

Abstract
The regulation of Cl- transport into and out of cells plays a critical role in the maintenance of intracellular volume and the excitability of GABA responsive neurons. The molecular determinants of these seemingly diverse processes are related ion cotransporters: Cl- influx is mediated by the Na-K-2Cl cotransporter NKCC1 and Cl- efflux via K-Cl cotransporters, KCC1 or KCC2. A Cl-/volume-sensitive kinase has been proposed to coordinately regulate these activities via altered phosphorylation of the transporters; phosphorylation activates NKCC1 while inhibiting KCCs, and dephosphorylation has the opposite effects. We show that WNK3, a member of the WNK family of serine-threonine kinases, colocalizes with NKCC1 and KCC1/2 in diverse Cl--transporting epithelia and in neurons expressing ionotropic GABAA receptors in the hippocampus, cerebellum, cerebral cortex, and reticular activating system. By expression studies in Xenopus oocytes, we show that kinase-active WNK3 increases Cl- influx via NKCC1, and that it inhibits Cl- exit through KCC1 and KCC2; kinase-inactive WNK3 has the opposite effects. WNK3's effects are imparted via altered phosphorylation and surface expression of its downstream targets and bypass the normal requirement of altered tonicity for activation of these transporters. Together, these data indicate that WNK3 can modulate the level of intracellular Cl- via opposing actions on entry and exit pathways. They suggest that WNK3 is part of the Cl-/volume-sensing mechanism necessary for the maintenance of cell volume during osmotic stress and the dynamic modulation of GABA neurotransmission.
Keywords: cell volume, GABA, ion transport, protein serine-threonine kinases
A major question in systems biology is how the functions of diverse elements that comprise a system, which may be distributed across different cell types and tissues, are integrated to achieve a coordinated and coherent response to physiologic perturbation. The maintenance of intra- and extracellular electrolyte homeostasis represents one such complex system, which is required for a wide range of essential physiologic processes, including general functions such as the maintenance of proper cell volume, specialized cell functions such as the control of neuronal excitability, and whole-organism properties such as the regulation of blood pressure. In each case, homeostasis is achieved via the regulated movement of Na+, K+, and Cl- across cell membranes. The numerous proteins that mediate the entry and exit of these ions across cell membranes have largely been identified. These include ion channels, cotransporters, exchangers, and pumps that execute transmembrane electrolyte flux. The physiological properties of these individual transport proteins are well known; however, the mechanisms that coordinate their actions to achieve and maintain homeostasis remain obscure.
Although the functions of different cells and epithelia are diverse, the mechanisms by which they achieve many aspects of electrolyte homeostasis are strikingly similar (1). The predominant mechanism by which intracellular volume is maintained in epithelial cells in response to changes in extracellular tonicity is the raising or lowering of intracellular Cl- concentration ([Cl-]i), thereby minimizing transmembrane water flux (2, 3). [Cl-]i is modulated by altering the balance between Cl- entry and exit. The major mediator of Cl- entry is the Na-K-2Cl cotransporter NKCC1; Cl- exit is largely mediated by the K-Cl cotransporter KCC1 (4-6). These cotransporters are potently regulated by extracellular tonicity: hypertonicity activates NKCC1 and inhibits KCC1, whereas hypotonicity has the opposite effect (7-9). A common mechanism has been proposed to coordinately regulate NKCC1 and KCC1 by a system of sensors and transducers that ultimately result in phosphorylation and dephosphorylation of these transporters (3). Although a volume/Cl- responsive kinase has been implicated in the regulation of the response to altered tonicity, the identity of this kinase and of the underlying signaling pathway responsible for the coordination of these homeostatic responses remains to be established (9).
An analogous system plays a key role in the control of neuronal excitability (10). In the adult brain, GABA is the major inhibitory neurotransmitter. GABA acts by opening Cl- channels on postsynaptic membranes; the ensuing electrical response is determined by the cell's electrochemical potential for Cl- (10). If [Cl-]i is below its equilibrium potential, Cl- enters the cell, resulting in hyperpolarization and inhibition. If [Cl-]i is above its equilibrium potential, GABA induces Cl- efflux, depolarization, and neuronal excitation. The importance of [Cl-]i regulation has been recognized with the discovery that GABA neurotransmission is not uniformly inhibitory; it is predominantly excitatory in the neonatal period (11, 12). Similarly, neurons of the suprachiasmatic nucleus show circadian variation in their response to GABA, demonstrating the ability to dynamically regulate [Cl-]i (13-15). Finally, GABA neurotransmission in the peripheral nervous system is predominantly excitatory (16). Variation in [Cl-]i in these neurons is determined by mechanisms highly similar to those governing cell volume. Cl- influx largely occurs via NKCC1, whereas Cl- efflux is mediated via the neuronal-specific K-Cl cotransporter KCC2 (11, 17-19). The importance of this regulation is underscored by the consequences of KCC2 deficiency in mouse, which reduces GABA's inhibitory signaling, resulting in motor defects, epilepsy, and anxiety-like behavior (20-22). The mechanism underlying the dynamic and coordinated modulation of NKCC1 and KCC2 activity in neurons is unknown.
The mediators of transcellular Cl- cotransport (Na-Cl cotransporter, NKCC1, NKCC2, KCC1, and KCC2) are all related members of the SLC12A family of cation/Cl- cotransporters (Fig. 6A, which is published as supporting information on the PNAS web site); each takes advantage of inward Na+ or outward K+ gradients to move Cl- into or out of cells, respectively. The importance of this family of transporters is underscored by their use as pharmacologic targets (thiazide diuretics act at NCC, and loop diuretics act at NKCC2), and that their mutation results in diverse diseases. Mutations in NCC and NKCC2 result in the NaCl wasting diseases Gitelman's (23) and Bartter's syndromes, respectively (24). Disruption of NKCC1 in mouse leads to hearing loss, altered pain perception, neuronal excitability, and altered blood pressure (25). Targeted disruption of KCC2 results in epilepsy (20, 21).
The members of this gene family all are known to be regulated by phosphorylation (9). Phosphorylation increases the activity of NCC, NKCC1, and NKCC2, whereas members of the KCC family are inhibited by phosphorylation (7, 8, 26, 27). Phosphorylation of paralagous threonines in the N terminus of NKCC1 and NKCC2 is essential for their activation; these sites are conserved in NCC (28, 29). The mechanisms underlying the coordinated regulation of these cotransporters are poorly understood.
In the accompanying paper (30), we have shown that the serine-threonine kinase WNK3 is a regulator of Cl- entry into renal epithelia; kinase-active WNK3 increases the surface expression and activity of the Na-K-2Cl cotransporter NKCC2, expressed in the renal thick ascending limb of Henle, and the Na-Cl cotransporter NCC, expressed in the distal convoluted tubule. The recognition that WNK3 shows highest expression in the brain has led us to investigate its localization in this and other extrarenal sites and to investigate its regulatory effects on other members of the SLC12A family.
Methods
cDNA Constructs. Wild-type pGH19-WNK3 or pGH19-WNK3 harboring the kinase-inactivating (D294A) or pseudohypoaldosteronism type II (PHAII)-like mutation (Q545E) were previously described (30). pol1-hNKCC1 (31), pSPORT1-hKCC1 (32), and pSPORT1-hKCC2 (32) were used for 86Rb+ influx studies.
Antibodies. Anti-WNK3 antibody was obtained from Alpha Diagnostics (San Antonio, TX). Other antibodies used were anti-GABAA β2/β3 receptor subunits (33), anti-NKCC1 (T4 antibody), and anti-Phospho NKCC1 (R5 antibody; ref. 28) (gifts of B. Forbush, Yale University School of Medicine), and affinity-purified secondary antibodies conjugated to the CY2, CY3, or CY5 fluors (Jackson ImmunoResearch).
In Situ Hybridization. RNA complementary to mouse WNK3 mRNA corresponding to bases 2685-3681 of human WNK3 (a unique stretch 5′ of the kinase domain) was prepared with incorporation of digoxigenin-UTP (34). Brains were fixed by intracardiac perfusion with 4% paraformaldehyde and 36-μm-thick sections were cut. In situ hybridization was performed and specific hybridization detected by using antidigoxin antibodies (35). Specificity of hybridization was demonstrated by the absence of signal after hybridization of WNK3 sense probes.
Immunolocalization Studies. Studies were approved by the Yale University Animal Care and Use Committee. Mice were killed by cervical dislocation. Tissues were prepared as described (36). Slides were processed with primary and secondary antibodies and visualized by immunofluorescence microscopy (36). Results were similar in threemice. Anti-WNK3 immunostaining was competed with a 3-fold molar excess of the immunizing peptide.
Functional Assays with Cation/Cl- Cotransporters. Xenopus laevis oocytes were harvested and injected with cRNA of NKCC1, KCC1, or KCC2 alone or together with cRNA of wild-type, kinase dead or PHAII-like mutant WNK3, as described (37). After 4 days of incubation, bumetanide-sensitive 86Rb+ influx (for NKCC1, ref. 38) and Cl--dependent 86Rb+ efflux (for KCC1 and KCC2, ref. 39) was determined. NKCC1 measurements were performed in isotonic conditions or after exposure to hypertonic medium (220-380 mM). KCC1 and KCC2 measurements were performed in isotonic conditions or after exposure to hypotonic conditions (110 mM). In each experiment, ≈15 oocytes were tested in each group; results were highly reproducible across at least four independent experiments for each condition. The significance of differences between groups of oocytes was assessed by two-tailed Student t test or one-way ANOVA with Bonferroni correction for multiple comparisons, as appropriate.
NKCC1 Phospho-Protein Studies. Oocytes injected with indicated constructs were incubated as above and exposed to extracellular tonicity ranging from 180 to 220 mM. Oocytes were immediately homogenized by pipetting in ice-cold antiphosphatase solution (150 mM NaCl/30 mM NaF/5 mM EDTA/15 mM Na2HPO4/15 mM pyrophosphate/20 mM Hepes, pH 7.2) with 1% Triton X-100 and a protease inhibitor mixture. The homogenate was cleared by centrifugation, and supernatants were subjected to Western blotting. The previously characterized anti-NKCC1 antibody T4 (40) and the anti-Phospho-NKCC1 antibody R5 (28) were used to detect total and phosphorylated NKCC1, respectively.
Results
WNK3 Expression in the Brain. In contrast to WNK1 and WNK4, WNK3 mRNA is most highly expressed in brain (41, 42). In situ hybridization with WNK3-specific antisense probes reveals prominent WNK3 expression in hippocampus and dentate gyrus; transcripts are also detected in cortical layers and in thalamic and hypothalamic nuclei (Fig. 1A). WNK3 also is strongly expressed in the supraoptic and suprachiasmatic nuclei (Fig. 1 A). Moving posteriorly, WNK3 transcripts are highly expressed in the dorsal raphe nucleus (Fig. 1C), the locus ceruleus (Fig. 1D), the Purkinje layer of the cerebellum (Fig. 1F), and the reticular formation (Fig. 1F). Expression of WNK3 is strongly developmentally regulated. In the hippocampus and cerebellum, it is virtually absent at postnatal day 10 and becomes highly expressed by postnatal day 21 (Fig. 1 A and E; see also Fig. 7, which is published as supporting information on the PNAS web site). Interestingly, the spatial and temporal expression pattern of WNK3 closely parallels that of KCC2, the neuron-specific K-Cl cotransporter, in the hippocampus (43). WNK3 transcripts are also abundant in the choroid plexus and the epithelial lining of the ventricles (epithelia that secrete and modify the cerebrospinal fluid, respectively; data not shown).
Fig. 1.

WNK3 expression in brain. (A-F) Antisense riboprobe to WNK3 was prepared, hybridized in situ to mouse brain sections, and visualized as described in Methods. Coronal sections at postnatal day 15 (P15; A-D) and P21 (E and F) are shown. (A) WNK3 expression is high in the hippocampus (Hp), thalamus (Th), layers of the cerebral (Cx), and entorhinal (En) cortices and in the supraoptic (SON) and suprachiasmatic nuclei (SCN). (B) Strong WNK3 expression is seen in hypothalamus (Hyp), Cx, and thalamus (Th). (C) WNK3 is expressed in the dorsal raphe nucleus (DR). (D) Intense WNK3 expression in the locus ceruleus (LC). (E) WNK3 expression is higher at P21, especially in Hp, Cx, and En. WNK3 expression in Hyp is also strong. (F) At P21, WNK3 is expressed in the Purkinje layer (arrow) of the cerebellum (Cb) and the medullary reticular formation (RF). (G-L) Expression in brain epithelia. Sections were stained with anti-WNK3 (red) and anti-zona-occludens-1 (green). WNK3 localizes to intercellular junctions in the epithelium (arrow) lining the third and lateral ventricles (Vn) of mouse brain. (Original magnification: G-I, ×200; J-L, ×630.) (M-O) Expression in brain parenchyma. Sections were stained with anti-WNK3 (red) and anti-GABAA receptor (green). A section of hippocampus is shown. WNK3 localizes to the cell bodies of neurons expressing the ionotropic GABAA receptor; similar results are seen in cerebellum and cortex. (Original magnification: ×350.)
Immunofluorescence microscopy with highly specific anti-WNK3 antibodies (30) shows that WNK3 localizes to intercellular junctions in the epithelium lining the third and lateral ventricles (Fig. 1 G-L), as well as the choroid plexus (data not shown). Within brain parenchyma, WNK3 localizes to the cell bodies of neurons expressing ionotropic GABAA receptor subunits (Fig. 1 M-O) but is not expressed in glia (marked by glial fibrillary acid protein GFAP; data not shown). These neurons are known to express NKCC1 and KCC2 (11).
WNK3 in Extrarenal Epithelia. WNK1 and WNK4 are expressed in a number of extrarenal epithelia (31, 36). A survey of WNK3's tissue expression by using immunofluoresence microscopy after staining tissue sections with anti-WNK3 antibody revealed sites that overlapped with WNK1 and WNK4 expression, including pancreatic ducts (Fig. 2 A-C), bile ducts (Fig. 2 D-F), and epididymis (not shown); in these epithelia, WNK3 localizes to intercellular junctions. In other tissues, the distribution of WNK3 is distinct from that of WNK1 and WNK4. For example, in the gastrointestinal tract, WNK3 is expressed in the small intestine (Fig. 2 G-I) and stomach (Fig. 2 J-L) but not the colon, a pattern opposite that of WNK1 and WNK4. In the small intestine, WNK3 localizes to intercellular junctions of the highly absorptive surface epithelium and enterocytes in the crypts of Lieberkuhn, glands that secrete a 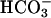 -rich protective mucus. In the stomach, WNK3 localizes to intercellular junctions of the Cl--secreting cells of the gastric glands.
-rich protective mucus. In the stomach, WNK3 localizes to intercellular junctions of the Cl--secreting cells of the gastric glands.
Fig. 2.

Localization of WNK3 in Cl--transporting extrarenal epithelia. Frozen mouse tissue sections were stained with anti-WNK3 (red) and anti-zona-occludens-1 (green) and analyzed with immunofluoresence light microscopy. Composite views (large) and staining by each antibody alone (small) are shown. (A-C) Pancreas. WNK3 localizes to intercellular junctions of large main exocrine pancreatic ducts. (D-F) Liver. WNK3 localizes to intercellular junctions of epithelial cells that line bile ducts. (G-I) Small intestine. WNK3 is in enterocytes in the crypts of Lieberkuhn. (J-L) Stomach. WNK3 is at intercellular junctions of HCl-secreting epithelial cells lining gastric glands. (Original magnification: ×630.)
WNK3 Activates NKCC1 and Inhibits KCC. The localization of WNK3 to diverse epithelia involved in electrolyte transport and to neurons that are known to modulate intracellular Cl- concentration, along with prior evidence that WNK3 regulates NKCC2 and NCC, motivated the examination of WNK3's effect on the activities of NKCC1 and members of the KCC family. In each case, wild-type WNK3 and WNK3 harboring the D294A mutation (kinase-dead WNK3) were separately tested for effects by coexpression studies in X. laevis oocytes. For some targets, we also tested the effect of a WNK3 mutant harboring the Q565E mutation, which mimics a mutation that causes PHAII in WNK4 (PHAII-like WNK3).
NKCC1 and KCC are normally regulated by extracellular osmolarity in mammalian cells and Xenopus oocytes (39). When expressed alone, NKCC1 is inactive under hypotonic conditions (≤180 mosM; Fig. 3A), partially active under isotonic conditions (200 mosM; Fig. 3A), and fully activated under hypertonic conditions (≥220 mosM; not shown). Coexpression with WNK3 had a striking effect on NKCC1 regulation, resulting in maximal activity regardless of tonicity (Fig. 3A). This stimulatory effect of WNK3 is opposite to WNK4's inhibition of NKCC1 (31). Conversely, coexpression of kinase-dead WNK3 with NKCC1 markedly inhibited 86Rb influx in all osmolarities (P < 0.0001, Fig. 3A). PHAII-like WNK3 had the same effect as wild-type WNK3 (Fig. 3A). These findings indicate that WNK3 potently regulates NKCC1 activity and eliminates the normal requirement of hypertonicity for NKCC1 activation in Xenopus oocytes.
Fig. 3.

WNK3 has opposing effects on NKCC1 and KCC1/KCC2. Xenopus oocytes were injected with cRNAs encoding either NKCC1 (A), KCC2 (B), or KCC1 (C) alone or in combination with wild-type or mutant WNK3. NKCC1-dependent 86Rb+ influx (A) or efflux (B and C) was determined as described in Methods. Data show the mean ± SE of 86Rb+ flux and are representative of at least four independent experiments. (A) WNK3 regulates NKCC1. NKCC1 normally shows negligible activity under hypotonic conditions (180 mosM) and is partially active in isotonic conditions (200 mM). WNK3 increases NKCC1 to maximal activity in both conditions. Conversely, kinase-dead WNK3 strongly inhibits NKCC1 activity. *, P < 0.0001 vs. NKCC1 alone. (B) WNK3 regulates KCC2. KCC2 alone is partially active under isotonic conditions and induced under hypotonic conditions. WNK3 inhibits KCC2 under both conditions. In contrast, kinase-dead WNK3 strongly activates KCC2 under both hypotonic and isotonic conditions. *, P < 0.0001 vs. KCC2 alone. (C) WNK3 regulates KCC1. KCC1 alone is inactive under isotonic conditions and induced under hypotonic conditions. WNK3 inhibits KCC1 under both conditions. In contrast, kinase-dead WNK3 strongly activates KCC1 under both hypotonic and isotonic conditions. *, P < 0.0001 vs. KCC1 alone.
In contrast to WNK3's activation of NCC, NKCC2 (30), and NKCC1, kinase-active WNK3 was a potent inhibitor of the neuron-specific K-Cl cotransporter KCC2 under hypotonic conditions in which it is normally maximally active (P < 0.0001, Fig. 3B). Kinase-dead WNK3 not only fails to display this inhibitory effect but actually further increases the activity of KCC2 when oocytes were incubated in hypotonic medium (P < 0.0001, Fig. 3B). When expressed in oocytes under isotonic conditions, KCC2 (unlike other KCCs) is partially active (32). WNK3 inhibited KCC2-induced 86Rb+ influx activity under these conditions (P < 0.0001, Fig. 3B). Most strikingly, however, kinase-dead WNK3 induced a dramatic 22-fold increase of KCC2 activity under isotonic conditions (P < 0.0001, Fig. 3B).
Similar results were obtained with the ubiquitously expressed K-Cl cotransporter KCC1, which normally requires hypotonic conditions for activation. Like KCC2, in hypotonic conditions, KCC1 is strongly inhibited by wild-type WNK3 (P < 0.0001, Fig. 3C). Conversely, kinase-dead WNK3 further increased activity of KCC1 (P < 0.0001 vs. KCC1, Fig. 3C). When oocytes were incubated in isotonic medium, KCC1 alone showed no significant activity. Similar to KCC2, however, kinase-dead WNK3 induced a dramatic increase in KCC1 activity under isotonic conditions (P < 0.0001, Fig. 3C), conditions under which KCC1 normally shows no activity. These observations that kinase-dead WNK3 can bypass the normal requirement of hypotonicity for activation of KCCs are analogous to the finding that kinase-active WNK3 eliminates the need for hypertonicity to activate NKCC1.
WNK3 Regulates NKCC1 Phosphorylation. The activation of NKCC1 requires phosphorylation of Thr-212 and Thr-217 in its cytoplasmic amino terminus (28). Phosphorylation of the paralagous residues are integral for NKCC2 activation and are conserved in other cation/Cl- cotransporters (29). We monitored phosphorylation of NKCC1 at these sites with anti-R5, an antibody that specifically recognizes phosphorylation of Thr-212 and Thr-217 (28). NKCC1 phosphorylation closely parallels its transport activity (ref. 28 and Fig. 4). In the absence of WNK3, NKCC1 phosphorylation increases from neglible levels under hypotonic conditions to high levels under hypertonic conditions. In contrast, coexpression with WNK3 results in robust NKCC1 phosphorylation under hypotonic and isotonic, as well as hypertonic, conditions (Fig. 4). Conversely, when kinase-dead WNK3 is expressed with NKCC1, there is a dramatic reduction in NKCC1 phosphorylation compared with the level seen with NKCC1 expression alone (Fig. 4). This effect can account for the observed inhibition of NKCC1 by kinase-dead WNK3.
Fig. 4.

WNK3 regulates the phosphorylation of NKCC1. Xenopus oocytes were injected with the indicated constructs and incubated at varying extracellular osmolarities. After incubation, oocytes were lysed, and Western blotting was performed by using the R5 (anti-Phospho-NKCC1) or T4 (anti-NKCC1) antibodies, as in Methods. Phosphorylation of NKCC1 normally increases from negligible levels in hypotonic conditions (180 mM) to complete phosphorylation in hypertonic conditions (220 mM). In contrast, coexpression of NKCC1 with kinase-active WNK3 results in robust phosphorylation of NKCC1 at all osmolarities. Conversely, expression of kinase-dead WNK3 results in marked reduction of NKCC1 phosphorylation.
These findings indicate that the effects of WNK3 on NKCC1 are associated with altered phosphorylation of its known regulatory sites. The reduced phosphorylation of NKCC1 induced by kinase-dead WNK3 is not simply due to loss of function, because the level of NKCC1 phosphorylation is lower than that seen in the absence of WNK3. Kinase-inactive WNK3's dominant negative effect could be accounted for by direction of a phosphatase activity to the target protein, inhibition of a kinase that normally maintains phosphorylation of NKCC1, or both.
Discussion
These findings establish that WNK3 has potent and opposing effects on mediators of Cl- entry (NKCC1) and Cl- exit (KCC1 and KCC2). The colocalization of WNK3 with NKCC1 and KCC in extrarenal epithelia and GABA-responsive neurons suggests the relevance of these effects in vivo. Like WNK3's effect on renal mediators of Cl- entry (NKCC2 and NCC), kinase-active WNK3 is a potent activator of NKCC1, whereas it is an equally potent inhibitor of KCC1 and KCC2, mediators of Cl- efflux. These combined effects are expected to increase intracellular Cl-. Interestingly, when WNK3's kinase activity is eliminated by point mutation, its effects at these targets are reversed, resulting in strong inhibition of Cl- entry and activation of Cl- exit. These divergent effects on mediators of Cl- entry and Cl- exit are strikingly different from the activities of WNK4, which generally inhibit members of the SLC12A cotransporter family (31).
The effects of WNK3 are mediated via altered phosphorylation and surface expression of its downstream targets. Kinase-active WNK3 increases phosphorylation at sites necessary for NKCC1's activation; in contrast, kinase-dead WNK3 decreases phosphorylation at these same sites. This altered phosphorylation can account for the observed changes in activity. Because NCC (26), NKCC1 (7), and NKCC2 (8) are all positively regulated by phosphorylation, whereas KCC1 and KCC2 are inhibited by phosphorylation (27), these observations suggest that kinase-dependent WNK3 effects lead to phosphorylation of downstream targets, whereas kinase-independent effects promote dephosphorylation. Because hypertonicity can lead to robust phosphorylation of NKCC1 in the absence of exogenous WNK3 in the oocyte system (see Fig. 4), and because of evidence that the kinase PASK may be responsible for the direct phosphorylation of regulatory sites of NKCC1 (44, 45), we presume that WNK3 is not directly phosphorylating its targets but instead is regulating downstream kinases/phosphatases that ultimately act at the target. WNK3's localization to intercellular junctions, distant from the apical sites of these transporters, is consistent with this speculation.
The observation that kinase-dead WNK3 has effects opposite those of kinase-active WNK3 at each target is intriguing. We presume that these mirror-image effects mimic a normal biochemical event in vivo for several reasons. First, the magnitudes of these effects are very large and uniform for all five members of the SLC12A family studied. Second, the biological simplicity of switching from promotion of increased [Cl-]i to decreased [Cl-]i via regulation of WNK3 kinase activity is intuitively attractive and matches functions that are known to modulate [Cl-]i in vivo. Third, mutations in the related protein WNK4 that cause the human disease PHAII (46) alter the balance between kinase-dependent and -independent functions of this protein, which alters the balance between NaCl reabsorption and K+ secretion, an analogous switch (37, 47). It is tempting to speculate that WNK3 is itself regulated by posttranslational modification that switches its function from promoting to inhibiting phosphorylation of downstream targets. In this regard, it is interesting that WNK1 is activated by both hypertonic and hypotonic stress, and that the autoinhibitory domain of WNK1 can inhibit the activity of a number of different WNK kinases (48, 49). This suggests that WNK kinases are components of the sensors that monitor changes in [Cl-]i and/or cell volume. Elucidation of the details of the upstream and downstream signaling pathways will require further investigation.
WNK3's discrete expression and its activity at specific targets suggest a role for this kinase in a number of fundamental extrarenal physiologic processes. For example, in neurons harboring ionotropic GABAA receptors, WNK3 kinase activity is inferred to increase Cl- entry and inhibit Cl- exit, thereby increasing [Cl-]i (Fig. 5A). Increased resting [Cl-]i would drive GABA signaling from inhibitory toward excitatory. In contrast to the predominant inhibitory effect of GABA signaling in the adult, excitatory GABA signaling occurs in the neonatal period, varies from inhibitory to excitatory with circadian rhythm in many brain centers, and is excitatory in the peripheral nervous system (13-16). In contrast, kinase-dead WNK3 inhibits NKCC1 and activates KCC2, which would reduce [Cl-]i and promote inhibitory GABA signaling. These observations suggest that WNK3 allows the dynamic modulation of [Cl-]i and plasticity in response to GABA. These observations have potential implications for diverse processes in the CNS, including the regulation of wakefulness and sleep in the reticular activating system (13, 15). Consistent with this possibility, WNK kinase gene expression shows circadian variation in Arabidopsis thaliana (50, 51). Further work will be required to evaluate a role of WNK3 in this process.
Fig. 5.

Proposed physiologic roles of WNK3. (A) WNK3 regulation of the neuronal response to GABA. Kinase-active WNK3 can increase [Cl-]i by increasing activity of NKCC1 and inhibiting KCC2, promoting an excitatory response to GABA. Kinase-dead WNK3 inhibits NKCC1 and activates KCC2, potentially promoting neruonal inhibition. (B) WNK3 regulation of cell volume. WNK3 can increase [Cl-]i by activating NKCC1 and inhibiting KCC1; kinase-inactive WNK3 could decrease [Cl-]i by inhibiting NKCC1 and activating KCC1. These are activities required to maintain cell volume in response to hyper- and hypoosmolar stress, respectively.
WNK3 also has attributes that suggest its role in the regulation of cell volume (Fig. 5B), suggesting that it is the (or one of the) long-sought Cl--responsive kinases that regulate cell volume in response to osmotic stress and/or changes in [Cl-]i (2, 6, 52). Hypertonicity increases the activity of NKCC1 and inhibits KCCs by stimulating the phosphorylation of both proteins, resulting in increased [Cl-]i. Conversely, hypotonicity has the opposite effect on phosphorylation and activity of these transporters, resulting in decreased [Cl-]i (9). These responses maintain intracellular volume in response to osmotic stress. The activities of kinase-active and -inactive WNK3 fit the profile expected of the Cl-/volume-sensitive kinase. Consistent with this notion is the observation that WNK3 eliminates the requirement for hypertonicity to activate NKCC1, and that kinase-dead WNK3 bypasses the requirement for hypotonicity to activate KCCs. These observations suggest that WNK3 is integral to the Cl-/volume-sensing mechanism and the regulation of cell volume.
Finally, the presence of WNK3 in diverse epithelia involved in Cl- flux (which also express NKCC1 and KCCs) suggests a broad role for WNK3 in regulating epithelial Cl- secretion.
Although much remains to be done to define the regulators of WNK3's activities and the details of the downstream pathway to its targets, it is anticipated that elucidation of these processes will have an impact on a broad range of topics in biological regulation.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health [Specialized Center of Research Grant in Hypertension (to R.P.L.), Grant DK-36803 (to S.C.H. and G.G.), and Grant DK-64635 (to G.G.)] and the Wellcome Trust [Grant GR070159MA (to G.G.)]. K.T.K. is a trainee of the National Institutes of Health Medical Scientist Training Program. I.G. is a Ramón y Cajal Investigator of the Spanish Ministry of Education and Science. R.P.L. is an Investigator of the Howard Hughes Medical Institute.
Author contributions: K.T.K., J.R., A.L., G.G., I.G., and R.P.L. designed research; K.T.K., J.R., P.d.l.H., A.L., P.M., N.V., and I.G. performed research; K.T.K., J.R., A.L., S.C.H., G.G., I.G., and R.P.L. contributed new reagents/analytic tools; K.T.K., J.R., P.d.l.H., A.L., S.C.H., G.G., I.G., and R.P.L. analyzed data; and K.T.K., J.R., A.L., G.G., I.G., and R.P.L. wrote the paper.
Conflict of interest statement: No conflicts declared.
Abbreviations: PHAII, pseudohypoaldosteronism type II; WNK, with no lysine (K).
References
- 1.Boron, W. F. & Boulpaep, E. L. (2005) Medical Physiology: A Cellular and Molecular Approach (Elsevier Saunders, Philadelphia).
- 2.Lang, F., Busch, G. L., Ritter, M., Volkl, H., Waldegger, S., Gulbins, E. & Haussinger, D. (1998) Physiol. Rev. 78, 247-306. [DOI] [PubMed] [Google Scholar]
- 3.Parker, J. (1994) in Cellular and Molecular Physiology of Cell Volume Regulation, ed. Strange, K. (CRC, Boca Raton, FL), pp. 311-321.
- 4.Lytle, C. (1998) Am. J. Physiol. 274, 1002-1010. [Google Scholar]
- 5.Adragna, N. C., Fulvio, M. D. & Lauf, P. K. (2004) J. Membr. Biol. 201, 109-137. [DOI] [PubMed] [Google Scholar]
- 6.Lytle, C. & McManus, T. (2002) Am. J. Physiol. 283, C1422-C1431. [DOI] [PubMed] [Google Scholar]
- 7.Haas, M. & Forbush, B., III (2000) Annu. Rev. Physiol. 62, 515-534. [DOI] [PubMed] [Google Scholar]
- 8.Hebert, S. C., Mount, D. B. & Gamba, G. (2004) Pflügers Arch. 447, 580-593. [DOI] [PubMed] [Google Scholar]
- 9.Gamba, G. (2005) Physiol. Rev. 85, 423-493. [DOI] [PubMed] [Google Scholar]
- 10.Jentsch, T. J., Hubner, C. A. & Fuhrmann, J. C. (2004) Nat. Cell Biol. 6, 1039-1047. [DOI] [PubMed] [Google Scholar]
- 11.Delpire, E. (2000) News Physiol. Sci. 15, 309-312. [DOI] [PubMed] [Google Scholar]
- 12.Rivera, C., Voipio, J. & Kaila, K. (2005) J. Physiol. 562, 27-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner, S., Castel, M., Gainer, H. & Yarom, Y. (1997) Nature 387, 598-603. [DOI] [PubMed] [Google Scholar]
- 14.Lundkvist, G. B., Kristensson, K. & Hill, R. H. (2002) J. Biol. Rhythms 17, 40-51. [DOI] [PubMed] [Google Scholar]
- 15.Shimura, M., Akaike, N. & Harata, N. (2002) Am. J. Physiol. 282, C366-C373. [DOI] [PubMed] [Google Scholar]
- 16.Sung, K. W., Kirby, M., McDonald, M. P., Lovinger, D. M. & Delpire, E. (2000) J. Neurosci. 20, 7531-7538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., Pirvola, U., Saarma, M. & Kaila, K. (1999) Nature 397, 251-255. [DOI] [PubMed] [Google Scholar]
- 18.Yamada, J., Okabe, A., Toyoda, H., Kilb, W., Luhmann, H. J. & Fukuda, A. (2004) J. Physiol. 557, 829-841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu, L., Lovinger, D. & Delpire, E. (2005) J. Neurophysiol. 93, 1557-1568. [DOI] [PubMed] [Google Scholar]
- 20.Hubner, C. A., Stein, V., Hermans-Borgmeyer, I., Meyer, T., Ballanyi, K. & Jentsch, T. J. (2001) Neuron 30, 515-524. [DOI] [PubMed] [Google Scholar]
- 21.Woo, N. S., Lu, J., England, R., McClellan, R., Dufour, S., Mount, D. B., Deutch, A. Y., Lovinger, D. M. & Delpire, E. (2002) Hippocampus 12, 258-268. [DOI] [PubMed] [Google Scholar]
- 22.Tornberg, J., Voikar, V., Savilahti, H., Rauvala, H. & Airaksinen, M. S. (2005) Eur. J. Neurosci. 21, 1327-1337. [DOI] [PubMed] [Google Scholar]
- 23.Simon, D. B., Nelson-Williams, C., Bia, M. J., Ellison, D., Karet, F. E., Molina, A. M., Vaara, I., Iwata, F., Cushner, H. M., Koolen, M., et al. (1996) Nat. Genet. 12, 24-30. [DOI] [PubMed] [Google Scholar]
- 24.Simon, D. B., Karet, F. E., Hamdan, J. M., DiPietro, A., Sanjad, S. A. & Lifton R. P. (1996) Nat. Genet. 13, 183-188. [DOI] [PubMed] [Google Scholar]
- 25.Delpire, E., Lu, J., England, R., Dull, C. & Thorne, T. (1999) Nat. Genet. 22, 192-195. [DOI] [PubMed] [Google Scholar]
- 26.Knepper, M. A. & Brooks, H. L. (2001) Curr. Opin. Nephrol. Hypertens. 10, 655-659. [DOI] [PubMed] [Google Scholar]
- 27.Mount, D. B. & Gamba, G. (2001) Curr. Opin. Nephrol. Hypertens. 10, 685-691. [DOI] [PubMed] [Google Scholar]
- 28.Flemmer, A. W., Gimenez, I., Dowd, B. F., Darman, R. B. & Forbush, B. (2002) J. Biol. Chem. 277, 37551-37558. [DOI] [PubMed] [Google Scholar]
- 29.Gimenez, I. & Forbush, B. (2003) J. Biol. Chem. 278, 26946-26951. [DOI] [PubMed] [Google Scholar]
- 30.Rinehart, J., Kahle, K. T., de los Heros, P., Vazquez, N., Meade, P., Wilson, F. H., Hebert, S. C., Gimenez, I., Gamba, G. & Lifton, R. P. (2005) Proc. Natl. Acad. Sci. USA 102, 16777-16782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kahle, K. T., Gimenez, I., Hassan, H., Wilson, F. H., Wong, R. D., Forbush, B., Aronson, P. S. & Lifton, R. P. (2004) Proc. Natl. Acad. Sci. USA 101, 2064-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song, L., Mercado, A., Vazquez, N., Xie, Q., Desai, R., George, A. L., Jr., Gamba, G. & Mount, D. B. (2002) Brain Res. Mol. Brain Res. 103, 91-105. [DOI] [PubMed] [Google Scholar]
- 33.Richards, J. G., Schoch, P., Haring, P. Takacs, B. & Mohler, H. (1987) J. Neurosci. 7, 1866-1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Louvi, A. & Wassef, M. (2000) Development (Cambridge, U.K.) 127, 4061-4071. [DOI] [PubMed] [Google Scholar]
- 35.Tole, S. & Patterson, P. H. (1995) J. Neurosci. 15, 970-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choate, K. A., Kahle, K. T., Wilson, F. H., Nelson-Williams, C. & Lifton, R. P. (2003) Proc. Natl. Acad. Sci. USA 100, 663-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kahle, K. T., Wilson, F. H., Leng, Q., Lalioti, M. D., O'Connell, A. D., Dong, K., Rapson, A. K., MacGregor, G. G., Giebisch, G., Hebert, S. C., et al. (2003) Nat. Genet. 35, 372-376. [DOI] [PubMed] [Google Scholar]
- 38.Plata, C., Meade, P., Vazquez, N., Hebert, S. C. & Gamba, G. (2002) J. Biol. Chem. 277, 11004-11012. [DOI] [PubMed] [Google Scholar]
- 39.Mercado, A., de los Heros, P., Vazquez, N., Meade, P., Mount, D. B. & Gamba G. (2001) Am. J. Physiol. 281, C670-C680. [DOI] [PubMed] [Google Scholar]
- 40.Lytle, C., Xu, J. C., Biemesderfer, D. & Forbush, B., III (1995) Am. J. Physiol. 269, C1496-C1505. [DOI] [PubMed] [Google Scholar]
- 41.Holden, S., Cox, J. & Raymond, F. L. (2004) Gene 335, 109-119. [DOI] [PubMed] [Google Scholar]
- 42.Verissimo, F. & Jordan, P. (2001) Oncogene 20, 5562-5569. [DOI] [PubMed] [Google Scholar]
- 43.Lu, J., Karadsheh, M. & Delpire, E. (1999) J. Neurobiol. 39, 558-568. [PubMed] [Google Scholar]
- 44.Piechotta, K., Garbarini, N., England, R. & Delpire, E. (2003) J. Biol. Chem. 278, 52848-52856. [DOI] [PubMed] [Google Scholar]
- 45.Dowd, B. F. & Forbush, B. (2003) J. Biol. Chem. 278, 27347-27353. [DOI] [PubMed] [Google Scholar]
- 46.Wilson, F. H., Disse-Nicodeme, S., Choate, K. A., Ishikawa, K., Nelson-Williams, C., Desitter, I., Gunel, M., Milford, D. V., Lipkin, G. W., Achard, J. M., et al. (2001) Science 293, 1107-1112. [DOI] [PubMed] [Google Scholar]
- 47.Wilson, F. H., Kahle, K. T., Sabath, E., Lalioti, M. D., Rapson, A. K., Hoover, R. S., Hebert, S. C., Gamba, G. & Lifton, R. P. (2003) Proc. Natl. Acad. Sci. USA 100, 680-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu, B., English, J. M., Wilsbacher, J. L., Stippec, S., Goldsmith, E. J. & Cobb, M. H. (2000) J. Biol. Chem. 275, 16795-16801. [DOI] [PubMed] [Google Scholar]
- 49.Lenertz, L. Y., Lee, B. H., Min, X., Xu, B. E., Wedin, K., Earnest, S., Goldsmith, E. J. & Cobb, M. H. (2005) J. Biol. Chem. 280, 26653-26658. [DOI] [PubMed] [Google Scholar]
- 50.Nakamichi, N., Murakami-Kojima, M., Sato, E., Kishi, Y., Yamashino, T. & Mizuno, T. (2002) Biosci. Biotechnol. Biochem. 11, 2429-2436. [DOI] [PubMed] [Google Scholar]
- 51.Murakami-Kojima, M., Nakamichi, N., Yamashino, T. & Mizuno, T. (2002) Plant Cell Physiol. 6, 675-683. [DOI] [PubMed] [Google Scholar]
- 52.Hebert, S. C. (1987) Semin. Nephrol. 7, 48-60. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}