

Abstract
Keratinolytic proteases secreted by dermatophytes are likely to be virulence-related factors. Microsporum canis, the main agent of dermatophytosis in dogs and cats, causes a zoonosis that is frequently reported. Using Aspergillus fumigatus metalloprotease genomic sequence (MEP) as a probe, three genes (MEP1, MEP2, and MEP3) were isolated from an M. canis genomic library. They presented a quite-high percentage of identity with both A. fumigatus MEP and Aspergillus oryzae neutral protease I genes. At the amino acid level, they all contained an HEXXH consensus sequence, confirming that these M. canis genes (MEP genes) encode a zinc-containing metalloprotease gene family. Furthermore, MEP3 was found to be the gene encoding a previously isolated M. canis 43.5-kDa keratinolytic metalloprotease, and was successfully expressed as an active recombinant enzyme in Pichia pastoris. Reverse transcriptase nested PCR performed on total RNA extracted from the hair of M. canis-infected guinea pigs showed that at least MEP2 and MEP3 are produced during the infection process. This is the first report describing the isolation of a gene family encoding potential virulence-related factors in dermatophytes.
Dermatophytes are fungi that have the ability to invade keratinized structures, such as the superficial cornified skin layers, hair, and nails, causing a superficial cutaneous infection called dermatophytosis (48). Microsporum canis is the main agent of dermatophytosis in dogs and cats (41) and causes a zoonosis that has increased in several European countries (23). Furthermore, this zoophilic dermatophyte is the most frequently isolated agent in human tinea capitis in Belgium (20) and in Italy (38). The cat, considered as the natural host and the main reservoir of M. canis, is the principal source of human contamination (41). In addition, the existence of feline asymptomatic infection and the lack of efficient immunoprophylaxis are responsible for the high frequency of endemic M. canis dermatophytosis in catteries (41). In this context, studies on vaccination prophylaxis are recommended by the World Health Organization and the International Society of Human and Animal Mycology (50).
Pathophysiological mechanisms of dermatophytosis, including M. canis infection, are poorly understood. Among potential fungal virulence factors, attention has been paid to proteases for their potential role in the nutrition of the fungi (2), in tissue invasion (1), and in the control of host defense mechanisms (8, 13). Given the ability of dermatophytes to invade and to be essentially confined to keratinized structures, it can be assumed that keratinolytic proteases (keratinases) might be significant virulence factors. Therefore, the characterization of keratinases seems to be a major step for a better understanding of dermatophytic infection pathogenesis and subsequently of the host-fungus relationship. Some keratinases have been isolated from Trichophyton rubrum (1, 3, 19, 25), Trichophyton mentagrophytes (46, 51, 52), and M. canis (5, 22, 29, 42, 43).
Recently, the present authors have purified and characterized two M. canis keratinases, a 31.5-kDa subtilisin-like protease (29) and a 43.5-kDa metalloprotease (5) produced in vitro in a minimal feline keratin-enriched medium. Although keratinases are supposed to be involved in dermatophyte pathogenicity, very few authors have assessed their in vivo production (21, 27, 28). Moreover, only one protease with keratinolytic activity, a recombinant T. rubrum protease, was characterized at the gene level (49). In fact, most authors have only reported enzymatic activities of dermatophyte proteases on different macromolecular substrates and related their in vitro keratinolytic activity to the ability of dermatophytes to invade keratinized structures in vivo. In order to better understand the role of keratinases in M. canis pathogenicity, the assessment of their in vivo production is required.
Despite the fact that the M. canis 31.5-kDa subtilisin-like protease was produced in vivo in naturally infected cats (28) and in experimentally infected guinea pigs (27), this protease does not appear as a major antigen in M. canis infection in these natural and experimental hosts (26, 27). However, cellular and humoral immune responses to a crude M. canis exo-antigen containing mainly the 31.5- and 43.5-kDa keratinases were demonstrated in both animals, suggesting that the 43.5-kDa metalloprotease could be a valuable candidate for further immunological studies, including vaccination trials. Such studies would imply the production of large amounts of protease, which could be facilitated by protein engineering.
This paper describes the isolation of a M. canis metalloprotease gene family (the MEP gene family) and the expression of the 43.5-kDa metalloprotease (MEP3) as a recombinant protein in P. pastoris and reports the in vivo production of two MEPs (MEP2 and MEP3) during experimental M. canis dermatophytosis in guinea pigs.
MATERIALS AND METHODS
Animals and experimental infection.
Two specific-pathogen-free 3-month-old female guinea pigs of the Hartley strain (B & K Universal Limited) were infected with M. canis as described by Van Cutsem (47). Briefly, inoculum consisting of mycelia and spores collected from a 13-day-old slope culture and suspended in a honey-water (2:1, vol/vol) mixture was applied on a 15-cm2 dorsal skin area previously clipped and scarified under general anesthesia. Negative controls consisted of two guinea pigs exposed to the same procedure except that the honey-water mixture did not contain any fungus. Animal infection was regularly monitored using clinical, Wood's light, and microscopic examinations. Animal experiments were approved by the local ethic committee (University of Liège).
Strains and plasmids.
Microsporum canis strain IHEM 15221 (Institute of Hygiene and Epidemiology-Mycology [IHEM], Brussels, Belgium) was used for the construction of the genomic library, the extraction of total RNA, and guinea pig infection. Escherichia coli LE392 (Promega) was used for the propagation of the bacteriophage λEMBL3 (Promega). Plasmid subcloning experiments were performed in E. coli DH5α (Amersham Pharmacia Biotech) or TOP10 (Invitrogen). Subcloning of DNA fragments from the genomic library was performed using plasmid pMTL21 (6). Cloning of MEP cDNAs was performed using plasmid pCR4Blunt-TOPO (Invitrogen) whereas shuttle vector pPICZαB (Invitrogen) and P. pastoris KM71H (Invitrogen) were used to express recombinant MEP3. Cloning of reverse transcriptase nested PCR (RT-nested PCR) products was performed using plasmid pUC18 (Amersham Pharmacia Biotech).
Growth media.
For the construction of the genomic DNA library, M. canis was cultivated in liquid Sabouraud medium for 7 days before DNA extraction. For RNA extraction, M. canis was cultivated for 12 days in the previously described minimal liquid medium containing feline keratin as the sole nitrogen source (29).
Construction of the genomic library.
Genomic DNA was obtained as described by Girardin et al. (12) and purified using Genomic Tips (Qiagen). The isolated DNA was partially digested with Sau3AI. DNA fragments ranging from 12 to 20 kb were isolated from low-melting-point agarose (Bio-Rad) and inserted into bacteriophages using the λEMBL3 BamHI arm cloning system (Promega).
Screening of the genomic library.
Approximately 3 × 104 recombinant plaques of the genomic library were immobilized on nylon membranes (Genescreen; NEN Life Science Products). The membranes were prehybridized for 45 min in 5× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) solution containing 20% formamide, 1% SDS, 10% dextran sulfate, and denatured salmon sperm DNA (100 μg/ml) at 42°C. Thereafter, the filters were probed using low-stringency conditions at 42°C for 12 h with an α-32P-labeled Aspergillus fumigatus DNA corresponding to the MEP gene. This probe was obtained by PCR using primers 5′-ATG CGC GGC CTT CTT CTG-3′ and 5′-TCA ACA GAC ACC ACT GGG-3′ and the plasmid pMTL21-H5 (15) as template DNA. The PCR product was labeled with [α-32P]dATP using a random-primed DNA labeling kit (Boehringer). The membranes were exposed to X-ray film after two 20-min washes in 2× SSC solution containing 1% SDS at 40°C. Positive plaques were purified, and the associated bacteriophage DNAs were isolated as described by Grossberger (14). Agarose gel electrophoresis of restricted recombinant bacteriophage λEMBL3 DNA and Southern blotting were performed according to standard protocols (39). Hybridizing fragments were subcloned into pMTL21 using standard procedures (39). Microsynth (Balgach, Switzerland) performed the sequencing of the MEP genes.
RT-PCR.
PCRs and RT-PCRs were performed with a Biozym PTC 200 Thermal Cycler. M. canis total RNA was isolated from a 12-day-old culture using the filamentous fungi protocol of a Qiagen RNeasy total RNA purification kit. RT-PCRs were performed on 1 μg of total RNA using a Qiagen One-Step RT-PCR kit, according to the manufacturer's instructions except that 1 U of Pfu DNA polymerase (Promega) was added. Primers used for MEP1, MEP2, and MEP3 were primers 1 and 2, primers 5 and 6, and primers 9 and 10, respectively (Table 1), at a concentration of 40 μM each. The reaction mixtures were incubated at 50°C for 30 min and 95°C for 15 min and then were subjected to 35 cycles of 1 min at 94°C, 1 min at 55°C, and 4 min at 68°C and finally were incubated for 10 min at 68°C. The PCR products were visualized on a 0.8% agarose gel.
TABLE 1.
Primers used for isolation of M. canis IHEM 15221 MEP cDNAs by RT-PCR and for assessment of in vivo production of M. canis MEPs by RT-nested PCR
| Gene | Primer no. | Oligonucleotide primer sequence | Orientation | Encoded amino acid sequencea | Template | PCR product length (bp) (cloning sites) |
|---|---|---|---|---|---|---|
| MEP1 | 1 | CTT CTT TTC (C/T)C GAG AAA AGA CAC CCC CAG CCG TCG AGC ACA AGC | Senseb | (L)(L)(F)(L)(E)(K)(R)HPQPSSTS | Total RNA | 1,883 (PstI-NotI) |
| 2 | GTT GTT GTT G(C/G)GC CGC CTT GCA CTC CTT TGG AAC CTC GTT GC | Antisenseb | NEVPKECK(A)(A)(A)(T)(T) | |||
| 3 | GAG ATG ATG AAG CAG CCC | Sensec | EMMKQP | cDNA or pP-cMEP1 | 384 | |
| 4 | CTC GTC CAT GTT TTC AAC | Antisensec | VENMDE | |||
| MEP2 | 5 | GA AGT TCG T(C/T) GCA GTC CAC CCA GCC CGT CCA CAA ACT G | Senseb | (S)(S)(S)(A)(V)HPARPQT | Total RNA | 1,873 (XhoI-NotI) |
| 6 | GTT GTT GTT G(C/G)GCCGC GCA GCC AGA GGG CAT GTC GAA G | Antisenseb | FDMPSGC(A)(A)(A)(T)(T) | |||
| 7 | AAC GAG CAC AAG ACC ACC | Sensec | NEHKTT | cDNA or pP-cMEP2 | 663 | |
| 8 | GTC CTT GAT CAT GGA GGG | Antisensec | PSMIKD | |||
| MEP3 | 9 | GTT (C/T)GGAG CAC CCT GCT GAG CAG CAG ACC | Senseb | (V)(L)(E)HPAEQQT | Total RNA | 1,868 (PstI-NotI) |
| 10 | GTT G(C/G)GC CGC TTA GCA GCC TGG TGG AGG TTG | Antisenseb | QPPPGC STOP | |||
| 11 | AC CCA ACC AGC TTC CGT G | Sensec | PTSFR | cDNA or pP-cMEP3 | 857 | |
| 12 | GTC AGC ACT GTA GGT ACC | Antisensec | GTYSAD |
In parentheses are shown amino acids encoded by the restriction enzyme site sequences and amino acids encoded by the nucleic sequences added to those corresponding to both N-terminal and C-terminal extremities of MEPs.
Primer used for RT-PCR.
Internal primer used for nested PCR
Construction of expression plasmid.
RT-PCR products were incubated for 40 min at 68°C with 1 U of Pfu DNA polymerase and were further purified using a PCR product purification kit (Boehringer). The isolated cDNAs were inserted into plasmid pCR4Blunt-TOPO according to the instructions of the manufacturer, generating the plasmids pT-cMEP1, pT-cMEP2, and pT-cMEP3. These plasmids were, respectively, digested by PstI-NotI, XhoI-NotI, and PstI-NotI. Digestion products corresponding to cDNA inserts were gel purified, and ligation into the expression shuttle vector pPICZαB, previously digested with the appropriate enzymes, was performed using standard protocols (39), generating plasmids pP-cMEP1, pP-cMEP2, and pP-cMEP3. Eurogentec (Liège, Belgium) performed the sequencing of MEP cDNAs.
Expression of recombinant M. canis MEP3.
P. pastoris KM71H was transformed by electroporation with 10 μg of pP-cMEP3 linearized by DraI, using the following parameters: 1,000 V, 329 Ω, and 40 μF. As a negative control, 10 μg of pPICZαB linearized by DraI was used. Transformants were selected on yeast extract peptone dextrose agar containing Zeocin (100 μg/ml; Invitrogen) after incubation at 30°C for 3 days. Twenty transformants were used to inoculate 40 ml of buffered glycerol complex medium (BMGY) (Invitrogen) (0.1 M potassium phosphate buffer at pH 6.0 containing 1% [wt/vol] yeast extract, 2% [wt/vol] peptone, 1.34% [wt/vol] yeast nitrogen base without amino acids, 1% [vol/vol] glycerol, and 4 × 10−5% [wt/vol] biotin). Cultures were grown at 30°C in a shaking incubator (260 rpm) until the cultures reached an absorbance of 4 at 600 nm. Cells were harvested by centrifugation (1,500 × g for 10 min) and resuspended in 8 ml of buffered methanol complex medium (same medium as BMGY except that glycerol was replaced by methanol [0.5%, vol/vol]) and incubated for 6 days. Expression of MEP3 was induced by adding once a day a volume of methanol to a final concentration of 0.5% (vol/vol).
Purification procedure and N-terminal sequencing.
To identify clones expressing MEP3 and monitor its time course production, samples were harvested once a day, subjected to trichloroacetic precipitation, and loaded on a 12% polyacrylamide gel. SDS-polyacrylamide gel electrophoresis (PAGE) was performed using the method of Laemmli (18). Culture supernatant was separated from cells by centrifugation (1,500 × g for 10 min), concentrated by ultrafiltration on Amicon YM 10 (Amicon, Beverly, Mass.), dialysed against phosphate buffer (25 mM NaH2PO4, pH 6), and applied onto a carboxymethyl-agarose column previously equilibrated with the same buffer. Elution was performed with a linear gradient of salt (40 ml of phosphate buffer; 40 ml of phosphate buffer plus 0.1 M NaCl, pH 6). Electroblotting to an Immobilon membrane (Millipore) was performed according to standard protocols (45). After staining with Coomassie brilliant blue R-250 (Bio-Rad), the bands were cut and subjected to N-terminal sequencing on a Perkin-Elmer sequencer type Procise.
Proteolytic assays.
Collagenolytic activity was measured using azocoll (Sigma) as a substrate. Briefly, 200 μl of P. pastoris supernatant was added to 800 μl of 50 mM Tris-HCl-2 mM CaCl2, pH 7.5 (Tris buffer) and incubated with 10 mg of azocoll at 37°C for 30 min under continuous shaking. The degradation of the substrate was estimated by measuring the absorbance of the supernatant at 520 nm after centrifugation. Keratinolytic activity was measured using keratin azure (Sigma), essentially as previously described (29). Briefly, 200 μl of sample supernatant was added to 800 μl of Tris buffer and incubated with 5 mg of keratin azure at 37°C for 24 h. Keratin degradation was estimated by measuring the absorbance of the supernatant at 595 nm after centrifugation.
RNA extraction from M. canis-infected hairs.
Wood's light-positive M. canis-infected hairs, located in the scarification area, were collected from guinea pigs on days 14 and 21 postinoculation. Wood's light-negative noninfected control hairs were plucked from outside the scarification area of infected guinea pigs and from inside the scarification area of control animals. Approximately 20 hairs of each animal's sample were ground under liquid nitrogen with a pestle and a mortar, and total RNA was extracted by using the filamentous-fungus protocol of a Qiagen RNeasy Total RNA purification kit. Total RNA was subsequently treated with RNase-free DNase according to the manufacturer's instructions (Promega).
RT-nested PCR.
Ten microliters of freshly DNase-treated total RNA were used for RT-PCRs using a Qiagen One-Step RT-PCR kit. Primers 1 and 2 for MEP1, primers 5 and 6 for MEP2, and primers 9 and 10 for MEP3 (Table 1) were added at a concentration of 40 μM each. The reaction mixtures were incubated at 50°C for 30 min and 95°C for 15 min; subjected to 47 cycles of 1 min at 94°C, 1 min at 55°C, and 2 min at 72°C; and finally were incubated for 10 min at 72°C. RT-PCR products were then used as a template for a subsequent PCR using a Promega PCR Core system kit. RT-PCR products (5 to 10 μl) were added to 5 μl of 10× PCR buffer (100 mM Tris-HCl [pH 9], 500 mM KCl, 15 mM MgCl2, 1% Triton X-100); 2 μl of deoxynucleoside mix (containing 10 mM of each deoxynucleoside triphosphate); 1 μl of internal primers 3 and 4 for MEP1, 7 and 8 for MEP2, and 11 and 12 for MEP3 (Table 1), at a concentration of 40 μM each; and 2.5 U of Taq DNA polymerase. The reaction mixtures were incubated at 95°C for 5 min; subjected to 32 cycles of 1 min at 94°C, 1 min at 55°C, and 2 min at 72°C; and finally were incubated for 10 min at 72°C.
Cloning of RT-nested PCR fragments.
RT-PCR fragments were purified using a PCR product purification kit (Boehringer). They were subjected to a mung bean nuclease treatment according to the manufacturer's instructions (Promega). After ligation in plasmid pUC18 previously linearized with SmaI, RT-nested PCR fragments were sequenced using a Perkin-Elmer sequencer.
Nucleotide sequence accession numbers.
The sequences of the M. canis genes MEP3 (exons 1 to 5), MEP1 (exons 1 to 5), and MEP2 (exons 1 and 2) have been submitted to the EMBL nucleotide sequence database under accession numbers AJ490183, AJ190184, and AJ190185, respectively.
RESULTS
Cloning of three M. canis metalloprotease genes.
A screening of a M. canis λEMBL3 genomic library was performed using the A. fumigatus MEP gene as a probe and under low-stringency conditions of hybridization. Among 3 × 104 individual recombinant bacteriophage plaques, about 60 hybridizing clones were purified. Restriction enzyme analysis with EcoRI of purified bacteriophage DNA revealed three different groups of clones carrying similar, but nonidentical DNA fragments. By Southern analysis with the same probe and under low-stringency conditions, MEP1 and MEP3 genes were assigned to a 4.3-kb XhoI fragment and a 4.0-kb BamHI fragment, respectively. The MEP2 gene was located on two XhoI fragments of 2 and 5 kb. All these genomic fragments were subcloned into plasmid pMTL21. Sequencing revealed three long open reading frames of 2,172, 1,957, and 2,186 bp, for MEP1, MEP2, and MEP3, respectively. The amino acid sequences deduced from these three genes showed a quite high percentage of sequence identity between them (Table 2) and with both A. fumigatus MEP (15, 24, 31) and Aspergillus oryzae neutral protease I (NP I) (10) (Fig. 1). They all contained an HEXXH motif (Fig. 1). The amino acid sequence analysis suggested that M. canis MEP1, MEP2, and MEP3 would be synthesized as preproproteins with prosequences of 243, 244, and 246 amino acid residues, respectively. The mature protein sequence generated after cleavage of the prosequences predicted a molecular mass of 43.1 kDa for MEP1 and 42.5 kDa for MEP2 and MEP3 (Table 3).
TABLE 2.
Pairwise amino acid sequence comparisons between M. canis IHEM 15221 MEPs, A. fumigatus MEP, and A. oryzae NP I
| Enzyme | % similarity or identitya to:
|
||||
|---|---|---|---|---|---|
| A. fumigatus MEP | A. oryzae NP I |
M. canis metalloprotease:
|
|||
| MEP1 | MEP2 | MEP3 | |||
| A. fumigatus MEP | 84.5 | 72.9 | 70.5 | 73.9 | |
| A. oryzae NP I | 77.6 | 72.6 | 71.4 | 73.9 | |
| M. canis MEP1 | 63 | 62.4 | 68.9 | 74.4 | |
| M. canis MEP2 | 60.4 | 60 | 57.5 | 78.6 | |
| M. canis MEP3 | 63 | 64.2 | 64 | 68.3 | |
The percent similarity (top right-hand corner) and percent identity (bottom left-hand corner) values were obtained with the program Vector NTI (InforMax).
FIG. 1.

Alignment of deduced amino acid sequences of the M. canis IHEM 15221 metalloproteases (MEP1, MEP2, and MEP3) with A. fumigatus MEP (16, 24, 31) and A. oryzae NP I (10). Identical amino acids are shown on a black background, and homologous amino acids are shown on a gray background. The putative signal peptidase cleavage sites are indicated by a vertical arrowhead. An arrow delimits the N-terminal positions of the mature proteases. The sequence motif HEXXH found in zinc-dependent metalloproteases is boxed. The alignment was performed with Vector NTI (InforMax, Bethesda, Md.) software and reformatted with Boxshade software (version 3.31).
TABLE 3.
Molecular characteristics of M. canis IHEM 15221 MEPs
| Characteristic | Value for M. canis metalloprotease
|
||
|---|---|---|---|
| MEP1 | MEP2 | MEP3 | |
| Preproprotein length (amino acids) | 632 | 632 | 633 |
| Mature domain of the protein (amino acids) | 389 | 388 | 387 |
| Theoretical molecular mass of the polypeptide chain of the mature domain (kDa)a | 43.1 | 42.5 | 42.5 |
| Apparent molecular mass determined by SDS-PAGE (kDa)b | NDc | ND | 43.5 |
| No. of putative glycosylation sites | 2 | 1 | 2 |
| Calculated pIa (measured pIb) | 8.69 | 4.9 | 8.43 (7.7) |
| Optimum pH of activity experi- mentally determined | ND | ND | 8b |
The theoretical molecular mass of the mature domain and the pI were calculated with the software Vector NTI.
The pI, optimum pH, and apparent molecular mass of native MEP3 were previously reported (5).
ND, not done.
RT-PCR.
RT-PCR products, corresponding to MEP cDNAs, were visualized as bands of approximately 1,900 bp on an agarose gel. Sequencing of plasmids pP-cMEP1, pP-cMEP2, and pP-cMEP3 confirmed previous genomic DNA sequencing. Molecular characteristics of MEPs are described in Table 3.
Production of MEP3 as recombinant protease.
SDS-PAGE of culture supernatants of P. pastoris transformed with plasmid pP-cMEP3 showed three bands of approximately 44.5, 42.5, and 25 kDa after the chromatographic step on carboxymethyl-agarose (Fig. 2). Supernatants of P. pastoris transformed with only the parent vector showed neither major bands nor proteolytic activity, while both keratinolytic and collagenolytic activities were detected when P. pastoris was transformed with plasmid pP-cMEP3 (data not shown). N-terminal amino acid sequencing revealed that both bands of 44.5 and 42.5 kDa had the same N-terminal sequence (AEFKV) as that of the native protease, whereas the N-terminal sequence of the 25-kDa product was determined to be FSKEDD, which corresponded to amino acids of the MEP3 prosequence. The yield of recombinant MEP3 production was around 40 μg/ml.
FIG. 2.
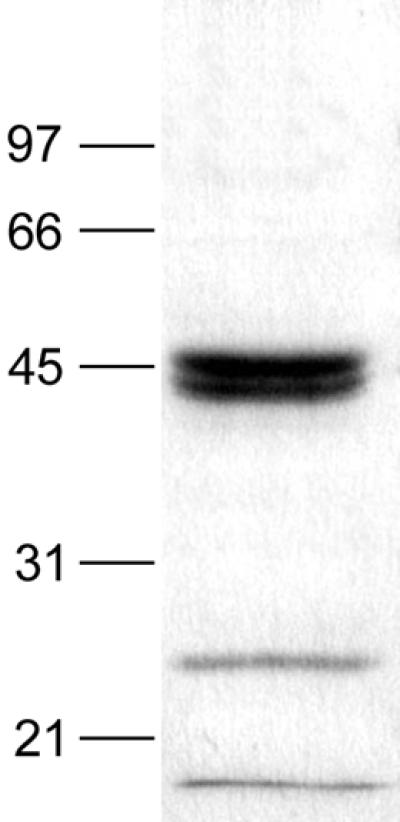
Expression of M. canis IHEM 15221 recombinant MEP3. The purified supernatant of P. pastoris transformed with pP-cMEP3 was loaded on an SDS-12% polyacrylamide gel, which was stained with Coomassie brilliant blue R-250. Molecular mass standards (in kilodaltons) are shown on the left. The stained band lower than 21 kDa is the running front.
In vivo expression of MEPs.
RT-nested PCR products, obtained from total RNA extracted from guinea pigs' M. canis-infected hairs, were visualized on an agarose gel as bands of approximately 650 bp for MEP2 and 850 bp for MEP3, while no band was observed for MEP1 (Fig. 3). The same result was obtained in both M. canis-infected guinea pigs on days 14 and 21 postinoculation, while no signal was observed in control animals (Fig. 3). Sequencing of plasmids pUC18 containing RT-nested PCR fragments showed that they were specific to MEP2 and MEP3 cDNAs.
FIG. 3.

Assessment of the in vivo production of M. canis IHEM 15221 MEPs. Lane 1, molecular weight marker (1-kb ladder; Gibco); lanes 2, 5, and 8, RT-nested PCR products obtained from hairs from M. canis-infected guinea pigs, corresponding to internal cDNA fragments of MEP1, MEP2, and MEP3, respectively; lanes 3, 6, and 9, RT-nested PCR products obtained from hairs from M. canis-noninfected guinea pigs; lanes 4, 7, and 10, PCRs performed on plasmids pP-cMEP1, pP-cMEP2, and pP-cMEP3. Primers used for PCRs were primers 3 and 4 for MEP1, primers 7 and 8 for MEP2, and primers 11 and 12 for MEP3 (Table 1). MEP1, MEP2, and MEP3 internal cDNA products were 384, 663, and 857 bp, respectively.
DISCUSSION
Three genes (named MEP1, MEP2, and MEP3) were isolated from a λEMBL3 M. canis genomic library using a PCR-generated probe from the A. fumigatus MEP gene, under low-stringency conditions of hybridization. One of these genes (MEP3) was found to be the one encoding a previously described 43.5-kDa M. canis keratinolytic metalloprotease (5). The deduced amino acid sequence analysis of the three M. canis MEPs showed that they all contained an HEXXH motif and disclosed a high percentage of sequence identity between them and with A. fumigatus MEP (15, 24, 31) and A. oryzae NP I (10). These last two enzymes are zinc-containing metallopeptidases of the M36 peptidase family (fungalysin family) containing fungal metalloproteases, with A. fumigatus MEP (fungalysin) as the type example (37). The M36 family belongs to the metallopeptidase clan MA(E), also called the clan of gluzincins, in which are classified metallopeptidases containing a zinc ion ligated by two histidines within the motif HEXXH and by a glutamic acid residue (HEXXH+E) (36, 37). In order to determine if other conserved motifs than HEXXH were present in M. canis MEPs, several sequence alignments were performed with other metalloproteases. An alignment with metalloproteases of the M4 family (thermolysin family) showed that M. canis MEPs, A. fumigatus MEP, and A. oryzae NP I presented three conserved amino acid residues (a glycine, a glutamic acid, and an aspartic acid residue) about twenty residues C-terminal to the HEXXH motif (Fig. 4), which seems to correspond to the second zinc-binding region of the thermolysin family (GXXNEXXSD) in which the glutamic acid is the third zinc ligand (30). This suggests that M. canis MEP1, MEP2, and MEP3 can be assigned to the M36 peptidase family. The determination of the three-dimensional structures and the use of molecular biological techniques to identify residues involved in zinc coordination or in catalysis will further confirm or deny the roles of these putative zinc-binding regions in M. canis MEPs and Aspergillus metalloproteases. Despite a high percentage of sequence isology between M. canis MEP3 and Aspergillus metalloproteases, they possess different proteolytic activities. Indeed, A. fumigatus MEP cleaves collagen but cleaves neither keratin (M. Monod, personal communication) nor elastin Congo red (31), even if elastinolytic activity was reported by one author (24), and A. oryzae NP I cleaves collagen but cleaves neither keratin nor elastin (M. Monod, personal communication), whereas M. canis MEP3 showed collagenolytic, elastinolytic, and keratinolytic activities (5). To date, differences in proteolytic activities among M. canis MEP3, A. fumigatus MEP, and A. oryzae NP I cannot be attributed to a particular change in amino acid composition.
FIG. 4.

Alignment of bacterial metalloproteases of the M4 peptidase family (thermolysin family) with A. fumigatus MEP, A. oryzae NP I, and M. canis MEPs showing the conserved HEXXH motif characteristic of zinc metallopeptidases and the three conserved amino acid residues (G, E, and D) belonging to the second zinc binding motif of metalloproteases of the M4 family. Sequences: 1, Bacillus thermoproteolyticus thermolysin (EC 3.4.24.27); 2, Bacillus subtilis neutral protease B; 3, Clostridium perfringens lambda toxin; 4, Legionella pneumophila zinc metalloprotease; 5, Pseudomonas aeruginosa elastase (EC 3.4.24.26); 6, A. fumigatus MEP; 7, A. oryzae NP I; 8, M. canis MEP1; 9, M. canis MEP2; 10, M. canis MEP3. Conserved amino acids are shown on a black background.
Other homologies between M. canis and Aspergillus sp. proteases have already been reported. Based on N-terminal amino acid sequencing, the 31.5-kDa M. canis keratinolytic subtilisin-like protease purified by Mignon et al.(29) disclosed marked similarities with other subtilisin-like proteases from A. fumigatus (16), A. oryzae (44), Aspergillus nidulans (17), and Aspergillus flavus (35). Moreover, the recent molecular characterization of the 31.5-kDa M. canis keratinase gene showed that it was homologous to the A. fumigatus alkaline protease gene (ALP) (Descamps et al., unpublished results). These results strengthen the hypothesis (5) according to which these fungi, belonging to the Plectomycetes class, would possess fundamental similarities in their proteolytic system, even if they produce proteinases with specificities related to the substrates they hydrolyze and tissues these fungi can invade.
Although some microbial metalloproteases were shown to be involved in bacterial virulence, such as the lethal factor of Bacillus anthracis (11) or the 35-kDa metalloprotease of Listeria monocytogenes (7), this is not the case so far with any fungal metalloprotease (30). To date, no correlation was clearly demonstrated between the proteolytic activity of A. fumigatus, which relies mainly on its ALP and MEP, and its capacity to invade the pulmonary tract. Indeed, no difference in pathogenicity was observed between wild-type and alp mep double mutant A. fumigatus strains in a murine model of experimental invasive aspergillosis (15). However, while only one MEP was described in A. fumigatus, the existence of a metalloprotease gene family in M. canis could suggest the potentially important role of these enzymes in fungus metabolism and pathogenicity. Indeed, the production of several proteases encoded by a gene family is thought to be related to virulence, as it has been suggested for acid proteases (Saps) of Candida albicans (4, 32) and Candida tropicalis (53). Moreover, it is now well established that several different Saps are involved in the pathogenesis of C. albicans infection (9, 40).
A major step in the evaluation of the role of the M. canis MEPs in virulence is to assess their in vivo production in skin structures. Consequently, RT-nested PCRs, using specific primers designed for each MEP, were performed on total RNA extracted from experimentally infected guinea pig hairs. This method was used rather than immunochemistry in order to prevent possible antibody-antigen cross-reactions among M. canis MEPs. Under the experimental conditions used, MEP2 and MEP3 but not MEP1 were shown to be produced during M. canis dermatophytosis at days 14 and 21 postinoculation, which corresponded to the period of lesion extension. The absence of signal for MEP1 was not related to a lack of specificity of MEP1 primers as shown by positive signals obtained when using the same PCR parameters with pP-cMEP1 as a template (Fig. 3). As the in vivo detection of gene products can suffer from a lack of sensitivity (33, 34), additional PCR cycles were performed but did not give any positive signal for MEP1 (data not shown). However, since only days 14 and 21 postinoculation were checked, the possibility that MEP1 is produced at other moments during infection cannot be excluded.
Besides the characterization of fungal virulence factors, the determination of antigenic epitopes able to induce an immune protective response against natural infection is another important investigation field in the comprehension of the host-fungus relationship. Both cellular and humoral immune responses against a crude M. canis exo-antigen were reported in naturally infected cats (26) and in experimentally infected guinea pigs (27). Despite the fact that the 31.5-kDa subtilisin-like protease is a major component of this exo-antigen (26) and is produced in vivo in naturally infected cats and experimentally infected guinea pigs, it seems not to be a major antigen in M. canis infection (27, 28). The 43.5-kDa keratinolytic metalloprotease, which was shown to be another major component of this exo-antigen (5), could be a more valuable candidate for further immunological studies, including vaccination trials. However, since the production of the native MEP3 leads to small amounts of pure protease and is time-consuming, the use of protein engineering was required. Recombinant MEP3 was therefore produced in P. pastoris at a quite-high rate of 40 μg/ml. After a chromatographic step on carboxymethyl-agarose, the SDS-PAGE pattern presented three major bands of 44.5, 42.5, and 25 kDa. N-terminal amino acid sequencing showed that the two higher bands had the same amino acid sequence, identical to that of the native protease, while the band at 25 kDa presented a sequence identical to a part of MEP3 prosequence. The presence of two different bands of 44.5 and 42.5 kDa could be related to a difference in protein glycosylation or a degradation at the carboxy-terminal extremity. The band at 25 kDa reflected a proteolysis product. Recombinant MEP3 showed collagenolytic and keratinolytic activities under the same conditions as the native protein (5), indicating an apparently correct folding of the enzyme.
This is, to our knowledge, the first report describing the isolation of genes encoding potential virulence-related factors in M. canis. Furthermore, these genes belong to a gene family from which at least 2 members are produced during the infection process in a guinea pig experimental model. These results, together with the keratinolytic activity of at least MEP3, reinforce the necessity to demonstrate whether or not these proteases are involved in M. canis pathogenicity. The role of MEPs in fungal virulence should definitively be ascertained by the construction of MEP mutants by gene-targeting disruption, and by comparing the pathogenicities of mutant and wild-type M. canis strains in experimental animal infection models. In the near future, the in vivo production of MEPs in naturally acquired feline dermatophytosis will be assessed and compared to the results obtained in experimentally infected guinea pigs. Furthermore, as native MEP1 and MEP2 have never been isolated so far, their production as recombinant enzymes will be performed both to evaluate their proteolytic activities and to use them in immunological studies. These studies would certainly lead to a better comprehension of the host-fungus relationship and provide information for the design of new prophylactic tools against M. canis dermatophytosis.
Acknowledgments
We thank Barbara Léchenne, Christophe Zaugg, Didier Baar, Jacques Detry, and Humbert Gianfreda for excellent technical assistance.
This work was supported by grant 3.4534.01 from the Fonds de la Recherche Scientifique Médicale (FRSM) and by grant 3200-063697.00 from the Fonds National Suisse pour la Recherche Scientifique. Frédéric Brouta and Frédéric Descamps are recipients of a studentship of FRIA (Fonds pour la Formation à la Recherche dans l'Industrie et dans l'Agriculture, Brussels, Belgium).
Editor: T. R. Kozel
REFERENCES
- 1.Apodaca, G., and J. H. McKerrow. 1989. Purification and characterization of a 27,000-Mr extracellular proteinase from Trichophyton rubrum. Infect. Immun. 57:3072-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Apodaca, G., and J. H. McKerrow. 1989. Regulation of Trichophyton rubrum proteolytic activity. Infect. Immun. 57:3081-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asahi, M., R. Lindquist, K. Fukuyama, G. Apodaca, W. L. Epstein, and J. H. McKerrow. 1985. Purification and characterization of major extracellular proteinases from Trichophyton rubrum. Biochem. J. 232:139-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borg-von Zepelin, M., S. Beggah, K. Boggian, D. Sanglard, and M. Monod. 1998. The expression of the secreted aspartyl proteinases Sap4 to Sap6 from Candida albicans in murine macrophages. Mol. Microbiol. 28:543-554. [DOI] [PubMed] [Google Scholar]
- 5.Brouta, F., F. Descamps, T. Fett, B. Losson, C. Gerday, and B. Mignon. 2001. Purification and characterization of a 43.5 kDa keratinolytic metalloprotease from Microsporum canis. Med. Mycol. 39:269-275. [DOI] [PubMed] [Google Scholar]
- 6.Chambers, S. P., S. E. Prior, D. A. Barstow, and N. P. Minton. 1988. The pMTL nic− cloning vectors. Improved pUC polylinker regions to facilitate the use of sonicated DNA for nucleotide sequencing. Gene 68:139-149. [DOI] [PubMed] [Google Scholar]
- 7.Coffey, A., B. van den Burg, R. Veltman, and T. Abee. 2000. Characteristics of the biologically active 35-kDa metalloprotease virulence factor from Listeria monocytogenes. J. Appl. Microbiol. 88:132-141. [DOI] [PubMed] [Google Scholar]
- 8.Collins, J. P., S. F. Grappel, and F. Blank. 1973. Role of keratinases in dermatophytosis. II. Fluorescent antibody studies with keratinase II of Trichophyton mentagrophytes. Dermatologica 146:95-100. [PubMed] [Google Scholar]
- 9.De Bernardis, F., S. Arancia, L. Morelli, B. Hube, D. Sanglard, W. Schafer, and A. Cassone. 1999. Evidence that members of the secretory aspartyl proteinase gene family, in particular SAP2, are virulence factors for Candida vaginitis. J. Infect. Dis. 179:201-208. [DOI] [PubMed] [Google Scholar]
- 10.Doumas, A., R. Crameri, B. Léchenne, and M. Monod. 1999. Cloning of the gene encoding neutral protease I of the koji mold Aspergillus oryzae and its expression in Pichia pastoris. J. Food Mycol. 2:271-279. [Google Scholar]
- 11.Duesbery, N. S., C. P. Webb, S. H. Leppla, V. M. Gordon, K. R. Klimpel, T. D. Copeland, N. G. Ahn, M. K. Oskarsson, K. Fukasawa, K. D. Paull, and G. F. Vande Woude. 1998. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280:734-737. [DOI] [PubMed] [Google Scholar]
- 12.Girardin, H., and J.-P. Latgé. 1994. DNA extraction and quantitation, p. 5-9. In B. Maresca and G. S. Kobayashi (ed.), Molecular biology of pathogenic fungi: a laboratory manual. Telos Press, New York, N.Y.
- 13.Grappel, S. F., and F. Blank. 1972. Role of keratinases in dermatophytosis. I. Immune responses of guinea pigs infected with Trichophyton mentagrophytes and guinea pigs immunized with keratinases. Dermatologica 145:245-255. [PubMed] [Google Scholar]
- 14.Grossberger, D. 1987. Minipreps of DNA from bacteriophage lambda. Nucleic Acids Res. 15:6737.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaton-Ogay, K., S. Paris, M. Huerre, M. Quadroni, R. Falchetto, G. Togni, J. P. Latgé, and M. Monod. 1994. Cloning and disruption of the gene encoding an extracellular metalloprotease of Aspergillus fumigatus. Mol. Microbiol. 14:917-928. [DOI] [PubMed] [Google Scholar]
- 16.Jaton-Ogay, K., M. Suter, R. Crameri, R. Falchetto, A. Fatih, and M. Monod. 1992. Nucleotide sequence of a genomic and cDNA clone encoding an extracellular alkaline protease of Aspergillus fumigatus. FEMS Microbiol. Lett. 92:163-168. [DOI] [PubMed] [Google Scholar]
- 17.Katz, M. E., R. N. Rice, and B. F. Cheetham. 1994. Isolation and characterization of an Aspergillus nidulans gene encoding an alkaline protease. Gene 150:287-292. [DOI] [PubMed] [Google Scholar]
- 18.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 19.Lambkin, I., A. J. Hamilton, and R. J. Hay. 1996. Purification and characterisation of a novel 34,000-Mr cell-associated proteinase from the dermatophyte Trichophyton rubrum. FEMS Immunol. Med. Microbiol. 13:131-140. [DOI] [PubMed] [Google Scholar]
- 20.Lateur, N. 2000. Tinea capitis chez l'enfant: diagnostic, étiologie et traitement. Percentile 5:100-103. [Google Scholar]
- 21.Lee, K. H., H. W. Park, and J. B. Lee. 1991. Localization of keratinolytic proteinase in skin tissue sections of guinea pigs with Microsporum canis infection by immunoperoxidase technique in electron microscopy. Ann. Dermatol. 3:1-4. [Google Scholar]
- 22.Lee, K. H., K. K. Park, S. H. Park, and J. B. Lee. 1987. Isolation, purification and characterization of keratinolytic proteinase from Microsporum canis. Yonsei Med. J. 28:131-138. [DOI] [PubMed] [Google Scholar]
- 23.Lunder, M., and M. Lunder. 1992. Is Microsporum canis infection about to become a serious dermatological problem? Dermatology 184:87-89. [DOI] [PubMed] [Google Scholar]
- 24.Markaryan, A., I. Morozova, H. Yu, and P. E. Kolattukudy. 1994. Purification and characterization of an elastinolytic metalloprotease from Aspergillus fumigatus and immunoelectron microscopic evidence of secretion of this enzyme by the fungus invading the murine lung. Infect. Immun. 62:2149-2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meevootisom, V., and D. J. Niederpruem. 1979. Control of exocellular proteases in dermatophytes and especially Trichophyton rubrum. Sabouraudia 17:91-106. [DOI] [PubMed] [Google Scholar]
- 26.Mignon, B., F. Coignoul, T. Leclipteux, C. Focant, and B. Losson. 1999. Histopathological pattern and humoral immune response to a crude exo-antigen and purified keratinase of Microsporum canis in symptomatic and asymptomatic infected cats. Med. Mycol. 37:1-9. [PubMed] [Google Scholar]
- 27.Mignon, B., T. Leclipteux, C. Focant, A. Nikkels, G. Pierard, and B. Losson. 1999. Humoral and cellular immune response to a crude exo-antigen and purified keratinase of Microsporum canis in experimentally infected guinea pigs. Med. Mycol. 37:123-129. [PubMed] [Google Scholar]
- 28.Mignon, B., A. Nikkels, G. Pierard, and B. Losson. 1998. The in vitro and in vivo production of a 31.5 kDa keratinolytic subtilase from Microsporum canis and the clinical status in naturally infected cats. Dermatology 196:438-441. [DOI] [PubMed] [Google Scholar]
- 29.Mignon, B., M. Swinnen, J. P. Bouchara, M. Hofinger, A. Nikkels, G. Pierard, C. Gerday, and B. Losson. 1998. Purification and characterization of a 31.5 kDa keratinolytic subtilisin-like serine protease from Microsporum canis and evidence of its secretion in naturally infected cats. Med. Mycol. 36:395-404. [PubMed] [Google Scholar]
- 30.Miyoshi, S., and S. Shinoda. 2000. Microbial metalloproteases and pathogenesis. Microbes Infect. 2:91-98. [DOI] [PubMed] [Google Scholar]
- 31.Monod, M., S. Paris, D. Sanglard, K. Jaton-Ogay, J. Bille, and J. P. Latgé. 1993. Isolation and characterization of a secreted metalloprotease of Aspergillus fumigatus. Infect. Immun. 61:4099-4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monod, M., G. Togni, B. Hube, and D. Sanglard. 1994. Multiplicity of genes encoding secreted aspartic proteinases in Candida species. Mol. Microbiol. 13:357-368. [DOI] [PubMed] [Google Scholar]
- 33.Naglik, J. R., G. Newport, T. C. White, L. L. Fernandes-Naglik, J. S. Greenspan, D. Greenspan, S. P. Sweet, S. J. Challacombe, and N. Agabian. 1999. In vivo analysis of secreted aspartyl proteinase expression in human oral candidiasis. Infect. Immun. 68:2482-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okeke, C. N., R. Tsuboi, M. Kawai, M. Hiruma, and H. Ogawa. 2001. Isolation of an intron-containing partial sequence of the gene encoding dermatophyte actin (ACT) and detection of a fragment of the transcript by reverse transcription-nested PCR as a means of assessing the viability of dermatophytes in skin scales. J. Clin. Microbiol. 39:101-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramesh, M. V., T. Sirakova, and P. E. Kolattukudy. 1994. Isolation, characterization, and cloning of cDNA and the gene for an elastinolytic serine proteinase from Aspergillus flavus. Infect. Immun. 62:79-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rawlings, N. D., and A. J. Barrett. 1995. Evolutionary families of metallopeptidases. Methods Enzymol. 248:183-228. [DOI] [PubMed] [Google Scholar]
- 37.Rawlings, N. D., and A. J. Barrett. 2000. MEROPS: the peptidase database. Nucleic Acids Res. 28:323-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Romano, C. 1999. Tinea capitis in Siena, Italy. An 18-year survey. Mycoses 42:559-562. [DOI] [PubMed] [Google Scholar]
- 39.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 40.Schaller, M., H. C. Korting, W. Schafer, J. Bastert, W. Chen, and B. Hube. 1999. Secreted aspartic proteinase (Sap) activity contributes to tissue damage in a model of human oral candidosis. Mol. Microbiol. 34:169-180. [DOI] [PubMed] [Google Scholar]
- 41.Scott, D. W., W. H. Miller, and C. E. Griffin. 1995. Fungal skin diseases, p. 329-391. In G. H. Muller (ed.), Small animal dermatology, 5th ed. W. B. Saunders, Philadelphia, Pa.
- 42.Takiuchi, I., D. Higuchi, Y. Sei, and K. Koga. 1982. Isolation of an extracellular proteinase (keratinase) from Microsporum canis. Sabouraudia 20:281-288. [PubMed] [Google Scholar]
- 43.Takiuchi, I., Y. Sei, H. Tagaki, and M. Negi. 1984. Partial characterization of the extracellular keratinase from Microsporum canis. Sabouraudia 22:219-224. [PubMed] [Google Scholar]
- 44.Tatsumi, H., M. Ohsawa, R. F. Tsuji, S. Murakami, E. Nakano, H. Motai, A. Masaki, Y. Ishida, K. Murakami, H. Kawabe, and H. Arimura. 1988. Cloning and sequence of the alkaline protease cDNA from Aspergillus oryzae. Agric. Biol. Chem. 52:1887-1888. [Google Scholar]
- 45.Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsuboi, R., I. J. Ko, K. Takamori, and H. Ogawa. 1989. Isolation of a keratinolytic proteinase from Trichophyton mentagrophytes with enzymatic activity at acidic pH. Infect. Immun. 57:3479-3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Cutsem, J. 1989. Animal models for dermatomycotic infections, p. 1-35. In M. R. McGinnis and M. Borgers (ed.), Current topics in medical mycology, vol. 3. Springer-Verlag, New York, N.Y. [DOI] [PubMed]
- 48.Weitzman, I., and R. C. Summerbell. 1995. The dermatophytes. Clin. Microbiol. Rev. 8:240-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woodfolk, J. A., L. M. Wheatley, R. V. Piyasena, D. C. Benjamin, and T. A. Platts-Mills. 1998. Trichophyton antigens associated with IgE antibodies and delayed type hypersensitivity. Sequence homology to two families of serine proteinases. J. Biol. Chem. 273:29489-29496. [DOI] [PubMed] [Google Scholar]
- 50.World Health Org./ISHAM Liaison Committee. 1987. Proceedings of the ISHAM consultation on prevention and control of dermatophytosis with special reference to immunity and immunization, p. 1-40. National Veterinary Institute, Oslo, Norway.
- 51.Yu, R. J., S. R. Harmon, and F. Blank. 1968. Isolation and purification of an extracellular keratinase of Trichophyton mentagrophytes. J. Bacteriol. 96:1435-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu, R. J., S. R. Harmon, S. F. Grappel, and F. Blank. 1971. Two cell-bound keratinases of Trichophyton mentagrophytes. J. Investig. Dermatol. 56:27-32. [DOI] [PubMed] [Google Scholar]
- 53.Zaugg, C., M. Borg-Von Zepelin, U. Reichard, D. Sanglard, and M. Monod. 2001. Secreted aspartic proteinase family of Candida tropicalis. Infect. Immun. 69:405-412. [DOI] [PMC free article] [PubMed] [Google Scholar]