 5; see Fig. 1Da for quantification). We confirmed that W391Δ substantially reduced binding to 35S-eIF2β (Fig. 1C, lane 6); the stronger effect of W391Δ compared with E396Δ is apparently due to deleting a part of helix 8 next to the C-terminal tail. We judged that the effect of this mutation on the integrity of the CTD structure was minimal, because GST-eIF5 carrying W391Δ bound eIF3c at 50% efficiency (Fig. 1Db, row 4), whereas GST-eIF5 carrying other Ts– mutations almost eliminated eIF3c as well as eIF2β binding (11). We then examined the acidic area adjacent to E396. The AN1 mutation altering D354, E358, E359, and E360 (shown in red in Fig. 1 A and B) to polar serine residues decreased GST-eIF5 binding to 35S-eIF2β and -eIF3c to 20% and 70% of the wild-type GST-eIF5 level, respectively (Fig. 1C top and second gels, lane 4), implicating the area altered by AN1 specifically in eIF2β binding. Together these results indicate that the acidic area I, constituted by AA-box amino acids, is the primary eIF2β K box-binding face, confirming the prediction made in Boesen et al. (15). The site of this interaction is indicated in Fig. 1Eb, in the schematic model of the eIF5-CTD.
Identification of the eIF3c/eIF1-Binding Face. To examine the eIF5-specific basic area II, we created two mutations, BN1 and BN2, altering basic surface amino acids to polar glutamine residues. As illustrated in Fig. 1 A and B, BN1 alters R382 in helix 8 and 5 lysine residues in the preceding loop, whereas BN2 changes H336 and K337 in the loop between helices 5 and 6. Remarkably, BN1 abolished GST-eIF5 binding to 35S-eIF3c and 35S-eIF1 without altering its binding to 35S-eIF2β. This defines area II as the primary eIF3c- and eIF1-binding face (Fig. 1C, lanes 8). Likewise, BN2 reduced GST-eIF5 binding to 35S-eIF1 and -eIF3c by 2-fold without affecting binding to 35S-eIF2β (lanes 7; see Fig. 1Db for quantification), indicating that the eIF1- and eIF3c-binding face extends to the outskirt of area II. Because eIF3c enhances the interaction between eIF1 and eIF5-CTD by mutual cooperativity effects (2), we suggest that eIF3c and eIF1 can bind eIF5-CTD at area II simultaneously, as illustrated in Fig. 1Ec.
Effect of HEAT Domain Surface Mutations on eIF4G Binding in Vitro. Because eIF5-CTD binding to eIF4G is competitive with its binding to eIF2β in vitro (13), we anticipated that the eIF4G-binding face on eIF5-CTD might overlap substantially with the K box interface at the acidic area I. Consistent with this prediction, AN1 reduced the interaction between GST-eIF5 and 35S-eIF4G2 C-terminal HEAT domain segment (14, 18) by 3-fold (Fig. 1C, fourth gel, lane 4). The partial inhibition of eIF4G binding by AN1 also suggests that the eIF4G interface may include an area larger than area I. Because BN1 and BN2 also reduced the interaction with eIF4G (Fig. 1C, fourth gel, lanes 7 and 8), the eIF4G interface seems to encompass the basic surface as well. We previously showed that the eIF4G–eIF5 interaction occurs simultaneously with the eIF5–eIF3c interaction (13). Based on this observation and the one mentioned earlier, we suggest that the eIF4G interface overlaps with the eIF2β interface at the acidic area, whereas simultaneous eIF5–CTD interactions with eIF4G and eIF3c contribute to the overall stability of the scanning preinitiation complex. Thus, we propose that the MFC rearranges on or before 48S complex formation, as illustrated in Fig. 1E.
Effect of HEAT Domain Surface Mutations on MFC Assembly in Vivo. In yeast, eIF5 is encoded by the single-copy essential gene, TIF5. We introduced each of the four mutant tif5 alleles, AN1, E396Δ, BN1, and BN2, into different yeast strains, creating eIF5 mutants as listed in Table 3. We found that derivatives of KAY23 carrying AN1, E396Δ, and BN1 modestly reduced yeast growth in rich yeast extract/peptone/dextrose medium (but not in minimal medium) at a high temperature of 36°C (data not shown), suggesting that the acidic and basic areas of eIF5-CTD promote optimal yeast growth at a restrictive temperature when nutrients are plentiful.
Yeast strains encoding tagged subunits (Table 3, column 3 or 4) allowed us to study the in vivo interaction of eIF5 with HA-eIF3 or FL-eIF2. Anti-HA coimmunoprecipitation indicates that BN1 substantially reduced HA-eIF3 binding to eIF5 (Fig. 2A, lane 8, fourth gel), demonstrating that the basic area II of eIF5 is a major eIF3-binding face in vivo. Conversely, BN1 did not affect HA-eIF3 binding to eIF1 (Fig. 2A, lane 8, third gel), indicating that direct eIF1 contact with eIF3 via the eIF3c (16) and/or eIF3a (3) subunit is sufficient for stable eIF1–eIF3 interaction. BN1 eliminated HA-eIF3 binding to eIF2α (Fig. 2A, lane 8, fifth gel), suggesting dissociation of eIF2 from the MFC. Anti-FLAG coimmunoprecipitation indicates that BN1 substantially reduced eIF5 binding to FL-eIF2 (Fig. 2B, third gel, lane 8). Together, our results indicate that alteration of the eIF1- and eIF3c-binding surface (area II in Fig. 1A) of eIF5 promotes its dissociation from the eIF1–eIF3 complex (Fig. 2A), as suggested by the in vitro binding studies (Fig. 1 C and D). Dissociation of eIF2 from eIF5 (Fig. 2B) suggests that the disruption of two of the three MFC partner interactions was severe enough to eliminate mutual cooperativity effects for MFC assembly. The effect of BN1 on MFC assembly in vivo is summarized in Fig. 2Db.
Fig. 2.

Effect of eIF5-CTD mutations on MFC formation in vivo. (A) Coimmunoprecipitation of HA-eIF3. Whole-cell extracts prepared from KAY37 (TIF34 TIF5; Control), KAY113 (TIF34-HA TIF5: TIF5), and its derivatives listed in Table 3 with indicated mutations were used for immunoprecipitation with anti-HA affinity resin. The entire pellet fractions (P) were analyzed together with 10% input (I) and 10% supernatant (S) fractions by immunoblotting with antibodies indicated to the right (see Supporting Text). TIF34-HA encodes the HA-tagged eIF3i subunit. (B) Coimmunoprecipitation of FLAG-eIF2. KAY17 (SUI3 TIF5; Control) (4), KAY128 (FL-SUI3 TIF5; TIF5), and its derivatives listed in Table 3 with indicated mutations were used for immunoprecipitation with anti-FLAG affinity resin, and the immune complex was analyzed as in A. Numbers below anti-eIF5 blots of the P fractions indicate percent of eIF5 found in these fractions, as measured with nih image software (National Institutes of Health). (C) The fraction of eIF5 found in the FL-eIF2 pellet fractions from indicated strains was compared with that of eIF5 found in the pellet fraction from the wild-type strain. Average (filled box) and SD (empty box) from at least three independent experiments are presented. (D) Models of MFC assembly in the strains tested. Circles, individual eIFs. Filled circle, mutant eIF5. Thick solid or dotted lines, MFC partner interfaces strongly or weakly impaired by the mutation introduced, respectively, as judged by in vitro binding studies. Direct contact, strong interactions. No contact, defective interactions, as judged by coimmunoprecipitation studies. Thin lines, interaction eliminated in the coimmunoprecipitation via HA-eIF3 but not affected in that via FL-eIF2.
The other three mutations, BN2, AN1 and E396Δ, eliminated eIF2α from the HA–eIF3 complex (Fig. 2A, fifth gel) without altering eIF5 (Fig. 2A, fourth gel) or eIF1 (Fig. 2A, third gel) binding to HA-eIF3, again suggesting dissociation of eIF2 from the MFC. However, these mutations did not disrupt eIF5 binding to eIF2 when the complex was precipitated via FL-eIF2 (Fig. 2B). Coimmunoprecipitation experiments can result in dissociation of peripheral components of the multiprotein complex that are distant from the epitope, due to the washing step of the method (19). Because such dissociation was not observed with wild-type MFC, we hypothesize that this observed dissociation of eIF2 from the HA–eIF3 complex occurred during the washing step because of the instability of MFC carrying the eIF5-CTD mutations.
Having observed little effect of AN1 and E396Δ on FL-eIF2–eIF5 interaction in vivo, we first tested the effect of higher temperature. Consistent with the in vitro binding studies, the interaction of the AN1 mutant form of eIF5 with FL-eIF2 was significantly reduced when yeast was precultured at 35°C (data not shown). To further test this surface site, we created the double mutant carrying both AN1 and E396Δ. As expected, this double mutation further reduced in vitro interaction of GST-eIF5 with eIF2β without affecting interaction with eIF3c (Fig. 1C, lanes 9–11). Moreover, the double mutant reduced FL-eIF2 binding to eIF5 in vivo by 2-fold (Fig. 2 A and B, lanes 19–21; see Fig. 2C for quantification). These results confirm that D354, E358, E359, and E360 (altered by AN1) and the C-terminal tail (residues 396–405, removed by E396Δ) are involved in eIF2 binding. As mentioned above, W391Δ strongly reduced eIF2 interaction without greatly affecting eIF3 binding (5). Accordingly, we propose that residues 391–395, deleted by W391Δ but not deleted by E396Δ, are additionally required for eIF5 binding to eIF2. Perhaps solvent-exposed hydrophobic and acidic residues W391 and E393, respectively, are involved in the interaction with a lysine residue in eIF2β K boxes and a conserved hydrophobic amino acid flanking each of the K boxes (4).
Genetic Evidence for Defective MFC Assembly by HEAT Domain Surface Mutations. We then investigated whether the eIF5 mutations display general control phenotypes as evidence for impairing preinitiation complex assembly. To test whether eIF5-CTD mutants derepress general control (Gcd– phenotypes), we examined their effect on growth in a gcn2Δ background on media containing 3-aminotriazole (3AT), a histidine biosynthetic enzyme inhibitor used to induce amino acid starvation. Yeast gcn2Δ strains cannot derepress general control and therefore fail to grow in 3AT-containing media, as shown in Fig. 3Ab, row 1. As expected, the gcn2Δ strain carrying tif5-AN1 is 3AT-resistant due to a partial derepression of the general control response (Gcd– phenotype) (Fig. 3Ab, row 9). GCN4-lacZ reporter assay, in a GCN2+ strain, confirmed a 2-fold increase in GCN4 expression based on comparison of β-galactosidase activity in the absence of 3AT (Fig. 4A, compare lines 1 and 5). In addition, the gcn2Δ tif5-E396Δ strain partially derepressed general control (weak Gcd– phenotype) (Fig. 3Ab, row 7). Thus, the acidic site mutations apparently delay TC binding to the 40S subunit in vivo.
Fig. 3.

Effect of eIF5-CTD mutations on general control phenotypes. (A) Gcd– phenotype test. The same amount of the overnight culture of transformants of gcn2Δ strains (KAY24 derivatives in Table 3) carrying the indicated TRP1 and URA3 plasmids, grown in SC-ura-trp, and their 1/10 and 1/100 dilutions were spotted onto SD medium containing required supplements with (b and d) or without (a and c) 30 mM 3AT and incubated for 4 or 3 days, respectively. TRP1 plasmids used are: YEplac112 (Vector, odd-numbered rows) and YEpW-SUI1 (eIF1, even-numbered rows). URA3 plasmids used are YEplac195 (Vector, a and b) and p1780-IMT (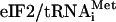 , c and d) (see Table 1). (B) Gcn– phenotype test. TIF5 GCN2 strains with indicated mutations (Table 3 and isogenic tif5-F364S strain KAY315 in row 2; ref. 11) were grown in yeast extract/peptone/dextrose, diluted and spotted as in A, onto SC-his medium with (c and e) or without (a, b, and d) 50 mM 3AT and 40 mM leucine. SC-his plates were incubated for 2 days at indicated temperatures, and SC-his containing 3AT were incubated for 4 and 6 days at 33°C and 36°C, respectively.
, c and d) (see Table 1). (B) Gcn– phenotype test. TIF5 GCN2 strains with indicated mutations (Table 3 and isogenic tif5-F364S strain KAY315 in row 2; ref. 11) were grown in yeast extract/peptone/dextrose, diluted and spotted as in A, onto SC-his medium with (c and e) or without (a, b, and d) 50 mM 3AT and 40 mM leucine. SC-his plates were incubated for 2 days at indicated temperatures, and SC-his containing 3AT were incubated for 4 and 6 days at 33°C and 36°C, respectively.
Fig. 4.

Effect of eIF5-CTD mutations on GCN4 translational control. Transformants of KAY314 (WT), KAY364 (BN1), KAY351 (AN1), and KAY405 (AN1 E396Δ) carrying the indicated GCN4-lacZ plasmid (Table 1) were grown in the following media and assayed for β-galactosidase activity, as described and expressed in ref. 11. Bars indicate β-galactosidase activities from several experiments by using at least two independent transformants, with SD in lines. Schematics at the top of A–D depict the GCN4 leader structure drawn to scale, with positive regulatory uORF1 (filled boxes), other negative regulatory uORFs (open boxes), and the GCN4 coding region (gray boxes). In the table to the left, column 1 (temp.) indicates temperature at which yeast cultures were incubated, column 2 (TIF5) indicates TIF5 mutations, and column 3 (ind.) indicates the ratio of LacZ activity in the presence of 3AT to that in the absence of 3AT. (A and B) Transformants were grown in SC-his-ura medium supplemented with (gray bars) or without 10 mM 3AT (filled bars). (C and D) Transformants were grown in SC-ura medium.
Although the gcn2Δ strains carrying tif5-BN1 or -BN2 were 3AT-sensitive (Gcd+, Fig. 3Ab, row 3 or 5), high-copy (hc) eIF1 turned them into Gcd– mutants (Fig. 3Ab, row 4 or 6), with tif5-BN2 altered to the weakest Gcd– mutant. hc eIF1 also enhanced the Gcd– phenotypes of tif5-E396Δ and -AN1 (rows 8 and 10). A similar enhancement of the general control response by hc eIF1 was observed with all of the previously characterized Ts– eIF5-CTD mutations (11), including tif5-W391Δ (Fig. 3A, rows 13 and 14). This effect is not seen in the presence of the wild-type TIF5 allele (row 2). It was hypothesized that, in the presence of eIF5-CTD mutations that only weakly impair MFC formation, excess eIF1 present in the hc eIF1 strains binds MFC partners in an uncoordinated manner, resulting in formation of inhibitory complexes. These complexes sequester TC, thereby reducing the level of functional TC in favor of GCN4 translation reinitiation and the Gcd– phenotype (5). All of the observed Gcd– phenotypes were suppressed by increasing the level of eIF2 TC by plasmid p1780-IMT (Fig. 3Ad), in agreement with the idea that the phenotypes are due to a delay in TC binding to the ribosome. Therefore the observed effects of BN1, BN2, AN1, and E396Δ on GCN4 translational control provide physiological evidence for their defects in MFC assembly (Fig. 2 A and B).
The tif5-AN1 E396Δ double mutation displayed a Gcd– phenotype comparable to (or slightly weaker than) that of tif5-AN1 alone (compare Fig. 3Ab, rows 9 and 11, and Fig. 4A, rows 5 and 7). Based on the weaker eIF2/eIF5 association observed with this double mutation, it was conceivable that the mutant would display a Gcd– phenotype stronger than the tif5-AN1 mutant due to further delay in TC binding to the 40S subunit. We reasoned that this discrepancy is due to the inability of the double mutant to more than partially derepress GCN4 translation. This idea is supported by data described below.
Some Basic and Acidic Surface Mutants Cannot Derepress the General Control Response. To examine whether the surface eIF5-CTD mutations impair the general control derepression on amino acid starvation (Gcn– phenotype), we tested the growth of GCN2+ eIF5-CTD mutant strains on 3AT medium. The GCN2+ strain carrying the wild-type eIF5 allele is 3AT-resistant due to the normal general control response (Fig. 3B, row 1). As expected, the BN1 and the AN1 E396Δ double mutants are 3AT-sensitive and hence Gcn–, with the phenotype observed at 33°C and enhanced at 36°C (Fig. 3B c and e, rows 3 and 7). These phenotypes are just as strong as the previously reported phenotypes with tif5-W391Δ and -F364S (Fig. 3B c and e, rows 2 and 8). tif5-E396Δ and -AN1, which alter the acidic interfaces, also showed a weak Gcn– phenotype at 36°C (Fig. 3Be, rows 5 and 6).
As shown in Fig. 4A, the wild-type strain induced GCN4 expression 3-(36°C, rows 17 and 18) to 9-fold (30°C, rows 1 and 2) in response to 3AT-induced amino acid starvation, as examined by β-galactosidase activity expressed from the wild-type GCN4-lacZ reporter plasmid p180 (see column 3 for level of increase by 3AT). Higher temperatures elevate GCN4 expression in the TIF5+ cells, resulting in a smaller response to 3AT. Consistent with their strong Gcn– phenotypes (Fig. 3B), tif5-BN1 and -AN1 E396Δ impaired GCN4 derepression upon amino acid starvation at all of the temperatures tested, with 3AT induction further reduced at 36°C (Fig. 4A). We also observed a partial defect in 3AT-induced GCN4 derepression by AN1 (Fig. 4A), consistent with its weak Gcn– phenotype (Fig. 3B). The tif5-BN1, -AN1, and -AN1 E396Δ alleles did not appear to affect transcription of the GCN4-lacZ reporter, because neither affected GCN4-lacZ expression from p227 lacking all uORFs (Fig. 4B). These results indicate that both the basic and acidic surfaces of eIF5-CTD are similarly important for positive regulation of GCN4 translation reinitiation.
Both the Acidic and Basic Surfaces of eIF5-HEAT Domain Are Important for the Scanning or AUG Recognition Process. Finally, we wished to understand the mechanism of the strong Gcn– phenotypes of the BN1 and AN1 E396Δ mutants. If uORF1 cannot be translated because of a postassembly defect in initiation, the ribosomes would not be committed to reinitiation of downstream cistrons in GCN4 mRNA (Model I). On the other hand, a postassembly defect in the scanning process might impair the ability of the committed ribosomes (i.e., those that have translated uORF1) to reinitiate translation (Model II). Both of these mechanisms would result in the inability to derepress GCN4 translation upon amino acid starvation, explaining their Gcn– phenotypes. To test these models, we used derivatives of the GCN4-lacZ plasmid, pM226 and pM199 (20). pM226 encodes a normal uORF1 start site, but frameshift mutations extend uORF1 downstream of the GCN4 start codon. In this construct, GCN4 can be translated only by the ribosomes that scan past (or “leaky scan”) uORF1. Therefore, an increased expression from this reporter would indicate increased frequency of uORF1 leaky scanning (in support of Model I). pM199 contains uORF1 only and is used to measure the efficiency of reinitiation of GCN4 translation following uORF1 translation (and thereby test Model II). As shown in Fig. 4C, tif5-BN1 and -AN1 E396Δ increased the GCN4-LacZ expression from pM226 by 2- to 4-fold at 33°C and 36°C, the temperatures at which Gcn– phenotypes were observed. These results support Model I. In contrast, these mutations did not affect GCN4 expression from pM199, as shown in Fig. 4D. Thus, the strong Gcn– phenotypes of tif5-BN1 and -AN1 E396Δ are at least in part due to leaky scanning of uORF1. These results provide strong genetic evidence that both the acidic and basic areas of eIF5-CTD are critical for the integrity of preinitiation complex during scanning and/or AUG recognition.
Discussion
In this report, we studied the structure–function relationship of eIF5-CTD based on its homology-modeled structure (Figs. 5 and 6) (15). In vitro binding studies define the acidic area I and basic area II as the primary eIF2β and eIF3c/eIF1-binding sites, respectively (Fig. 1). In vivo binding studies (Fig. 2) and general control phenotypes of the created mutants (Figs. 3 and 4) support the idea that these interfaces are critical for the preinitiation complex assembly and function.
The positions on the 40S subunit of eIF1, the 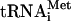 moiety of eIF2 TC, and eIF3 have been determined from different structural methods in combination with complementary biological verification (3, 21). However, the linkage on the ribosome of these factors among themselves and with other factors at different stages of the initiation reaction remains to be elucidated. Based on the mutual exclusivity/cooperativity tests on various interactions involving eIF5-CTD (2, 13, 14), we deduce that MFC is reorganized or “isomerized” to allow eIF4G binding and subsequent factor release, as illustrated in Fig. 1Ed. In this model, the preinitiation complex stability depends more on the eIF5 area II than at earlier stages because of a low affinity for the eIF4G/eIF5 interaction (14); this would explain the strong impact of BN1 on the scanning/AUG recognition process in vivo (Fig. 4C). It is also noteworthy that the AN1 E396Δ double mutation leads to a strong Gcn– phenotype by impairing the scanning/AUG recognition process (Fig. 4C), but that its effect on the MFC assembly was minor (Fig. 2). This could also be explained by a difference in factor interactions between the two stages. It is possible that the double mutation has a stronger impact on factor interactions in the scanning ribosome than in the MFC, by collectively affecting eIF5-CTD interactions with eIF1, eIF3c, and eIF4G.
moiety of eIF2 TC, and eIF3 have been determined from different structural methods in combination with complementary biological verification (3, 21). However, the linkage on the ribosome of these factors among themselves and with other factors at different stages of the initiation reaction remains to be elucidated. Based on the mutual exclusivity/cooperativity tests on various interactions involving eIF5-CTD (2, 13, 14), we deduce that MFC is reorganized or “isomerized” to allow eIF4G binding and subsequent factor release, as illustrated in Fig. 1Ed. In this model, the preinitiation complex stability depends more on the eIF5 area II than at earlier stages because of a low affinity for the eIF4G/eIF5 interaction (14); this would explain the strong impact of BN1 on the scanning/AUG recognition process in vivo (Fig. 4C). It is also noteworthy that the AN1 E396Δ double mutation leads to a strong Gcn– phenotype by impairing the scanning/AUG recognition process (Fig. 4C), but that its effect on the MFC assembly was minor (Fig. 2). This could also be explained by a difference in factor interactions between the two stages. It is possible that the double mutation has a stronger impact on factor interactions in the scanning ribosome than in the MFC, by collectively affecting eIF5-CTD interactions with eIF1, eIF3c, and eIF4G.
How does eIF5-CTD promote scanning or AUG recognition by interacting with eIF3 and other partners? It is conceivable that eIF5-CTD promotes these processes by controlling the eIF5 GAP function mediated by its N-terminal domain (22). Alternatively, eIF5-CTD might indirectly control the 40S subunit by affecting eIF3 and/or eIF1, the direct partners of the 40S subunit (see above). It is possible that defects in eIF5–CTD interactions might shift the 40S subunit conformation to one unfavorable for the scanning process, perhaps from the closed form to the open form in the mRNA-binding cleft as identified in elongating ribosomes (23). This could lead to dissociation of mRNA or, alternatively, failure to promote GTP hydrolysis (22), or a proper ribosomal conformational change (24) upon AUG selection.
Finally, can the MFC isomerization model in Fig. 1E explain the sequence of events from AUG recognition to 40S initiation complex formation? The release of eIF1 from the P site that is proposed to occur on AUG recognition (24) would easily trigger dissociation of eIF5 and eIF4G that bind together in the configuration depicted in Fig. 1E. Subsequent hydrolysis of eIF2-bound GTP would eject eIF2-GDP from the 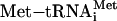 linked to the ribosomal P site (22). Although eIF3 may remain associated with the 40S subunit immediately following GTP hydrolysis (25), this association would be relatively weak due to the absence of eIF5 and eIF2 that might otherwise enhance the affinity of eIF3a/c subcomplex for the ribosome (3) and that of eIF5-CTD for eIF3c (5), respectively. Thus, the model proposed in Fig. 1E might indeed favor a rapid factor release leading to the production of 40S initiation complexes.
linked to the ribosomal P site (22). Although eIF3 may remain associated with the 40S subunit immediately following GTP hydrolysis (25), this association would be relatively weak due to the absence of eIF5 and eIF2 that might otherwise enhance the affinity of eIF3a/c subcomplex for the ribosome (3) and that of eIF5-CTD for eIF3c (5), respectively. Thus, the model proposed in Fig. 1E might indeed favor a rapid factor release leading to the production of 40S initiation complexes.
HEAT and ARM domains play crucial roles in protein–protein interactions in eukaryotes (26). They were originally identified in Huntingtin, elongation factor 3, the A subunit of protein phosphatase 2A, and the target of rapamycin kinase, as well as in the Drosophila protein Armadillo. The originally described HEAT and ARM domains contain 4–20 repeats of two and three α-helices, respectively. Subsequently, such domains were found in karyopherins, whose binding to the cognate nuclear export or import signal is controlled by a small GTPase Ran (reviewed in ref. 27). In the case of the eIF5 HEAT domain, its interaction with eIF3c is strongly enhanced by its association with eIF2β K boxes (5). To understand the molecular basis of this phenomenon as well as mutual cooperativity effects by many interactions linking MFC partners, it will be essential to study the structure of eIF5-CTD in complex with different partners found in the MFC and the 43S and 48S complexes.
Supplementary Material
Acknowledgments
We thank Alan Hinnebusch, Rob Denell, and Beth Montelone for critical reading of the manuscript. This work was supported by the National Institutes of Health COBRE award 1 P20 RR15563, matching support from the state of Kansas and Kansas State University, and National Institutes of Health Grant R01GM64781 (to K.A.), American Cancer Society Grant RPG-97-061-01-NP (to E.M.H.), and National Institutes of Health Grant R01 CA68262 (to G.W.).
Author contributions: G.W. and K.A. designed research; Y.Y., C.R.S., A.M., and N.S.H. performed research; A.M. analyzed data; E.M.H. contributed new reagents/analytic tools; and K.A. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: eIF, eukaryotic initiation factor; AA box, aromatic/acidic box; K box, lysine box; CTD, C-terminal domain; MFC, multifactor complex; TC, ternary complex; uORF, upstream ORF; Gcd, general control derepressed; Gcn, general control nonderepressible; FL, FLAG; HA, hemagglutinin; Ts–, temperature sensitive; 3AT, 3-aminotriazole.
References
- 1.Hershey, J. W. B. & Merrick, W. C. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J. W. B. & Mathews, M. B. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 33–88.
- 2.Asano, K., Clayton, J., Shalev, A. & Hinnebusch, A. G. (2000) Genes Dev. 14, 2534–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valásek, L., Mathew, A. A., Shin, B. S., Nielsen, K. H., Szamecz, B. & Hinnebusch, A. G. (2003) Genes Dev. 17, 786–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asano, K., Krishnamoorthy, T., Phan, L., Pavitt, G. D. & Hinnebusch, A. G. (1999) EMBO J. 18, 1673–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh, C. R., Yamamoto, Y. & Asano, K. (2004) J. Biol. Chem. 279, 49644–49655. [DOI] [PubMed] [Google Scholar]
- 6.Dever, T. E. (2002) Cell 108, 545–556. [DOI] [PubMed] [Google Scholar]
- 7.Natarajan, K., Meyer, M. R., Jackson, B. M., Slade, D., Roberts, C., Hinnebusch, A. G. & Marton, M. J. (2001) Mol. Cell. Biol. 21, 4347–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hinnebusch, A. G. (1997) J. Biol. Chem. 272, 21661–21664. [DOI] [PubMed] [Google Scholar]
- 9.Valásek, L., Nielsen, K. H., Zhang, F., Fekete, C. A. & Hinnebusch, A. G. (2004) Mol. Cell. Biol. 24, 9437–9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh, C. R., Hui, H., Ii, M., Yamamoto, Y. & Asano, K. (2004) J. Biol. Chem. 279, 31910–31920. [DOI] [PubMed] [Google Scholar]
- 11.Singh, C. R., Curtis, C., Yamamoto, Y., Hall, N. S., Kruse, D. S., Hannig, E. M. & Asano, K. (2005) Mol. Cell. Biol. 25, 5480–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nielsen, K. H., Szamecz, B., Valasek, L., Jivotovskaya, A., Shin, B. S. & Hinnebusch, A. G. (2004) EMBO J. 23, 1166–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Asano, K., Shalev, A., Phan, L., Nielsen, K., Clayton, J., Valasek, L., Donahue, T. F. & Hinnebusch, A. G. (2001) EMBO J. 20, 2326–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He, H., von der Haar, T., Singh, R. C., Ii, M., Li, B., McCarthy, J. E. G., Hinnebusch, A. G. & Asano, K. (2003) Mol. Cell. Biol. 23, 5441–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boesen, T., Mohammad, S. S., Pavitt, G. D. & Andersen, G. R. (2004) J. Biol. Chem. 279, 10584–10592. [DOI] [PubMed] [Google Scholar]
- 16.Asano, K., Phan, L., Anderson, J. & Hinnebusch, A. G. (1998) J. Biol. Chem. 273, 18573–18585. [DOI] [PubMed] [Google Scholar]
- 17.Schwede, T., Kopp, J., Guex, N. & Peitsch, M. C. (2003) Nucleic Acids Res. 31, 3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcotrigiano, J., Lomakin, I. B., Sonenberg, N., Pestova, T. V., Hellen, C. U. T. & Burley, S. K. (2001) Mol. Cell 7, 193–203. [DOI] [PubMed] [Google Scholar]
- 19.Natarajan, K., Jackson, B. M., Rhee, E. & Hinnebusch, A. G. (1998) Mol. Cell 2, 683–692. [DOI] [PubMed] [Google Scholar]
- 20.Grant, C. M., Miller, P. F. & Hinnebusch, A. G. (1994) Mol. Cell. Biol. 14, 2616–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lomakin, I. B., Kolupaeva, V. G., Marintchev, A., Wagner, G. & Pestova, T. V. (2003) Genes Dev. 17, 2786–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang, H., Yoon, H., Hannig, E. M. & Donahue, T. F. (1997) Genes Dev. 11, 2396–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spahn, C. M., Beckmann, R., Eswar, N., Penczek, P. A., Sali, A., Blobel, G. & Frank, J. (2001) Cell 107, 373–386. [DOI] [PubMed] [Google Scholar]
- 24.Maag, D., Fekete, C. A., Gryczynski, Z. & Lorsch, J. R. (2005) Mol. Cell 17, 265–275. [DOI] [PubMed] [Google Scholar]
- 25.Unbehaun, A., Borukhov, S. I., Hellen, C. U. & Pestova, T. V. (2004) Genes Dev. 18, 3078–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andrade, M. A., Petosa, C., O'Donoghue, S. I., Muller, C. W. & Bork, P. (2001) J. Mol. Biol. 309, 1–18. [DOI] [PubMed] [Google Scholar]
- 27.Cook, A., Fernandez, E., Lindner, D., Ebert, J., Schlenstedt, G. & Conti, E. (2005) Mol. Cell 18, 355–367. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.