

Abstract
The molecule α calcium calmodulin kinase II (αCaMKII) is known to play a fundamental role in the induction of many forms of synaptic plasticity. A major theory of αCaMKII function proposes that autophosphorylation of the molecule mediates not only the induction but also the maintenance of synaptic plasticity. To test this hypothesis, we assessed ocular dominance plasticity in genetically engineered mice that carry a mutation preventing autophosphorylation of αCaMKII. These mutant mice are deficient in plasticity after monocular deprivation, but a sufficiently long period of monocular deprivation will induce ocular dominance plasticity. After induction of ocular dominance plasticity, the stability of the induced changes was assayed after binocular deprivation. Plasticity in homozygous mutant animals was as stable as that measured in WT littermates; also, response characteristics did not differ between the two groups. Our results suggest that αCaMKII autophosphorylation is required for the induction of ocular dominance plasticity but is not needed for its stable maintenance thereafter.
Keywords: binocular deprivation, monocular deprivation, visual cortex
The molecule α calcium calmodulin kinase II (αCaMKII) plays a fundamental role in activity-dependent synaptic plasticity. Blockade of αCaMKII, by pharmacological or genetic means, prevents long-term potentiation in hippocampal and cortical neurons, impairs performance in spatial learning tasks and impedes plasticity in primary visual and somatosensory cortices (1-5).
In its resting state, αCaMKII activation requires Ca2+/calmodulin binding for activation (6). During synaptic depolarization leading to plasticity, Ca2+ rises to levels that are sufficient to locally activate a population of αCaMKII molecules. When activated, αCaMKII translocates rapidly to the postsynaptic density at which it binds the N-methyl-d-aspartate receptor and may phosphorylate substrate molecules, including the AMPA receptor (7-9). Phosphorylation of such substrate molecules leads to increases in synaptic strength, in some cases directly. For example, αCaMKII phosphorylation of the GluR1 subunit of the AMPA receptor causes conformational changes in the channel that lead to increased conductance (10).
These data have established a critical role for αCaMKII in the induction of synaptic plasticity. Based largely on theoretical studies, αCaMKII has been proposed to play an additional role in maintaining existing synaptic changes (11). After activation, αCaMKII is capable of autophosphorylation, which renders the kinase activity of the molecule Ca2+/calmodulin-independent, prolonging the duration of the activated state of the molecule and enabling kinase activity to outlast typically fleeting Ca2+ transients (12). The autophosphorylation occurs through an intramolecular reaction (13). This activated, Ca2+-autonomous state constitutes a mechanism through which αCaMKII might act as a molecular switch, with stable “on” and “off” states. A stably activated, dendritically localized population of αCaMKII could maintain the elevated synaptic strength of a potentiated synapse by chronically signaling a “potentiated” state to target molecules such as ion channels (11).
The unique structure and function of αCaMKII make this hypothesis attractive. By using mice that were genetically engineered to carry αCaMKII incapable of autophosphorylation, we tested this hypothesis in a model of synaptic change in vivo, ocular dominance plasticity. Ocular dominance plasticity is a well characterized activity-dependent form of synaptic plasticity, in which αCaMKII autophosphorylation has been shown to play a key role (14). Competitive interactions resulting from an imbalance in the activity of thalamocortical afferents subserving the two eyes drive synaptic change in this paradigm (15). Suturing one eye shut (monocular deprivation, MD) induces this imbalance and thus serves as the stimulus for plasticity. The simplicity of this manipulation makes ocular dominance plasticity an ideal tool with which to dissect the mechanisms underlying the induction of synaptic change from mechanisms underlying maintenance because the timing of MD is easy to control.
Materials and Methods
Experimental Subjects. Genetically modified animals that were used in these experiments carry αCaMKII incapable of autophosphorylation, generated through substitution of alanine for threonine 286 of αCaMKII (T286A) (16). A total of 22 mice, which included homozygous mutants (T286A+/+), heterozygous (Het) mutants, and WT animals, were used in this study. All experiments were carried out in accord with National Institutes of Health guidelines for the care of experimental animals and approved by the University of California, San Francisco, Institutional Animal Care and Use Committee.
Deprivation Protocols. Deprivations were performed according to published protocols (2), except 3% isofluorane (Abbott) in oxygen was used for anesthesia. All long-term MDs (LTMDs) were initiated during or before the peak of the normal critical period for ocular dominance plasticity, extending from postnatal day 26-30. LTMD averaged 20 ± 6 days for WT and Het mutant mice (range, 12-27 days) and 17 ± 6 days for T286A+/+ animals (range, 10-26 days). In all experiments, results from WT and Het mutant mice were grouped because there were no significant differences between them (P > 0.05, for all).
LTMD drives plasticity by conferring a competitive advantage on the afferents serving the nondeprived eye. If αCaMKII autophosphorylation were necessary for the maintenance of plasticity, equalizing activity in the two eyes of T286A+/+ mice (and, thereby, removing the stimulus-driving plasticity) would be expected to result in the loss of the induced synaptic changes, and a gradual reversion of cortical responses to the baseline state. Then, in a second group of animals, the stability of ocular dominance plasticity induced by an initial period of LTMD was assayed by measuring the balance of the cortical input of the two eyes after a second interval of long-term binocular deprivation (LTBD). For T286A+/+ animals, LTMD averaged 17 ± 6 days and LTBD averaged 21 ± 6 days. For WT and Het mutant mice, LTMD averaged 22 ± 5 days and LTBD averaged 23 ± 6 days.
Single-Unit Recording. In all experiments, recordings were performed blind to the genotype of the studied animal. Mice were anesthetized for electrophysiological recording with a combination of Nembutal (50 mg/kg; Abbott) and chlorprothixene (0.2 mg; Sigma) according to standard protocols (2). In each mouse, single units were isolated at intervals of ≥60 μm by using lacquer-coated tungsten electrodes in the binocular region of the primary visual cortex (V1) contralateral to the deprived eye. A hand lamp was used to project moving bars or squares onto a tangent screen to drive neuronal responses. The balance of the input of the two eyes to each unit was scored on the 1-7 ocular-dominance scale of Hubel and Wiesel (17), in which a value of 1 indicates complete domination by the contralateral eye and a value of 7 indicates that input arises from the ipsilateral eye only. For each mouse, these ocular-dominance scores were used to compute a single contralateral bias index (CBI) according to the formula
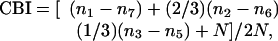 |
where n equals the total number of cells, and nx equals the number of cells with ocular-dominance scores equal to x. CBI scores range from 0 (exclusively ipsilateral input) to 1 (exclusively contralateral input). Most units were scored also for habituation and responsivity on scales of 0 (no habituation/sluggish responsivity) to 3 (prolonged and rapid habituation/brisk firing). Units were scored for the presence of orientation selectivity and, when present, orientation tuning. Because orientation tuning is broad in mice, tuning was measured to the nearest 45°.
Statistics. Student's t test was used for all statistical comparisons.
Results
LTMD Drives Plasticity in T286A+/+ Mice. In a previous study, we showed that a brief period (4 days) of MD, which produces near-saturating plasticity in WT mice, induced a substantially smaller ocular-dominance shift in T286A+/+ mice (14). Although small, this shift was significant (P < 0.05, vs. baseline CBI). Therefore, in these studies, we first used LTMD (see Materials and Methods) to determine whether a stronger stimulus for plasticity (see deprivation paradigms in Fig. 1A) could drive a more robust shift in ocular dominance in the mutant animals. Indeed, extending the length of the deprivation period eventually induced substantial plasticity in T286A+/+ mice, resulting in a shift in responses toward the nondeprived eye that was much stronger than that found with brief MD (CBI = 0.49 ± 0.05 vs. 0.64 ± 0.07 for LTMD and brief MD, respectively; P < 0.01) and indistinguishable from that seen in WT/Het mutant littermates after brief MD (Fig. 2; P > 0.05). After LTMD, the distribution of responses in T286A+/+ mice was, on balance, larger for the nondeprived ipsilateral eye than the deprived contralateral eye. This distribution is in marked contrast to responses recorded in the baseline condition (CBI = 0.74 ± 0.07; P < 0.001) and after brief MD; in both of these cases, responses from the contralateral eye predominate. As shown in ref. 18, LTMD also induced significantly greater plasticity in WT/Het mutant mice than did brief MD (P < 0.05).
Fig. 1.

Experimental deprivation paradigm. (A) LTMD paradigm used to induce ocular dominance plasticity in T286A+/+ mice (Upper) and WT and Het mutant littermates (Lower). Horizontal bars show duration of LTMD for individual animals. Vertical gray strip indicates the peak of the critical period (postnatal days 26-30). (B) LTMD (gray bars) plus LTBD (black bars) paradigm used to assess stability of existing ocular dominance plasticity.
Fig. 2.

LTMD drives robust plasticity in T286A+/+ mice. Baseline CBI values are similar in T286A+/+ mice and WT littermates (No MD; P > 0.05). The effects of the brief 4-day MD were near saturating in WT/Het mutant mice but less pronounced in T286A+/+ mice (Brief MD; P = 0.03 for T286A+/+ mice, baseline vs. brief MD). LTMD drives vigorous plasticity in T286A+/+ animals (LTMD; P < 0.01 for T286A+/+ mice, brief MD vs. LTMD). Each symbol represents a CBI value derived from recordings made in a single animal. Horizontal bars indicate average CBI values for each group. Note the inverted y-axis values. Gray shading indicates baseline CBI range (mean ± SD). Data in the “No MD” and “Brief MD” categories were reproduced here from ref. 14, for comparison.
Ocular Dominance Plasticity Induced in T286A+/+ Mice Is Stable After LTBD. Having induced robust synaptic plasticity in T286A+/+ mice, we sought to determine whether this change in synaptic strength was stably maintained in the absence of the stimulus driving plasticity or whether the defect in sustained αCaMKII phosphorylation also allowed the plasticity to decay back to baseline. For this purpose, we subjected a second group of T286A+/+ mice to LTMD, followed by LTBD of comparable length (i.e., the previously nondeprived eye was also sutured; see deprivation paradigms in Fig. 1B). Ocular dominance plasticity is driven by an imbalance in the activity of thalamocortical afferents. Then, LTBD serves to remove this stimulus for plasticity by equalizing activity in competing sets of afferents.
Responses in T286A+/+ mice after LTMD plus LTBD were nearly identical to responses recorded after LTMD alone (CBI = 0.49 ± 0.05 vs. 0.44 ± 0.07 for LTMD and LTMD plus LTBD, respectively; P > 0.05). In both cases, responses to stimulation of the ipsilateral eye were more robust than those recorded in response to stimulation of the contralateral eye. Distributions of ocular-dominance scores were similarly biased toward high values in both groups (Fig. 3C Upper vs. Lower). If changes induced by LTMD remained labile in T286A+/+ mice, CBI scores after LTMD plus LTBD treatment would be expected to drift back toward baseline values in which responses driven by the contralateral eye predominate. This change did not occur; responses that were dominated by the ipsilateral eye were more common after LTMD plus LTBD (49% of all responses in CBI categories 5-7, vs. 27% in CBI categories 1-3: P < 0.05) than after LTMD alone (41% of all responses in CBI categories 5-7, vs. 42% in CBI categories 1-3: P > 0.05). Thus, T286A+/+ mice, despite showing severe impairments in the induction of ocular dominance plasticity, possess stable and lasting maintenance of this plasticity in the absence of αCaMKII autophosphorylation.
Fig. 3.

Ocular dominance plasticity in T286A+/+ mice is stable in the absence of αCaMKII autophosphorylation. (A) Mean CBI values are unchanged in both WT/Het mutant mice and T286A+/+ after LTMD plus LTBD, relative to LTMD treatment alone (P > 0.05). (B and C) Distribution of ocular dominance values is similar for both genotypic groups after LTMD (Upper) and after LTMD plus LTBD (Lower).
WT and Het mutant mice were similarly resistant to LTBD treatment. CBI scores after LTMD plus LTBD were statistically indistinguishable from those recorded after LTMD alone (CBI = 0.36 ± 0.07 vs. 0.37 ± 0.08 for LTMD plus LTBD and LTMD, respectively; P > 0.05).
Receptive Field Properties in T286A+/+ Mice Are Normal After LTMD Plus LTBD. Plasticity in T286A+/+ animals was stable after LTMD plus LTBD. However, it remained possible that other receptivefield properties might in some way depend on αCaMKII autophosphorylation. Studies in other genetically altered mice have shown that maturation of receptive field properties may depend on molecular mechanisms that are distinct from those underlying ocular dominance plasticity (19). A similar possibility holds for the maintenance of activity-dependent response properties. To more comprehensively characterize cortical responses in these mice, we assayed orientation selectivity, orientation tuning, neural responsivity, and habituation of firing, as well as the distribution of recording depths in T286A+/+ mice and WT/Het mutant littermates after LTMD and LTMD plus LTBD treatments. In no case did response properties differ after LTMD and LTMD plus LTBD treatments for either genotype (Fig. 4 A-C; P > 0.5, for all). Units were encountered over a similar distribution of depths for all combinations of genotypes and deprivation paradigms (Fig. 4D; P > 0.5, for all), with the bulk of all units recorded in presumptive layer IV. Last, none of the properties characterized in T286A+/+ animals following longterm deprivation paradigms differed significantly from those measured in nondeprived WT animals (data not shown; P > 0.05, for all).
Fig. 4.

Response properties are normal in T286A+/+ mice. (A-C) Response properties are similar in WT/Het mutant mice and T286A+/+ mutants after LTMD and LTMD plus LTBD (all P > 0.05). (D) Single units were encountered over similar depths for both genotypic groups and deprivation paradigms. Gray indicates presumptive layer IV. The number of units is as shown in Fig. 3.
Discussion
Previous results from our laboratory showed that αCaMKII autophosphorylation is required for normal ocular dominance plasticity after brief MD (14). Results reported here extend this finding in two ways. First, deficits in plasticity observed in T286A+/+ mice can be compensated for by extending the period of MD. Second, plasticity induced by LTMD is resistant to degradation when activity levels in the two eyes are equalized during an extended period of BD. These results have important implications for understanding the role of αCaMKII autophosphorylation in synaptic plasticity. They suggest that αCaMKII autophosphorylation acts primarily to accelerate the rate at which synaptic change is induced; this model is supported by considerations of the biochemical effects of autophosphorylation, which sustains the molecule in its active conformation. Our results do not support a role for αCaMKII autophosphorylation in maintaining synapses in a potentiated state, because increases in the strength of nondeprived eye inputs are preserved in T286A+/+ animals, even over a period of weeks.
Our experimental approach relies on using LTMD to drive ocular dominance plasticity in T286A+/+ mice. A potential complication of this approach is that residual plasticity in these mutants might be mechanistically different from that occurring in WT animals. For example, PKA-dependent pathways are known to contribute to ocular dominance plasticity (20) and could be activated in parallel with αCaMKII. Thus, PKA activation could set in motion a non-CaMKII-dependent pathway for plasticity, which could drive ocular dominance plasticity in T286A+/+ animals. We cannot rule out the possibility that this or a similar mechanism occurs; even so, our results demonstrate that αCaMKII autophosphorylation is not an absolute requirement for stably maintained ocular dominance plasticity. Also, biochemical studies suggest that even in T286A+/+ mice, the induction of plasticity may be αCaMKII-dependent and mechanistically similar to plasticity occurring in WT animals. Preventing autophosphorylation traps αCaMKII in a Ca2+/calmodulin-dependent state, and consequently the kinetics of the kinase are tightly coupled to those of the rapid and fleeting Ca2+ transients required for its activation. However, in this state, the enzymatic activity of the molecule is not completely blocked. Mutant T286A αCaMKII can still phosphorylate substrate molecules and translocate to the postsynaptic density after Ca2+ influx, albeit transiently (21). Thus, Ca2+/calmodulin-dependent αCaMKII may be able drive ocular dominance plasticity but at a much reduced pace. We would therefore expect that providing a stronger stimulus for plasticity would allow synaptic changes to accrue incrementally, consistent with our findings here.
Our data are consistent with a recent study (22) that demonstrated that αCaMKII autophosphorylation is required for rapid learning in aversive-conditioning paradigms but not for the long-term storage or recall of learned information. In this study, learning (i.e., induction of plasticity) and memory (i.e., maintenance and/or recall of existing plasticity) were tested in T286A+/+ mice and WT littermates by using three aversive-conditioning paradigms. In each task, learning was impaired in T286A+/+ mice but could be compensated for with additional training. In subsequent tests of memory recall, T286A+/+ mice performed identically to WT controls. Combined with our own results, these data provide strong convergent evidence of dissociable molecular substrates for the induction of plasticity (in which αCaMKII autophosphorylation is necessary for both rapid ocular dominance plasticity and fear conditioning) and the maintenance of plasticity (in which αCaMKII autophosphorylation is not required).
We used BD in these experiments because it provided a simple means of terminating the stimulus for plasticity (i.e., the imbalance in retinal activity created by MD). Previous work using a similar paradigm with cats showed that the effects of an initial period of MD persist even after long periods of subsequent dark rearing (23). We extend those findings here by showing similar results for mice by using BD to suppress, rather than eliminate, visual input. An alternative approach would have been to simply open the previously deprived eye after MD, because this manipulation would also equalize activity levels between the two retinas. However, studies of this paradigm suggest that recovery of function during this period of binocular vision requires a second period of plasticity, which may be mechanistically distinct from that mediating ocular dominance plasticity; for example, recovery during normal vision is driven by absolute, rather than relative, levels of activity (24, 25). Thus, deficits in the maintenance of existing plasticity could be obscured by novel plasticity in distinct synapses. We chose to use BD as a simple and direct means of testing the stability of changes in synaptic strength.
Our data complement and extend in vitro evidence of a primary role for αCaMKII autophosphorylation in facilitating the induction of plasticity but not in maintaining it thereafter. Studies of long-term potentiation maintenance generally have shown that αCaMKII inhibitors fail to affect plasticity that has already been induced, although they block long-term potentiation if applied before the inductive stimulus (4, 26, 27). The limitations of the slice preparation necessarily limit conclusions that can be drawn from these studies. In particular, concerns have been raised that αCaMKII may be stable in its autophosphorylated state for as long as 1 day and, thus, could maintain synapses in a potentiated state for the lifetime of the slice (28, 29). The strength of our approach is that an in vivo test for plasticity allows maintenance to be tested over indefinitely long durations.
If αCaMKII autophosphorylation is not required for the maintenance of plasticity, what is? It is possible that other proposed molecular switches may contribute to ocular dominance plasticity maintenance. One such candidate molecule is PKMζ. Data from slice preparations and studies of Drosophila show that sustained activation of PKMζ is required for the maintenance of long-term potentiation and associative learning (30, 31). Another, more speculative candidate includes a member of the cytoplasmic polyadenylation element binding protein, which could be maintained in an active state by a prion-like conformational change (32). The roles of these and other “molecular-switch” proteins in ocular dominance plasticity have yet to be determined.
For many forms of synaptic change, including ocular dominance plasticity, structural changes in connectivity are likely to underlie long-lasting changes in synaptic strength. Ocular dominance plasticity is known to induce anatomical changes in thalamocortical projections to primary visual cortex and to require protein synthesis in cortical networks, which likely contributes to structural changes (18, 33). Indeed, optical imaging techniques have revealed dynamic modulation of dendritic organization in cortical neurons (34). The combination of rapid plasticity-induction mechanisms relying primarily on covalent modifications of molecules, and downstream slower, structural changes effected by rearrangements in axons and dendrites, may provide an appropriate means of balancing competing requirements for rapid plasticity and stability in neural networks.
Acknowledgments
We thank Dr. Jessica Hanover for help with some experiments and Prof. Alcino Silva for helpful discussions. This work was supported by grants to M.P.S. from the National Institutes of Health.
Author contributions: S.A.T. and M.P.S. designed research, analyzed data, and wrote the paper; and S.A.T. performed research.
Conflict of interest statement: No conflicts declared.
Abbreviations: αCaMKII, α calcium calmodulin kinase II; CBI, contralateral bias index; Het, heterozygous; BD, binocular deprivation; LTBD, long-term BD; MD, monocular deprivation; LTMD, long-term MD.
References
- 1.Glazewski, S., Chen, C. M., Silva, A. & Fox, K. (1996) Science 272, 421-423. [DOI] [PubMed] [Google Scholar]
- 2.Gordon, J. A., Cioffi, D., Silva, A. J. & Stryker, M. P. (1996) Neuron 17, 491-499. [DOI] [PubMed] [Google Scholar]
- 3.Kirkwood, A., Silva, A. & Bear, M. F. (1997) Proc. Natl. Acad. Sci. USA 94, 3380-3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malinow, R., Schulman, H. & Tsien, R. W. (1989) Science 245, 862-866. [DOI] [PubMed] [Google Scholar]
- 5.Silva, A. J., Paylor, R., Wehner, J. M. & Tonegawa, S. (1992) Science 257, 206-211. [DOI] [PubMed] [Google Scholar]
- 6.Soderling, T. R., Chang, B. & Brickey, D. (2001) J. Biol. Chem. 276, 3719-3722. [DOI] [PubMed] [Google Scholar]
- 7.Shen, K. & Meyer, T. (1999) Science 284, 162-166. [DOI] [PubMed] [Google Scholar]
- 8.Barria, A., Derkach, V. & Soderling, T. (1997) J. Biol. Chem. 272, 32727-32730. [DOI] [PubMed] [Google Scholar]
- 9.Bayer, K. U., De Koninck, P., Leonard, A. S., Hell, J. W. & Schulman, H. (2001) Nature 411, 801-805. [DOI] [PubMed] [Google Scholar]
- 10.Mammen, A. L., Kameyama, K., Roche, K. W. & Huganir, R. L. (1997) J. Biol. Chem. 272, 32528-32533. [DOI] [PubMed] [Google Scholar]
- 11.Lisman, J. E. (1994) Trends Neurosci. 17, 406-412. [DOI] [PubMed] [Google Scholar]
- 12.Miller, S. G. & Kennedy, M. B. (1986) Cell 44, 861-870. [DOI] [PubMed] [Google Scholar]
- 13.Hanson, P. I., Meyer, T., Stryer, L. & Schulman, H. (1994) Neuron 12, 943-956. [DOI] [PubMed] [Google Scholar]
- 14.Taha, S., Hanover, J. L., Silva, A. J. & Stryker, M. P. (2002) Neuron 36, 483-491. [DOI] [PubMed] [Google Scholar]
- 15.Wiesel, T. N. & Hubel, D. H. (1963) J. Neurophysiol. 26, 1003-1017. [DOI] [PubMed] [Google Scholar]
- 16.Giese, K. P., Fedorov, N. B., Filipkowski, R. K. & Silva, A. J. (1998) Science 279, 870-873. [DOI] [PubMed] [Google Scholar]
- 17.Hubel, D. H. & Wiesel, T. N. (1962) J. Physiol. 160, 106-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antonini, A., Fagiolini, M. & Stryker, M. P. (1999) J. Neurosci. 19, 4388-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fagiolini, M., Katagiri, H., Miyamoto, H., Mori, H., Grant, S. G., Mishina, M. & Hensch, T. K. (2003) Proc. Natl. Acad. Sci. USA 100, 2854-2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beaver, C. J., Ji, Q., Fischer, Q. S. & Daw, N. W. (2001) Nat. Neurosci. 4, 159-163. [DOI] [PubMed] [Google Scholar]
- 21.Shen, K., Teruel, M. N., Connor, J. H., Shenolikar, S. & Meyer, T. (2000) Nat. Neurosci. 3, 881-886. [DOI] [PubMed] [Google Scholar]
- 22.Irvine, E. E., Vernon, J. & Giese, K. P. (2005) Nat. Neurosci. 8, 411-412. [DOI] [PubMed] [Google Scholar]
- 23.Yinon, U. & Goshen, S. (1984) Brain Res. 318, 135-146. [DOI] [PubMed] [Google Scholar]
- 24.Mitchell, D. E. & Gingras, G. (1998) Curr. Biol. 8, 1179-1182. [DOI] [PubMed] [Google Scholar]
- 25.Mitchell, D. E., Gingras, G. & Kind, P. C. (2001) Proc. Natl. Acad. Sci. USA 98, 11662-11667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen, H. X., Otmakhov, N., Strack, S., Colbran, R. J. & Lisman, J. E. (2001) J. Neurophysiol. 85, 1368-1376. [DOI] [PubMed] [Google Scholar]
- 27.Otmakhov, N., Griffith, L. C. & Lisman, J. E. (1997) J. Neurosci. 17, 5357-5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lisman, J., Schulman, H. & Cline, H. (2002) Nat. Rev. Neurosci. 3, 175-190. [DOI] [PubMed] [Google Scholar]
- 29.Zhabotinsky, A. M. (2000) Biophys. J. 79, 2211-2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ling, D. S., Benardo, L. S., Serrano, P. A., Blace, N., Kelly, M. T., Crary, J. F. & Sacktor, T. C. (2002) Nat. Neurosci. 5, 295-296. [DOI] [PubMed] [Google Scholar]
- 31.Drier, E. A., Tello, M. K., Cowan, M., Wu, P., Blace, N., Sacktor, T. C. & Yin, J. C. (2002) Nat. Neurosci. 5, 316-324. [DOI] [PubMed] [Google Scholar]
- 32.Si, K., Lindquist, S. & Kandel, E. R. (2003) Cell 115, 879-891. [DOI] [PubMed] [Google Scholar]
- 33.Taha, S. & Stryker, M. P. (2002) Neuron 34, 425-436. [DOI] [PubMed] [Google Scholar]
- 34.Lendvai, B., Stern, E. A., Chen, B. & Svoboda, K. (2000) Nature 404, 876-881. [DOI] [PubMed] [Google Scholar]