

Abstract
Molecular dynamics was used to simulate the transition state for the first chemical reaction step (TS1) of cocaine hydrolysis catalyzed by human butyrylcholinesterase (BChE) and its mutants. The simulated results demonstrate that the overall hydrogen bonding between the carbonyl oxygen of (–)-cocaine benzoyl ester and the oxyanion hole of BChE in the TS1 structure for (–)-cocaine hydrolysis catalyzed by A199S/S287G/A328W/Y332G BChE should be significantly stronger than that in the TS1 structure for (–)-cocaine hydrolysis catalyzed by the WT BChE and other simulated BChE mutants. Thus, the transition-state simulations predict that A199S/S287G/A328W/Y332G mutant of BChE should have a significantly lower energy barrier for the reaction process and, therefore, a significantly higher catalytic efficiency for (–)-cocaine hydrolysis. The theoretical prediction has been confirmed by wet experimental tests showing an ≈(456 ± 41)-fold improved catalytic efficiency of A199S/S287G/A328W/Y332G BChE against (–)-cocaine. This is a unique study to design an enzyme mutant based on transitionstate simulation. The designed BChE mutant has the highest catalytic efficiency against cocaine of all of the reported BChE mutants, demonstrating that the unique design approach based on transition-state simulation is promising for rational enzyme redesign and drug discovery.
Keywords: molecular dynamics, rational design, transition-state stabilization, cocaine, enzyme–substrate binding
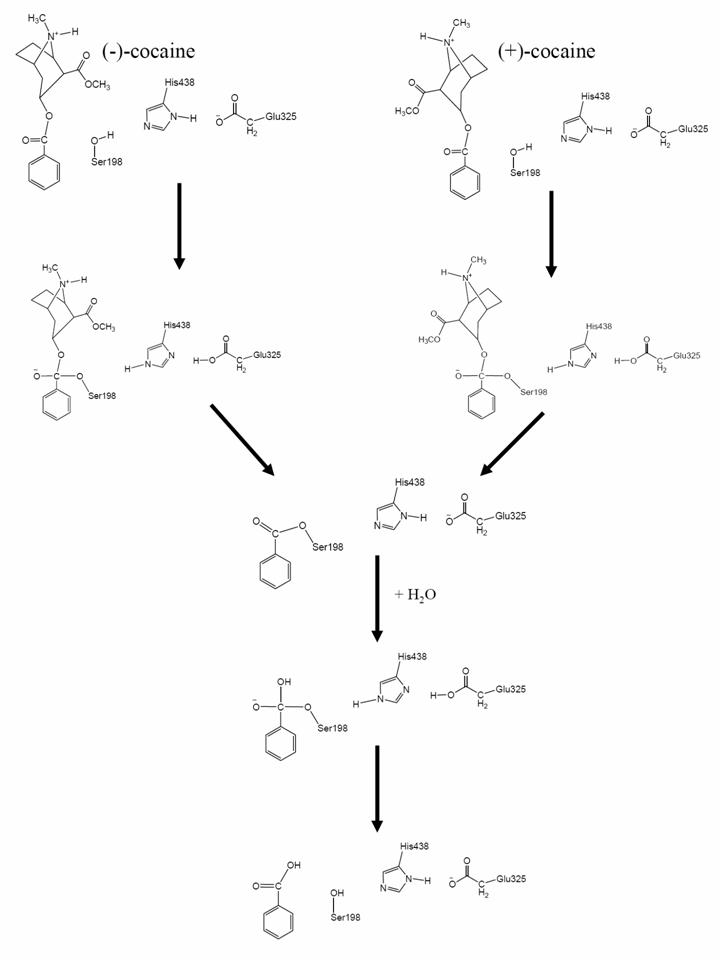
Cocaine is recognized as the most reinforcing of all drugs of abuse (1–3). The disastrous medical and social consequences of cocaine addiction have made the development of an effective pharmacological treatment a high priority (4–6). However, cocaine mediates its reinforcing and toxic effects by blocking neurotransmitter reuptake, and the classical pharmacodynamic approach has failed to yield small-molecule receptor antagonists because of the difficulties inherent in blocking a blocker (1–5). An alternative to receptor-based approaches is to interfere with the delivery of cocaine to its receptors or accelerate its metabolism in the body (5, 7–17). An ideal molecule for this purpose should be a potent enzyme catalyzing the hydrolysis of cocaine into biologically inactive metabolites. The dominant pathway for cocaine metabolism in primates is butyrylcholinesterase (BChE)-catalyzed hydrolysis at the benzoyl ester group (Fig. 3, which is published as supporting information on the PNAS web site), and the metabolites are all biologically inactive (5, 18). Clearly, BChE-catalyzed hydrolysis of cocaine at the benzoyl ester is the metabolic pathway most suitable for amplification. However, the catalytic activity of this plasma enzyme is ≈1,000-fold lower against the naturally occurring (–)-cocaine than that against the biologically inactive (+)-cocaine enantiomer (19–22). (+)-cocaine can be cleared from plasma in seconds, before partitioning into the CNS, whereas (–)-cocaine has a plasma half-life of ≈45–90 min, long enough for manifestation of the CNS effects, which peak in minutes (19, 20). Hence, BChE mutants with higher activity against (–)-cocaine are highly desirable for use as an exogenous enzyme in humans.
Generally speaking, for rational design of a mutant enzyme with a higher catalytic activity for a given substrate, one needs to design a mutation that can accelerate the rate-determining step of the entire catalytic reaction process while the other steps are not slowed down by the mutation. Reported computational modeling and experimental data indicated that the formation of the prereactive BChE–(–)-cocaine complex (the prereactive enzyme–substrate complex, ES) is the rate-determining step of (–)-cocaine hydrolysis catalyzed by WT BChE (23–27), whereas the rate-determining step of the corresponding (+)-cocaine hydrolysis is the chemical reaction process consisting of the acylation and deacylation stages, or four individual reaction steps (see Fig. 3) (26). This mechanistic understanding is consistent with the experimental observation (23) that the catalytic rate constant of WT BChE against (+)-cocaine is pH-dependent, whereas that of the same enzyme against (–)-cocaine is independent of the pH. The pH dependence of the rate constant for (+)-cocaine hydrolysis is clearly associated with the protonation of H438 residue in the catalytic triad (S198, H438, and E325). For the first and third steps of the reaction process, when H438 is protonated, the catalytic triad cannot function, and, therefore, the enzyme becomes inactive. The lower the pH of the reaction solution, the higher the concentration of the protonated H438 and the lower the concentration of the active enzyme. Hence, the rate constant was found to decrease with decreasing the pH of the reaction solution for the enzymatic hydrolysis of (+)-cocaine (23).
Based on the above mechanistic understanding, the previously reported efforts for rational design of the BChE mutants have focused on how to improve the ES formation process. Several BChE mutants, including A328W (25), A328W/Y332A (25), A328W/Y332G (27), and F227A/S287G/A328W/Y332M (28), have been found to have a significantly higher catalytic efficiency (kcat/Km) against (–)-cocaine; these mutants of BChE have an ≈9- to 34-fold improved catalytic efficiency against (–)-cocaine. Experimental observation (25) also indicated that the catalytic rate constant of A328W/Y332A BChE is pH-dependent for both (–)- and (+)-cocaine. The pH dependence reveals that for both (–)- and (+)-cocaine, the rate-determining step of the hydrolysis catalyzed by A328W/Y332A BChE should be either the first or the third step of the reaction process. Further, if the third step were rate-determining, then the catalytic efficiency of the A328W/Y332A mutant against (–)-cocaine should be as high as that of the same mutant against (+)-cocaine, because the (–)- and (+)-cocaine hydrolyses share the same third and fourth steps (i.e., the same deacylation process) (see Fig. 3). However, it has also been observed that the A328W/Y332A mutant only has an ≈9-fold improved catalytic efficiency against (–)-cocaine, whereas the A328W/Y332A mutation does not change the high catalytic activity against (+)-cocaine (25). This analysis of the experimental and computational data available in literature clearly shows that the rate-determining step of (–)-cocaine hydrolysis catalyzed by the A328W/Y332A mutant should be the first step of the chemical reaction process. Further, recently reported computational modeling also suggests that the formation of the ES is hindered mainly by the bulky side chain of Y332 residue in WT BChE but that the hindering can be removed by the Y332A or Y332G mutation (27). Therefore, starting from the A328W/Y332A or A328W/Y332G mutant, the rational design of further mutation(s) to improve the catalytic efficiency of BChE against (–)-cocaine can aim to decrease the energy barrier for the first reaction step without significantly affecting the ES formation and other chemical reaction steps.
Here, we report rational design of a high-activity mutant of BChE against (–)-cocaine based on detailed computational modeling of the transition state for the rate-determining step (i.e., the first step of the chemical reaction process). Molecular dynamics (MD) simulations were performed to model the protein environmental effects on the stabilization of the transition-state structure for BChE-catalyzed hydrolysis of (–)-cocaine. The simulated results indicate that the transition-state structure can be much better stabilized by the protein environment in A199S/S287G/A328W/Y332G BChE than that in WT BChE and in other BChE mutants examined. The computational modeling led to a prediction of the higher catalytic efficiency for the A199S/S287G/A328W/Y332G mutant against (–)-cocaine. The prediction has been confirmed by wet experimental tests showing that the A199S/S287G/A328W/Y332G mutant has a remarkably improved catalytic efficiency against (–)-cocaine. All of the obtained results clearly demonstrate that directly modeling the transition-state structure provides a reliable computational approach to the rational design of a high-activity mutant of BChE against (–)-cocaine.
Materials and Methods
MD Simulations. We must address a critical issue before describing how we performed any MD simulation on a transition state. In principle, MD simulation using a classical force field (molecular mechanics) can only simulate a stable structure corresponding to a local minimum on the potential energy surface, whereas a transition state during a reaction process is always associated with a first-order saddle point on the potential energy surface. Hence, MD simulation using a classical force field cannot directly simulate a transition state without any restraint on the geometry of the transition state. Nevertheless, if we can technically remove the freedom of imaginary vibration in the transition-state structure, then the number of vibrational freedoms (normal vibration modes) for a nonlinear molecule will decrease from 3N–6 to 3N–7 or less. The transition-state structure is associated with a local minimum on the potential energy surface within a subspace of the reduced vibrational freedoms, although it is associated with a first-order saddle point on the potential energy surface with all of the 3N–6 vibrational freedoms. Theoretically, the vibrational freedom associated with the imaginary vibrational frequency in the transition-state structure can be removed by appropriately freezing the reaction coordinate. The reaction coordinate corresponding to the imaginary vibration of the transition state is generally characterized by a combination of some key geometric parameters. These key geometric parameters are bond lengths of the forming and breaking covalent bonds for BChE-catalyzed hydrolysis of cocaine, as seen in Fig. 3. Thus, we just need to maintain the bond lengths of the forming and breaking covalent bonds during the MD simulation on a transition state. Technically, we can maintain the bond lengths of the forming and breaking covalent bonds by simply fixing all atoms within the reaction center, by using some constraints on the forming and breaking covalent bonds, or by redefining the forming and breaking covalent bonds. It should be pointed out that the only purpose of performing these types of MD simulations on a transition state is to examine the dynamic change of the protein environment surrounding the reaction center and the interaction between the reaction center and the protein environment. We are interested only in the simulated structures, because the total energies calculated in this way are meaningless. Transition-state modeling with empirical force fields was previously performed to study various organic reactions, and the theoretical justification of the transition-state modeling was discussed in detail by Eksterowicz and Houk (29).
The initial BChE structures used in the MD simulations were prepared based on our previous MD simulation (27) on the ES for WT BChE with (–)-cocaine in water by using the amber 7 package (30). Our previous MD simulations (27) on the ES started from the x-ray crystal structure (31) deposited in the Protein Data Bank (PDB ID code 1P0P) (32). The present MD simulation on the transition state for the first chemical reaction step (TS1) was performed in such a way that bond lengths of the partially formed and partially broken covalent bonds in the transition state were all constrained to be the same as those obtained from our previous ab initio reaction coordinate calculations on the model reaction system of WT BChE (24). For convenience, the partially formed and partially broken covalent bonds in the transition state will be called “transition” bonds (33, 34). A sufficiently long MD simulation with the transition bonds constrained should lead to a reasonable protein environment stabilizing the reaction center in the simulated transition-state structure. Further, the simulated TS1 structure for WT BChE with (–)-cocaine was used to build the initial structures of TS1 for the examined BChE mutants with (–)-cocaine; only the side chains of mutated residues needed to be changed.
The partial atomic charges for the nonstandard residue atoms, including cocaine atoms, in the TS1 structures were calculated by using the RESP protocol implemented in the antechamber module of the amber 7 package following electrostatic potential (ESP) calculations at ab initio HF/6-31G* level using the gaussian 03 program (35). The geometries used in the ESP calculations came from those obtained from the previous ab initio reaction coordinate calculations (26), but the functional groups representing the oxyanion hole were removed. Thus, residues G116, G117, and A199 were the standard residues as supplied by amber 7 in the MD simulations. The general procedure for carrying out the MD simulations in water is essentially the same as that used in our previously reported computational studies (26, 27, 36–39). Each aforementioned starting TS1 structure was neutralized by adding chloride counterions and was solvated in a rectangular box of TIP3P water molecules (40) with a minimum solute-wall distance of 10 Å. The total number of atoms in the solvated protein structures for the MD simulations is nearly 70,000, although the total number of atoms of BChE and (–)-cocaine is only 8,417 (for the WT BChE). All of the MD simulations were performed by using the sander module of the amber 7 package. The solvated systems were carefully equilibrated and fully energy-minimized. These systems were gradually heated from T = 10 K to T = 298.15 K in 30 ps before running the MD simulation at T = 298.15 K for 1 ns or longer, making sure that we obtained a stable MD trajectory for each of the simulated TS1 structures. The time step used for the MD simulations was 2 fs. Periodic boundary conditions in the NPT ensemble at T = 298.15 K with Berendsen temperature coupling (41) and P = 1 atm with isotropic molecule-based scaling (41) were applied. The SHAKE algorithm (42) was used to fix all covalent bonds containing hydrogen atoms. The nonbonded pair list was updated every 10 steps. The PME (particle mesh Ewald) method (43) was used to treat long-range electrostatic interactions. A residue-based cutoff of 10 Å was used for the noncovalent interactions. The coordinates of the simulated systems were collected every 1 ps during the production MD stages.
The above-described MD procedure was performed first for the TS1 structures of the WT, A328W/Y332A, and A328W/Y332G BChEs. Starting from the simulated TS1 structure for the A328W/Y332G mutant, we hoped to identify a mutant (with additional mutations) that possibly has a more stable TS1 structure. For this purpose, we particularly focused on the possible enhancement of the hydrogen bonding between the carbonyl oxygen of (–)-cocaine and the oxyanion hole of the enzyme, which made it necessary to examine the possible mutations on the amino acid residues within and nearby the oxyanion hole of the enzyme. The initial candidate mutants were chosen by simple geometric consideration of the possible modification of the TS1 structure; only an energy minimization was carried out in the simple geometric consideration of each possible mutant. Then, the MD simulations were performed only for the candidate mutants whose energy-minimized TS1 structures clearly suggested possibly stronger hydrogen bonding between the carbonyl oxygen of (–)-cocaine and the oxyanion hole of the enzyme. Only the most promising mutants identified by the MD simulations were tested by wet experimental studies (see below for the experimental procedure).
Most of the MD simulations were performed in parallel on a Hewlett–Packard supercomputer (a Superdome with 256 shared-memory processors) at the Center for Computational Sciences (University of Kentucky). Some of the computations were carried out on a 34-processor x335 Linux cluster (IBM, White Plains, NY) and SGI Fuel workstations (Silicon Graphics, Mountain View, CA) in our own laboratory.
Experimental Materials. Cloned pfu DNA polymerase and DpnI endonuclease were obtained from Stratagene. 3H-(–)-cocaine [50 Ci/mmol (1 Ci = 37 GBq)] was purchased from PerkinElmer. The expression plasmid pRc/CMV was a gift from O. Lockridge (University of Nebraska Medical Center, Omaha). All oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA). The QIAprep Spin Plasmid Miniprep Kit, plasmid purification kit, and QIAquick PCR purification kit were obtained from Qiagen (Valencia, CA). Human embryonic kidney 293T/17 cells were from American Type Culture Collection. DMEM was purchased from Fisher Scientific. Oligonucleotide primers were synthesized by Integrated DNA Technologies and the University of Kentucky analysis facility. 3,3′, 5,5′-Tetramethylbenzidine was obtained from Sigma. Anti-BChe (mouse monoclonal antibody, product no. HAH002-01) was purchased from AntibodyShop (Gentofte, Denmark), and goat anti-mouse IgG horseradish peroxidase conjugate was purchased from Zymed.
Site-Directed Mutagenesis, Protein Expression, and BChE Activity Assay. Site-directed mutagenesis of human BChE cDNA was performed by using the QuikChange method (44). Mutations were generated from WT human BChE in a pRc/CMV expression plasmid (45–49). Using plasmid DNA as template and primers with specific base pair alterations, mutations were made by PCR with Pfu DNA polymerase for replication fidelity. The PCR product was treated with DpnI endonuclease to digest the parental DNA template. Modified plasmid DNA was transformed into Escherichia coli, amplified, and purified. The DNA sequences of the mutants were confirmed by DNA sequencing. BChE mutants were expressed in human embryonic kidney cell line 293T/17. Cells were grown to 80–90% confluence in six-well dishes and then transfected by Lipofectamine 2000 complexes of 4 μg of plasmid DNA per well. Cells were incubated at 37°C in a CO2 incubator for 24 h, and cells were moved to 60-mm culture vessels and cultured for 4 more days. The culture medium (10% FBS in DMEM) was harvested for a BChE activity assay. To measure (–)-cocaine and benzoic acid, the product of (–)-cocaine hydrolysis catalyzed by BChE, we used sensitive radiometric assays based on toluene extraction of [3H]-(–)-cocaine labeled on its benzene ring (24). In brief, to initiate the enzymatic reaction, 100 nCi of [3H]-(–)-cocaine was mixed with 100 μl of culture medium. The enzymatic reactions proceeded at room temperature (25°C) for varying amounts of time. The reactions were stopped by adding 300 μl of 0.02 M HCl, which neutralized the liberated benzoic acid while ensuring a positive charge on the residual (–)-cocaine. [3H]Benzoic acid was extracted by 1 ml of toluene and measured by scintillation counting. Finally, the measured time-dependent radiometric data were fitted to the kinetic equation so that the catalytic efficiency (kcat/Km) was determined along with the use of an ELISA (50), described below.
ELISA. The ELISA buffers used in the present study are the same as those described in the literature (51). The coating buffer was 0.1 M sodium carbonate/bicarbonate buffer (pH 9.5). The diluent buffer (EIA buffer) was potassium phosphate monobasic/potassium phosphate monohydrate buffer (pH 7.5) containing 0.9% sodium chloride and 0.1% BSA. The washing buffer (PBS-T) was 0.01 M potassium phosphate monobasic/potassium phosphate monohydrate buffer (pH 7.5) containing 0.05% (vol/vol) Tween 20. All of the assays were performed in triplicate. Each well of an ELISA microtiter plate was filled with 100 μl of the mixture buffer, consisting of 20 μl of culture medium and 80 μl of coating buffer. The plate was covered and incubated overnight at 4°C to allow the antigen to bind to the plate. The solutions were then removed, and the wells were washed four times with PBS-T. The washed wells were filled with 200 μl of diluent buffer and kept shaking for 1.5 h at room temperature (25°C). After washing with PBS-T four times, the wells were filled with 100 μl of antibody (1:8,000) and incubated for 1.5 h, followed by washing four times. Then, the wells were filled with 100 μl of goat anti-mouse IgG horseradish peroxidase conjugate complex diluted to a final 1:3,000 dilution and were incubated at room temperature for 1.5 h, followed by washing four times. The enzyme reactions were started by addition of 100 μl of substrate (3,3′, 5,5′-tetramethylbenzidine) solution (51). The reactions were stopped after 15 min by the addition of 100 μl of 2 M sulfuric acid, and the absorbance was read at 460 nm by using a Bio-Rad ELISA plate reader.
Results and Discussion
Hydrogen Bonding. In the design of a high-activity mutant of BChE against (–)-cocaine, we aimed to predict some possible mutations that can lower the energy of TS1 and, therefore, lower the energy barrier for this critical reaction step. Apparently, a mutant associated with the stronger hydrogen bonding between the carbonyl oxygen of (–)-cocaine benzoyl ester and the oxyanion hole of the BChE mutant in the TS1 structure may potentially have a more stable TS1 structure and, therefore, a higher catalytic activity for (–)-cocaine hydrolysis. Hence, the hydrogen bonding with the oxyanion hole in the TS1 structure is a crucial factor affecting the transition-state stabilization and the catalytic activity. The possible effects of some mutations on the hydrogen bonding were examined by performing MD simulations on the TS1 structures for (–)-cocaine hydrolysis catalyzed by WT BChE and its various mutants.
The MD simulation in water was performed for 1 ns or longer to make sure we obtained a stable MD trajectory for each simulated TS1 structure with WT or mutant BChE. The MD trajectories actually became stable quickly, as did the H···O distances involved in the potential hydrogen bonds between the carbonyl oxygen of (–)-cocaine and the oxyanion hole of BChE. Depicted in Fig. 1 are plots of four important H···O distances in the MD-simulated TS1 structure versus the simulation time for (–)-cocaine hydrolysis catalyzed by A199S/S287G/A328W/Y332G BChE, along with the rms deviation of the simulated positions of backbone atoms from those in the corresponding initial structure. The H···O distances in the simulated TS1 structures for WT BChE and its three mutants are summarized in Table 1. The H···O distances between the carbonyl oxygen of (–)-cocaine and the peptidic NH hydrogen atoms of G116, G117, and A199 (or S199) of BChE are denoted by D1, D2, and D3, respectively, in Table 1 and Fig. 1. D4 in Table 1 and Fig. 1 refers to the H···O distance between the carbonyl oxygen of (–)-cocaine and the hydroxyl hydrogen of S199 side chain in the simulated TS1 structure corresponding to the A199S/S287G/A328W/Y332G mutant.
Fig. 1.

Plots of the key internuclear distances (in Å) versus the time in the simulated TS1 structure for (–)-cocaine hydrolysis catalyzed by A199S/S287G/A328W/Y332G BChE. Traces D1, D2, and D3 refer to the distances between the carbonyl oxygen of (–)-cocaine and the NH hydrogen of G116, G117, and S199, respectively. Trace D4 is the internuclear distance between the carbonyl oxygen of (–)-cocaine and the hydroxyl hydrogen of the S199 side chain in A199S/S287G/A328W/Y332G BChE. RMSD is the rms deviation (in Å) of the simulated positions of the protein backbone atoms from those in the initial structure.
Table 1. Summary of the MD-simulated key distance (in Å) and the calculated total HBEs (in kcal/mol) between the oxyanion hole and the carbonyl oxygen of (–)-cocaine benzoyl ester in TS1.
| Distance†
|
|||||
|---|---|---|---|---|---|
| Transition state | D1 | D2 | D3 | D4 | Total HBE‡ |
| TS1 structure for (—)-cocaine hydrolysis catalyzed by WT BChE | |||||
| Average | 4.59 | 2.91 | 1.92 | — | -5.5 (-4.6) |
| Maximum | 5.73 | 4.14 | 2.35 | — | |
| Minimum | 3.35 | 1.97 | 1.61 | — | |
| Fluctuation | 0.35 | 0.35 | 0.12 | — | |
| TS1 structure for (—)-cocaine hydrolysis catalyzed by A328W/Y332A mutant of BChE | |||||
| Average | 3.62 | 2.35 | 1.95 | — | -6.2 (-4.9) |
| Maximum | 4.35 | 3.37 | 3.02 | — | |
| Minimum | 2.92 | 1.78 | 1.61 | — | |
| Fluctuation | 0.23 | 0.27 | 0.17 | — | |
| TS1 structure for (—)-cocaine hydrolysis catalyzed by A328W/Y332G mutant of BChE | |||||
| Average | 3.60 | 2.25 | 1.97 | — | -6.4 (-5.0) |
| Maximum | 4.24 | 3.17 | 2.76 | — | |
| Minimum | 2.89 | 1.77 | 1.62 | — | |
| Fluctuation | 0.23 | 0.24 | 0.17 | — | |
| TS1 structure for (—)-cocaine hydrolysis catalyzed by A199S/S287G/A328W/Y332G mutant of BChE | |||||
| Average | 4.39 | 2.60 | 2.01 | 1.76 | -14.0 (-12.0) |
| Maximum | 5.72 | 4.42 | 2.68 | 2.50 | |
| Minimum | 2.87 | 1.76 | 1.62 | 1.48 | |
| Fluctuation | 0.48 | 0.36 | 0.17 | 0.12 | |
—, not applicable.
D1, D2, and D3 represent the internuclear distances between the carbonyl oxygen of cocaine benzoyl ester and the NH hydrogen of residues 116 (i.e., G116), 117 (i.e., G117), and 199 (i.e., A199 or S199) of BChE, respectively. D4 is the internuclear distance between the carbonyl oxygen of cocaine benzoyl ester and the hydroxyl hydrogen of the S199 side chain in the A199S/S287G/A328W/Y332G mutant.
The total HBE value is the average of the HBE values calculated by using the instantaneous distances in all of the snapshots. The value in parentheses is the total HBE value calculated by using the MD-simulated average distances.
As seen in Table 1, the simulated H···O distance D1 is always too long for the peptidic NH of G116 to form a N–H···O hydrogen bond with the carbonyl oxygen of (–)-cocaine in all of the simulated TS1 structures. In the simulated TS1 structure for WT BChE, the carbonyl oxygen of (–)-cocaine formed a firm N–H···O hydrogen bond with the peptidic NH hydrogen atom of the A199 residue; the simulated H···O distance (D3) was 1.61–2.35 Å, with an average D3 value of 1.92 Å. Meanwhile, the carbonyl oxygen of (–)-cocaine also had a partial N–H···O hydrogen bond with the peptidic NH hydrogen atom of the G117 residue; the simulated H···O distance (D2) was 1.97–4.14 Å (the average D2 value was 2.91 Å). The average D2 and D3 values became 2.35 and 1.95 Å, respectively, in the simulated TS1 structure for the A328W/Y332A mutant. These distances suggest a slightly weaker N–H···O hydrogen bond with A199 but a stronger N–H···O hydrogen bond with G117 in the simulated TS1 structure for the A328W/Y332A mutant than the corresponding N–H···O hydrogen bonds for the WT. The average D2 and D3 values (2.25 and 1.97 Å, respectively) in the simulated TS1 structure for the A328W/Y332G mutant are close to the corresponding distances for the A328W/Y332A mutant. The overall strength of the hydrogen bonding between the carbonyl oxygen of (–)-cocaine and the oxyanion hole of the enzyme is not expected to change considerably when WT BChE is replaced by the A328W/Y332A or A328W/Y332G mutant.
However, the story for the simulated TS1 structure for the A199S/S287G/A328W/Y332G mutant was remarkably different. As one can see from Table 1 and Figs. 1 and 2, when residue 199 becomes a serine (i.e., S199), the hydroxyl group on the side chain of S199 can also hydrogen-bond to the carbonyl oxygen of (–)-cocaine to form an O–H···O hydrogen bond, in addition to the two N–H···O hydrogen bonds with the peptidic NH of G117 and S199. The simulated average H···O distances with the peptidic NH hydrogen of G117, the peptidic NH hydrogen of S199, and the hydroxyl hydrogen of S199 are 2.60, 2.01, and 1.76 Å, respectively. Because of the additional O–H···O hydrogen bond, the overall strength of the hydrogen bonding with the modified oxyanion hole of A199S/S287G/A328W/Y332G BChE should be significantly stronger than that of WT, A328W/Y332A, and A328W/Y332G BChEs.
Fig. 2.

Schematic representation of the transition-state structure for the first reaction step for (–)-cocaine hydrolysis catalyzed by a BChE mutant with an A199S mutation.
To better represent the overall strength of hydrogen bonding between the carbonyl oxygen of (–)-cocaine and the oxyanion hole in a MD-simulated TS1 structure, we estimated the hydrogen-bonding energy (HBE) associated with each simulated H···O distance by using the empirical HBE equation implemented in the autodock 3.0 program suite (52). Based on the general HBE equation, we have 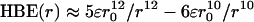 , in which r is the H···O distance in the considered hydrogen bond and r0 is the minimum value of the H···O distance for which the HBE equation can be used (33). We used r0 = 1.52 Å because it is the shortest H···O distance found in all of our MD simulations. The ε value was determined by using the condition that HBE(r) = –5.0 kcal/mol when r = 1.90 Å. Specifically, for each hydrogen bond with the carbonyl oxygen of (–)-cocaine, a HBE value can be evaluated with each snapshot of the MD-simulated structure. The final HBE of the MD-simulated hydrogen bond is considered to be the average HBE value of all snapshots taken from the stable MD trajectory. The estimated total HBE value for the hydrogen bonds between the carbonyl oxygen of (–)-cocaine and the oxyanion hole in each simulated TS1 structure is also listed in Table 1.
, in which r is the H···O distance in the considered hydrogen bond and r0 is the minimum value of the H···O distance for which the HBE equation can be used (33). We used r0 = 1.52 Å because it is the shortest H···O distance found in all of our MD simulations. The ε value was determined by using the condition that HBE(r) = –5.0 kcal/mol when r = 1.90 Å. Specifically, for each hydrogen bond with the carbonyl oxygen of (–)-cocaine, a HBE value can be evaluated with each snapshot of the MD-simulated structure. The final HBE of the MD-simulated hydrogen bond is considered to be the average HBE value of all snapshots taken from the stable MD trajectory. The estimated total HBE value for the hydrogen bonds between the carbonyl oxygen of (–)-cocaine and the oxyanion hole in each simulated TS1 structure is also listed in Table 1.
We also estimated the HBE for each hydrogen bond by using the MD-simulated average H···O distance. As seen in Table 1, the total HBEs (i.e., –4.6, –4.9, –5.0, and –12.0 kcal/mol for the WT, A328W/Y332A, A328W/Y332G, and A199S/S287G/A328W/Y332G BChEs, respectively) estimated in this way are systematically higher (i.e., less negative) than the corresponding total HBEs (i.e., –5.5, –6.2, –6.4, and –14.0 kcal/mol) estimated in the aforementioned way. However, the two sets of total HBE values are qualitatively consistent with each other in terms of the relative hydrogen-bonding strengths in the three simulated TS1 structures. In particular, the two sets of total HBE values consistently reveal that the overall strength of the hydrogen bonding between the carbonyl oxygen of (–)-cocaine and the oxyanion hole in the simulated TS1 structure for A199S/S287G/A328W/Y332G BChE is significantly higher than that for WT, A328W/Y332A, and A328W/Y332G BChEs.
Catalytic Activity. The computational results discussed above suggest that the TS1 of (–)-cocaine hydrolysis catalyzed by the A199S/S287G/A328W/Y332G mutant should be significantly more stable than that by the A328W/Y332A or A328W/Y332G mutant because of the significant increase of the overall hydrogen bonding between the carbonyl oxygen of (–)-cocaine and the oxyanion hole of the enzyme in the TS1 structure. The aforementioned analysis of the literature (24–26, 28) also indicates that the first chemical reaction step associated with TS1 should be the rate-determining step of (–)-cocaine hydrolysis catalyzed by a BChE mutant including the Y332A or Y332G mutation, although the formation of the ES is the rate-determining step for (–)-cocaine hydrolysis catalyzed by WT BChE. This mechanistic insight suggests a clear correlation between the TS1 stabilization and the catalytic activity of A328W/Y332A, A328W/Y332G, and A199S/S287G/A328W/Y332G BChEs for (–)-cocaine hydrolysis: The more stable the TS1 structure, the lower the energy barrier and the higher the catalytic activity. Thus, the MD simulations predict that A199S/S287G/A328W/Y332G BChE should have a higher catalytic activity than A328W/Y332A or A328W/Y332G BChE for (–)-cocaine hydrolysis.
The catalytic efficiency (kcat/Km) of A328W/Y332A BChE for (–)-cocaine hydrolysis was reported to be ≈8.6 × 106 M·min–1 (25), which is ≈9.4 times the kcat/K value (≈9.1 × 105 M·min–1 of WT BChE for (–)-cocaine hydrolysis. The catalytic efficiency of A328W/Y332G BChE was found to be slightly higher than that of A328W/Y332A BChE for (–)-cocaine hydrolysis (27). To examine our theoretical prediction of the higher catalytic activity for A199S/S287G/A328W/Y332G BChE, we produced the A328W/Y332A and A199S/S287G/A328W/Y332G mutants of BChE through site-directed mutagenesis. To minimize the possible systematic experimental errors of the kinetic data, we performed kinetic studies with the two mutants and WT BChE under the same conditions and compared the catalytic efficiency of A328W/Y332A and A199S/S287G/A328W/Y332G BChEs to that of the WT for (–)-cocaine hydrolysis at the benzoyl ester group. Based on the kinetic analysis of the measured time-dependent radiometric data and the ELISA data, the ratio of the kcat/Km value of A328W/Y332A BChE to the kcat/Km value of WT BChE for (–)-cocaine hydrolysis was determined to be ≈8.6. The determined catalytic efficiency ratio of ≈8.6 is in good agreement with the ratio of ≈9.4 determined by Sun et al. (25). Further, by using the same experimental protocol, the ratio of the kcat/Km value of A199S/S287G/A328W/Y332G BChE to the kcat/Km value of A328W/Y332A BChE for (–)-cocaine hydrolysis was determined to be ≈50.6. These data indicate that the ratio of the kcat/Km value of A199S/S287G/A328W/Y332G BChE to the kcat/Km value of WT BChE for (–)-cocaine hydrolysis should be ≈50.6 × 8.6 = ≈435 or ≈50.6 × 9.4 = ≈476. Thus, we can conclude that A199S/S287G/A328W/Y332G BChE has an ≈(456 ± 41)-fold improved catalytic efficiency against (–)-cocaine compared with the WT or that A199S/S287G/A328W/Y332G BChE has a kcat/Km value of ≈(4.15 ± 0.37) × 108 M·min–1 for (–)-cocaine hydrolysis. The catalytic efficiency of A199S/S287G/A328W/Y332G BChE against (–)-cocaine is significantly higher than that of AME-359 (i.e., F227A/S287G/A328W/Y332M BChE, for which kcat/Km = 3.1 × 107 M·min–1 and whose catalytic efficiency against (–)-cocaine is the highest of all of the previously reported BChE mutants) (28), which has an ≈34-fold improved catalytic efficiency against (–)-cocaine compared with WT BChE.
By using the designed A199S/S287G/A328W/Y332G BChE as an exogenous enzyme in humans, when the concentration of this mutant is kept the same as that of the WT BChE in plasma, the half-life of (–)-cocaine in plasma should be reduced from ≈45–90 min to only ≈6–12 s, considerably shorter than the time required for cocaine crossing the blood–brain barrier to reach the CNS. Hence, the outcome of this study could eventually result in a valuable, efficient anticocaine medication.
Conclusion
The transition-state simulations demonstrate that the overall hydrogen bonding between the carbonyl oxygen of (–)-cocaine benzoyl ester and the oxyanion hole of BChE in the TS1 structure for (–)-cocaine hydrolysis catalyzed by A199S/S287G/A328W/Y332G BChE should be significantly stronger than that in the TS1 structure for (–)-cocaine hydrolysis catalyzed by the WT BChE and the other simulated BChE mutants. Thus, the MD simulations predict that A199S/S287G/A328W/Y332G BChE should have a significantly lower energy barrier for the chemical reaction process and, therefore, a significantly higher catalytic efficiency (kcat/Km) for (–)-cocaine hydrolysis. The theoretical prediction has been confirmed by wet experimental tests that show an ≈(456 ± 41)-fold improved catalytic efficiency for A199S/S287G/A328W/Y332G BChE against (–)-cocaine compared with the WT BChE. The kcat/Km value determined for A199S/S287G/A328W/Y332G BChE is much higher than the kcat/Km value for AME-359 (i.e., F227A/S287G/A328W/Y332M BChE, whose catalytic efficiency against (–)-cocaine is the highest of all of the previously reported BChE mutants) (28), which has an ≈34-fold improved catalytic efficiency against (–)-cocaine compared with the WT BChE. The encouraging outcome of this study suggests that the transition-state simulation is a promising, unique approach for rational enzyme redesign and drug discovery.
Supplementary Material
Acknowledgments
We thank the Center for Computational Sciences (University of Kentucky) for providing supercomputing time on the Superdome (a shared-memory supercomputer with four nodes and 256 processors). This work was supported by National Institutes of Health/National Institute on Drug Abuse Grant R01 DA013930 (to C.-G.Z.).
Author contributions: C.-G.Z. designed research; Y.P., D.G., W.Y., H.C., and G.Y. performed research; H.-H.T. contributed new reagents/analytic tools; H.-H.T. and C.-G.Z. analyzed data; and C.-G.Z. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: TS1, transition state for the first chemical reaction step; BChE, butyrylcholinesterase; MD, molecular dynamics; HBE, hydrogen-bonding energy; ES, prereactive enzyme–substrate complex.
References
- 1.Mendelson, J. H. & Mello, N. K. (1996) N. Engl. J. Med. 334, 965–972. [DOI] [PubMed] [Google Scholar]
- 2.Singh, S. (2000) Chem. Rev. 100, 925–1024. [DOI] [PubMed] [Google Scholar]
- 3.Paula, S., Tabet, M. R., Farr, C. D., Norman, A. B. & Ball, W. J., Jr. (2004) J. Med. Chem. 47, 133–142. [DOI] [PubMed] [Google Scholar]
- 4.Sparenborg, S., Vocci, F. & Zukin, S. (1997) Drug Alcohol Depend. 48, 149–151. [DOI] [PubMed] [Google Scholar]
- 5.Gorelick, D. A. (1997) Drug Alcohol Depend. 48, 159–165. [DOI] [PubMed] [Google Scholar]
- 6.Redish, A. D. (2004) Science 306, 1944–1947. [DOI] [PubMed] [Google Scholar]
- 7.Landry, D. W., Zhao, K., Yang, G. X.-Q., Glickman, M. & Georgiadis, T. M. (1993) Science 259, 1899–1901. [DOI] [PubMed] [Google Scholar]
- 8.Carrera, M. R. A., Ashley, J. A., Parsons, L. H., Wirsching, P., Koob, G. F. & Janda, K. D. (1995) Nature 378, 727–730. [DOI] [PubMed] [Google Scholar]
- 9.Baird, T. J., Deng, S.-X., Landry, D. W., Winger, G. & Woods, J. H. (2000) J. Pharmacol. Exp. Ther. 295, 1127–1134. [PubMed] [Google Scholar]
- 10.Carrera, M. R. A., Ashley, J. A., Wirsching, P., Koob, G. F. & Janda, K. D. (2001) Proc. Natl. Acad. Sci. USA 98, 1988–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng, S.-X., de Prada, P. & Landry, D. W. (2002) J. Immunol. Methods 269, 299–310. [DOI] [PubMed] [Google Scholar]
- 12.Kantak, J. M. (2003) Expert Opin. Pharmacother. 4, 213–218. [DOI] [PubMed] [Google Scholar]
- 13.Gorelick, D. A., Gardner, E. L. & Xi, Z.-X. (2004) Drugs 64, 1547–1573. [DOI] [PubMed] [Google Scholar]
- 14.Carrera, M. R. A., Kaufmann, G. F., Mee, J. M., Meijler, M. M., Koob, G. F. & Janda, K. D. (2004) Proc. Natl. Acad. Sci. USA 101, 10416–10421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dickerson, T. J., Kaufmann, G. F. & Janda, K. D. (2005) Expert Opin. Biol. Ther. 5, 773–781. [DOI] [PubMed] [Google Scholar]
- 16.Meijler, M. M., Kaufmann, G. F., Qi, L. W., Mee, J. M., Coyle, A. R., Moss, J. A., Wirsching, P., Matsushita, M. & Janda, K. D. (2005) J. Am. Chem. Soc. 127, 2477–2484. [DOI] [PubMed] [Google Scholar]
- 17.Zhan, C.-G., Deng, S.-X., Skiba, J. G., Hayes, B. A., Tschampel, S. M., Shields, G. C. & Landry, D. W. (2005) J. Comput. Chem. 26, 980–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamendulis, L. M., Brzezinski, M. R., Pindel, E. V., Bosron, W. F. & Dean, R. A. (1996) J. Pharmacol. Exp. Ther. 279, 713–717. [PubMed] [Google Scholar]
- 19.Gateley, S. J. (1991) Biochem. Pharmacol. 41, 1249–1254. [DOI] [PubMed] [Google Scholar]
- 20.Gatley, S. J., MacGregor, R. R., Fowler, J. S., Wolf, A. P., Dewey, S. L. & Schlyer, D. J. (1990) J. Neurochem. 54, 720–723. [DOI] [PubMed] [Google Scholar]
- 21.Darvesh, S., Hopkins, D. A. & Geula, C. (2003) Nat. Rev. Neurosci. 4, 131–138. [DOI] [PubMed] [Google Scholar]
- 22.Giacobini, E., ed. (2003) Butyrylcholinesterase: Its Function and Inhibitors (Martin Dunitz, London).
- 23.Sun, H., Yazal, J. E., Lockridge, O., Schopfer, L. M., Brimijoin, S. & Pang, Y. P. (2001) J. Biol. Chem. 276, 9330–9336. [DOI] [PubMed] [Google Scholar]
- 24.Sun, H., Shen, M. L., Pang, Y. P., Lockridge, O. & Brimijoin, S. (2002) J. Pharmacol. Exp. Ther. 302, 710–716. [DOI] [PubMed] [Google Scholar]
- 25.Sun, H., Pang, Y. P., Lockridge, O. & Brimijoin, S. (2002) Mol. Pharmacol. 62, 220–224. [DOI] [PubMed] [Google Scholar]
- 26.Zhan, C.-G., Zheng, F. & Landry, D. W. (2003) J. Am. Chem. Soc. 125, 2462–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamza, A., Cho, H., Tai, H.-H. & Zhan, C.-G. (2005) J. Phys. Chem. B 109, 4776–4782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao, Y., Atanasova, E., Sui, N., Pancook, J. D., Watkins, J. D. & Brimijoin, S. (2005) Mol. Pharmacol. 67, 204–211. [DOI] [PubMed] [Google Scholar]
- 29.Eksterowicz, J. E. & Houk, K. N. (1993) Chem. Rev. 93, 2439–2461. [Google Scholar]
- 30.Case, D. A., Pearlman, D. A., Caldwell, J. W., Cheatham, T. E., III, Wang, J., Ross, W. S., Simmerling, C. L., Darden, T. A., Merz, K. M., Stanton, R. V., et al. (2002) amber 7 (Univ. of California, San Francisco).
- 31.Nicolet, Y., Lockridge, O., Masson, P., Fontecilla-Camps, J. C. & Nachon, F. (2003) J. Biol. Chem. 278, 41141–41147. [DOI] [PubMed] [Google Scholar]
- 32.Bernstein, F. C., Koetzle, T. F., Williams, G. J. B., Meyer, E. E., Jr., Brice, M. D., Rodgers, J. R., Kennard, O., Shimanouchi, T. & Tasumi, M. (1977) J. Mol. Biol. 112, 535–542. [DOI] [PubMed] [Google Scholar]
- 33.Gao, D. & Zhan, C.-G. (2005) Proteins, in press.
- 34.Zhan, C.-G. & Gao, D. (2005) Biophys. J., in press. [DOI] [PMC free article] [PubMed]
- 35.Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Montgomery, J. A., Jr., Vreven, T., Kudin, K. N., Burant, J. C., et al. (2003) gaussian 03, Revision A.1 (Gaussian, Pittsburgh).
- 36.Zhan, C.-G., Norberto de Souza, O., Rittenhouse, R. & Ornstein, R. L. (1999) J. Am. Chem. Soc. 121, 7279–7282. [Google Scholar]
- 37.Koca, J., Zhan, C.-G., Rittenhouse, R. & Ornstein, R. L. (2001) J. Am. Chem. Soc. 123, 817–826. [DOI] [PubMed] [Google Scholar]
- 38.Koca, J., Zhan, C.-G., Rittenhouse, R. C. & Ornstein, R. L. (2003) J. Comput. Chem. 24, 368–378. [DOI] [PubMed] [Google Scholar]
- 39.Hamza, A., Cho, H., Tai, H.-H. & Zhan, C.-G. (2005) Bioorg. Med. Chem. 13, 4544–4551. [DOI] [PubMed] [Google Scholar]
- 40.Jorgensen, W. L., Chandrasekhar, J., Madura, J. D. & Klein, M. L. (1983) J. Chem. Phys. 79, 926–935. [Google Scholar]
- 41.Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A. & Haak, J. R. (1984) J. Chem. Phys. 81, 3684–3690. [Google Scholar]
- 42.Ryckaert, J. P., Ciccotti, G. & Berendsen, H. J. C. (1977) J. Comp. Phys. 23, 327–341. [Google Scholar]
- 43.Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H. & Pedersen, L. G. (1995) J. Chem. Phys. 103, 8577–8593. [Google Scholar]
- 44.Braman, J., Papworth, C. & Greener, A. (1996) Methods Mol. Biol. 57, 31–44. [DOI] [PubMed] [Google Scholar]
- 45.Masson, P., Legrand, P., Bartels, C. F., Froment, M.-T., Schopfer, L. M. & Lockridge, O. (1997) Biochemistry 36, 2266–2277. [DOI] [PubMed] [Google Scholar]
- 46.Masson, P., Xie, W., Froment, M.-T., Levitsky, V., Fortier, P.-L., Albaret, C. & Lockridge, O. (1999) Biochim. Biophys. Acta 1433, 281–293. [DOI] [PubMed] [Google Scholar]
- 47.Xie, W., Altamirano, C. V., Bartels, C. F., Speirs, R. J., Cashman, J. R. & Lockridge, O. (1999) Mol. Pharmacol. 55, 83–91. [DOI] [PubMed] [Google Scholar]
- 48.Duysen, E. G., Bartels, C. F. & Lockridge, O. (2002) J. Pharmacol. Exp. Ther. 302, 751–758. [DOI] [PubMed] [Google Scholar]
- 49.Nachon, F., Nicolet, Y., Viguie, N., Masson, P., Fontecilla-Camps, J. C. & Lockridge, O. (2002) Eur. J. Biochem. 269, 630–637. [DOI] [PubMed] [Google Scholar]
- 50.Brock, A., Mortensen, V., Loft, A. G. R. & Nørgaard-Pedersen, B. (1990) J. Clin. Chem. Clin. Biochem. 28, 221–224. [DOI] [PubMed] [Google Scholar]
- 51.Khattab, A. D., Walker, C. H., Johnston, G., Siddiqui, M. K. & Saphier, P. W. (1994) Environ. Toxicol. Chem. 13, 1661–1667. [Google Scholar]
- 52.Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K. & Olson, A. J. (1998) J. Comput. Chem. 19, 1639–1662. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}