

Abstract
The C-terminal activation domain (C-TAD) of the hypoxia-inducible transcription factors HIF-1α and HIF-2α binds the CH1 domains of the related transcriptional coactivators CREB-binding protein (CBP) and p300, an oxygen-regulated interaction thought to be highly essential for hypoxia-responsive transcription. The role of the CH1 domain in vivo is unknown, however. We created mutant mice bearing deletions in the CH1 domains (ΔCH1) of CBP and p300 that abrogate their interactions with the C-TAD, revealing that the CH1 domains of CBP and p300 are genetically non-redundant and indispensable for C-TAD transactivation function. Surprisingly, the CH1 domain was only required for an average of ∼35–50% of global HIF-1-responsive gene expression, whereas another HIF transactivation mechanism that is sensitive to the histone deacetylase inhibitor trichostatin A (TSAS) accounts for ∼70%. Both pathways are required for greater than 90% of the response for some target genes. Our findings suggest that a novel functional interaction between the protein acetylases CBP and p300, and deacetylases, is essential for nearly all HIF-responsive transcription.
Keywords: CBP, CH1, HIF, hypoxia, p300
Introduction
The closely related HIF-1α and HIF-2α are crucial for the physiological adaptation to hypoxia that requires the increased expression of genes involved in glucose metabolism, angiogenesis, hematopoiesis, cell survival, invasion, and vascular tone (Giaccia et al, 2003, 2004; Semenza, 2003). The mechanism(s) enabling HIF-dependent stimulation of transcription in vivo is uncertain but is thought to chiefly involve the physical interaction of the C-terminal activation domain (C-TAD) of HIF-1α and HIF-2α with the CH1 (C/H1, TAZ1) domain of CREB-binding protein (CBP; Crebbp) and the closely related p300 (Ep300) (Dames et al, 2002; Freedman et al, 2002; Semenza, 2002). The NMR co-structures of the CH1 domains of CBP and p300 with the C-TAD of HIF-1α have revealed the specificity of this high-affinity (Kd∼7 nM) interaction, including how oxygen-dependent hydroxylation of HIF-1α Asn803 inhibits complex formation with CH1 (Dames et al, 2002; Freedman et al, 2002), which is believed to be important for inhibiting HIF activity under normoxia (Bruick, 2003; Giaccia et al, 2004; Poellinger and Johnson, 2004).
CBP and p300 are required for normal development (Tanaka et al, 2000; Alarcon et al, 2004; Kalkhoven, 2004; Kang-Decker et al, 2004; Korzus et al, 2004; Zhou et al, 2004; Wood et al, 2005), consistent with observations that they interact with ∼10% of the ∼2000 mammalian transcriptional regulatory proteins (PB, submitted) (Messina et al, 2004). CBP and p300 possess protein and histone acetyltransferase (PAT, HAT) activities, but it is largely unknown what roles their transcription factor-binding domains play in vivo (Goodman and Smolik, 2000). The CH1 domain is a zinc-containing structure that is highly conserved between CBP and p300, as well as in man, mice, nematodes, and flies (Figure 1A) (Goodman and Smolik, 2000). CH1 has transactivation function when fused to a heterologous DNA-binding domain, consistent with it having a role in the proposed adaptor functions of CBP and p300 (Newton et al, 2000; Zanger et al, 2001), but its main function is thought to involve binding to specific transcription factors in the recruitment of CBP and p300 to promoters. Indeed, 26 of the 37 transcriptional regulators that bind the CH1 region are essential in mice (Supplementary Table S1), but particular interest has focused on HIF-1α and HIF-2α because of the importance of HIF-1 and HIF-2 (a heterodimeric complex of ARNT with HIF-1α or HIF-2α, respectively) in mediating the transcriptional response to hypoxia.
Figure 1.
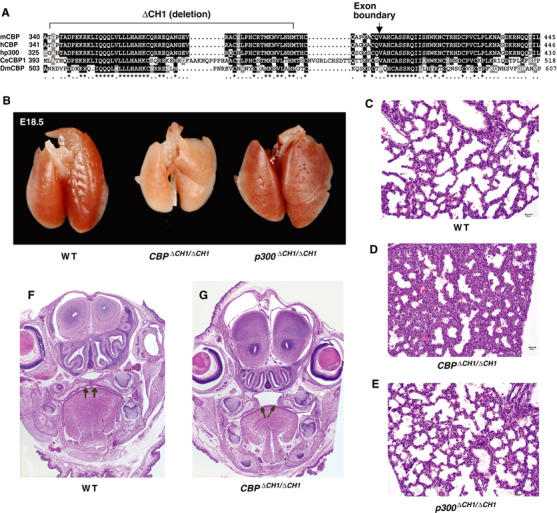
The CH1 domain of CBP is conserved and required for normal development. (A) CH1 domains of CBP and p300 span two exons and are conserved in mouse (m), human (h), Caenorhabditis elegans (Ce), and Drosophila (Dm). ΔCH1 deletion mutation, exon boundary, conserved residues, and relative amino-acid positions are indicated. (B) Grossly, CBPΔCH1/ΔCH1 E18.5 lungs are small compared to WT and p300ΔCH1/ΔCH1. (C–E) Microscopically, lungs from CBPΔCH1/ΔCH1 E18.5 embryos have thicker interstitial septa and decreased alveolar airspace. (F, G) Some CBPΔCH1/ΔCH1 E18.5 embryos have cleft palate (arrows).
Results
Genetically non-redundant roles for the CH1 domains of CBP and p300 in vivo
To test the requirements for the CH1 domain in vivo, we introduced the ΔCH1 mutation into one of the two exons encoding CH1 in CBP and p300 by homologous recombination in mouse embryonic stem (ES) cells (Figure 1A and Supplementary Figure S1). The deletions are essentially equivalent in CBP and p300 and remove more than 50% of the 88 largely conserved residues of CH1 (aa 329–379 deleted for p300, and aa 342–393 for CBP; an NheI site encoding a flexible Ala–Ser linker was inserted in-frame to facilitate identification of the mutant alleles) (Figure 1A). The ΔCH1 mutation removes critical components of the domain, including two of the four α-helices, five Cys and His residues that bind to two of the three zinc ions in the structure, eight of 14 residues that comprise the conserved hydrophobic core, and much of the binding surface for the HIF-1α C-TAD, including three residues (CBP Asp346, Lys349, Ile353) that contact Asn803 (Dames et al, 2002; Freedman et al, 2002). The structural integrity of CH1 is highly dependent on the hydrophobic core (e.g. p300 Leu344, Leu345), and bound zinc, which strongly indicates that the ΔCH1 mutation will block the interaction with most, if not all, CH1-binding partners (Newton et al, 2000; Gu et al, 2001; Matt et al, 2004).
Essentially normal adult mice heterozygous for p300ΔCH1 or CBPΔCH1 were generated at near the expected Mendelian frequency (there was a modest ∼30% decrease in the number of CBP+/ΔCH1 mice), indicating that ΔCH1 is not an overt dominant-negative mutation. Homozygous CBPΔCH1/ΔCH1 mice on a mixed 129 and C57BL/6 strain background typically died shortly after birth (one runted homozygous mutant survived to adulthood out of 651 mice derived from mating CBP+/ΔCH1 mice). In contrast, p300ΔCH1/ΔCH1 adult mice were overtly normal, although they were produced at about 50% of the expected frequency. Analysis of day 0.5 neonates and day 18.5 embryos revealed that CBPΔCH1/ΔCH1 and p300ΔCH1/ΔCH1 mice were present nearer the expected frequency. The rare survival of CBPΔCH1/ΔCH1 mice past the neonatal stage suggested that animals with hybrid vigor would have improved viability. Indeed, F1 hybrid CBPΔCH1/ΔCH1 offspring derived from interbreeding C57BL/6 and 129 congenic CBP+/ΔCH1 mice had markedly enhanced survival to adulthood (∼25% of the expected frequency), but were growth retarded and had craniofacial defects (to be described elsewhere). F1 hybrid CBP+/ΔCH1;p300+/ΔCH1 compound heterozygotes were also smaller than wild-type (WT) littermates and some had craniofacial defects (incomplete penetrance), indicating that the p300 CH1 domain has a role in normal development in the context of the CBPΔCH1 mutation (not shown). Craniofacial abnormalities are a hallmark of Rubinstein–Taybi syndrome, where CBP, and to a lesser degree p300, monoallelic mutations have been identified (Roelfsema et al, 2005); thus, our results suggest that CBP and p300 CH1 domain insufficiency is an important determinant in this human disease. Together, these results demonstrate that the CH1 domains of CBP and p300 are genetically non-redundant, with the CBP CH1 domain being especially important for normal mouse development.
Examination of day 18.5 embryos of mixed background revealed that CBPΔCH1/ΔCH1 embryos had lung defects not seen in p300ΔCH1/ΔCH1 embryos. CBPΔCH1/ΔCH1 lungs were smaller than WT and p300ΔCH1/ΔCH1 as a percentage of total body weight (4.1±0.4% for WT, 4.2±0.2 for p300ΔCH1/ΔCH1, and 3.1±0.1 for CBPΔCH1/ΔCH1, N=5–6, P=0.0014, t-test; Figure 1B). CBPΔCH1/ΔCH1 (Figure 1D) lungs had thickened interstitial septa and decreased alveolar air space, compared to WT (Figure 1C) and p300ΔCH1/ΔCH1 (Figure 1E) embryos. Some of the CBP+/ΔCH1 embryos also displayed a similar lung phenotype, possibly explaining the partially penetrant lethality (not shown). Cell proliferation, determined by immunostaining for Ki67, was significantly reduced in CBPΔCH1/ΔCH1 lungs, consistent with a delay in lung maturation, but not in p300ΔCH1/ΔCH1 lungs (25.0±0.7% for WT, 23.9±1.7 for p300ΔCH1/ΔCH1, 16.9±1.0 for CBPΔCH1/ΔCH1, P=1.5 × 10−7, N=5–6, t-test). HIF-2α has been implicated in lung development in mice, but we did not observe a synergistic genetic interaction in mice doubly heterozygous for an HIF-2α knockout allele and the CBPΔCH1 or p300ΔCH1 mutation (not shown) (Compernolle et al, 2002). Additionally, ∼50% of CBPΔCH1/ΔCH1 newborn mice had cleft palate (Figure 1F and G, arrows), indicative of a role for the CBP CH1 domain in palate morphogenesis. The relative expression of CBP and p300 does not obviously account for the differential effects of the ΔCH1 mutation, as each is expressed ubiquitously and at roughly comparable levels in the embryonic palate and lung (Naltner et al, 2000; Warner et al, 2002). CBPΔCH1/ΔCH1;p300ΔCH1/ΔCH1 compound homozygous mutant mice are not viable, as no such embryos were observed at day 14.5 of gestation (E14.5). However, CBP+/ΔCH1;p300ΔCH1/ΔCH1 and CBPΔCH1/ΔCH1;p300+/ΔCH1 viable embryos that retain one WT CBP or p300 allele, respectively, could be recovered at E14.5. These ‘triple-ΔCH1' embryos yielded primary mouse embryonic fibroblasts (MEFs) with growth and morphological characteristics comparable to WT MEFs (not shown).
CBPΔCH1 and p300ΔCH1 are hypomorphic proteins with specific defects in mediating HIF-dependent transcription
The mutant transcripts were correctly spliced, as determined by RT–PCR (Supplementary Figure S1A and B). The biochemical integrity of the CBPΔCH1 and p300ΔCH1 proteins was confirmed by examining their expression and acetyltransferase activities. Western blot established that CBP and p300 protein levels and stability were indistinguishable in WT, CBPΔCH1/ΔCH1, and p300ΔCH1/ΔCH1 MEFs (Figure 2A). HAT activities measured in vitro following immunoprecipitation of CBP and p300 with specific antibodies were comparable between the WT and mutant CBP and p300 (Figure 2B and C). Thus, CBPΔCH1 and p300ΔCH1 are normally expressed hypomorphic proteins, and their HAT domain is intact.
Figure 2.
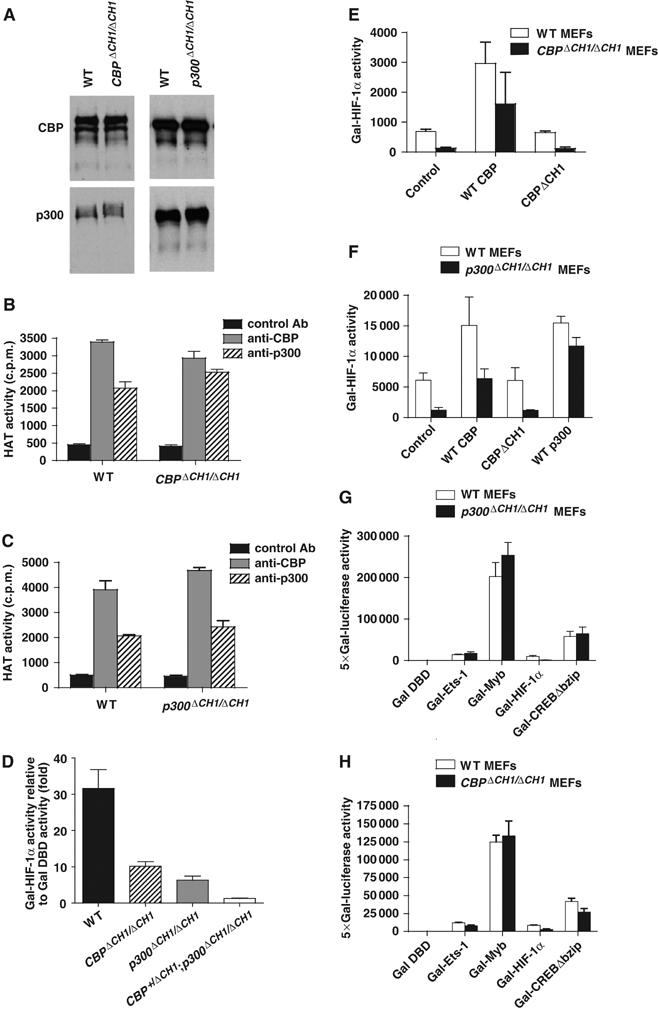
ΔCH1 mutation does not affect other domains or functions of CBP and p300. (A) Western blot of CBP and p300 showing normal protein levels in CBPΔCH1/ΔCH1 and p300ΔCH1/ΔCH1 MEFs. (B, C) Immunoprecipitation/HAT assay showing that HAT activity in CBPΔCH1/ΔCH1 (B) and p300ΔCH1/ΔCH1 (C) MEFs is comparable to WT MEFs (mean±s.e.m., N=2). (D–F) Transient transfection assays showing that Gal-HIF-1α function is reduced in MEFs with multiple ΔCH1 alleles (mean±s.e.m., N=3) (D), and can be rescued by WT CBP (E, F) or p300 (F), but not by CBPΔCH1 (E) (mean±s.e.m., N=2–4). (G, H) Transactivation by factors utilizing other domains of p300 and CBP or other coactivators is unimpaired (mean±s.d., N=4).
Transient transfection assays showed that the transactivation function of the HIF-1α C-TAD fused to the Gal4 DNA-binding domain (Gal-HIF-1α) was attenuated about 60–80% in CBPΔCH1/ΔCH1 and p300ΔCH1/ΔCH1 MEFs, and was reduced about 90% in triple-ΔCH1 (CBP+/ΔCH1;p300ΔCH1/ΔCH1) MEFs (Figure 2D). Overexpression of CBP (Figure 2E and F) or p300 (Figure 2F), but not CBPΔCH1, rescued Gal-HIF-1α activity; CBPΔCH1 overexpression did not affect Gal-HIF-1α activity in WT MEFs. Other activation domains fused to Gal4 (Myb, Ets-1, and CREB), which interact with other CBP and p300 domains, or with other coactivators, were not significantly affected by the ΔCH1 mutation (Figure 2G and H). Therefore, HIF-1α C-TAD activity is specifically attenuated by the ΔCH1 mutation, the combined dosage of CBP and p300 CH1 domains is crucial for C-TAD activity, and the CBPΔCH1 protein does not function as a dominant negative.
Remarkably, endogenous hypoxia-inducible gene expression was largely unaffected in CBPΔCH1/ΔCH1 and p300ΔCH1/ΔCH1 MEFs (not shown). As the CH1 domain may not be limiting for HIF function in such cells, we also analyzed endogenous gene expression using two types of triple-ΔCH1 mutant MEFs (CBP+/ΔCH1;p300ΔCH1/ΔCH1 and CBPΔCH1/ΔCH1;p300+/ΔCH1). Affymetrix microarrays showed that there was a modest average decrease in the expression levels of 111 hypoxia-inducible genes (not necessarily HIF targets; defined by 148 probe sets induced ⩾3-fold in WT MEFs) in CBP+/ΔCH1;p300ΔCH1/ΔCH1 MEFs compared to WT MEFs (best-fit line slope is less than one; Figure 3A). As a control, we examined non-hypoxia-inducible genes represented by 281 probe sets that differed no more than ±1% between normoxia and hypoxia in WT cells, which showed that transcription was not broadly affected in triple-ΔCH1 MEFs (best-fit line slope is close to one with minimal data scatter; Figure 3B). Quantitative real-time RT–PCR (qRT–PCR) analysis of RNA from both types of triple-ΔCH1 MEFs revealed strong to moderate dependence on the CH1 domain for selected hypoxia-inducible genes including placental growth factor (Pgf), vascular endothelial growth factor (Vegf), and glucose transporter-1 (Glut1 or Slc2a1), when normalized to β-actin mRNA (Figure 3C–E, data from 2–6 independent MEF lines for each genotype). Vegf and Slc2a1 are direct HIF targets, but it is unclear if Pgf is a direct or indirect target (Manalo et al, 2005). These three genes play important roles in angiogenesis or glucose metabolism, with Vegf levels being especially critical for angiogenesis and normal development, as Vegf+/− mice die at E11 to E12 (Ferrara et al, 1996). These results suggest that a moderate decrease in the expression of many HIF-target genes could have consequences in both neoplastic and normal cells carrying the ΔCH1 mutation. Interestingly, Vegf deficiency, but apparently not deficiency of the related protein Pgf, leads to defective lung development in mice (Compernolle et al, 2002).
Figure 3.
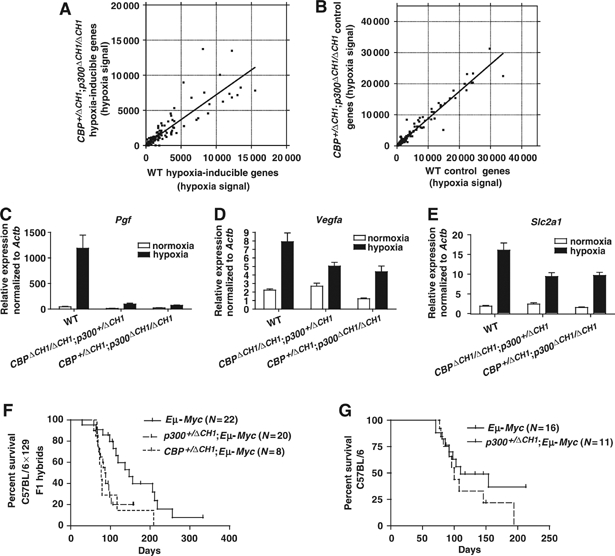
ΔCH1 mutation attenuates the expression of many endogenous hypoxia-inducible genes, but not Eμ-Myc-induced B-cell lymphomagenesis. (A, B) Affymetrix microarray analysis of hypoxia-inducible genes (A) (⩾3-fold induced by hypoxia in WT MEFs, probe sets scored as present in WT hypoxia sample) and non-hypoxia-responsive control genes (B) (±1% hypoxia/normoxia signal ratio in WT MEFs; probe sets scored as present in WT hypoxia and normoxia samples) in WT and CBP+/ΔCH1;p300ΔCH1/ΔCH1 MEFs. Each symbol represents hypoxia-dependent expression level for an Affymetrix probe set; note degree of data scatter and slope of the best-fit line. (C–E) qRT–PCR analysis of physiologically important hypoxia-inducible genes Pgf (C), Vegfa (D), and Slc2a1 (Glut1) (E) in WT, CBPΔCH1/ΔCH1;p300+/ΔCH1, and CBP+/ΔCH1;p300ΔCH1/ΔCH1 triple-ΔCH1 MEFs, normalized to β-actin mRNA (mean±s.e.m., N=6–34, data from 2–6 independent MEF lines for each genotype). Survival curves for C57BL/6 × 129 F1 hybrid (F) and C57BL/6 Eμ-Myc mice (G) with or without ΔCH1 mutant alleles (indicated) are shown.
The ΔCH1 mutation does not significantly attenuate tumorigenesis
The interaction of the CH1 domain with HIF is thought to be vital for tumorigenesis and has been proposed as a therapeutic target (Kung et al, 2000; Semenza, 2003). In this regard, the small molecule chetomin has been identified as a pharmacological agent that disrupts the CH1 structure and inhibits hypoxia-inducible transcription and tumor growth in vivo (Kung et al, 2004). The reduced hypoxia-dependent transcriptional response in triple-ΔCH1 MEFs predicts that tumorigenesis would be attenuated by the ΔCH1 mutation. To test this hypothesis, we introduced CBPΔCH1 and p300ΔCH1 alleles into mice carrying an Eμ-Myc transgene, which induces B-cell lymphoma, a highly vascularized solid mass tumor, with high penetrance and short latency. We found that a single CBPΔCH1 (N=8, P=0.014, log-rank test) or p300ΔCH1 (N=20, P=0.006) mutant allele decreased the median survival of C57BL/6 × 129 F1 hybrid Eμ-Myc mice by about 8–10 weeks, when compared to littermates carrying only the Eμ-Myc transgene (N=22; Figure 3F). The survival curves for Eμ-Myc;p300+/ΔCH1 mice on a C57BL/6 background (N=11, P=0.21) were indistinguishable from Eμ-Myc;p300+/+ mice (N=16), showing that strain background also contributes to tumor latency (Figure 3G). Thus, a reduction in the levels of the CH1 domain does not inhibit Myc-induced B-cell lymphomagenesis.
We also tested WT and triple-ΔCH1 MEFs (CBP+/ΔCH1;p300ΔCH1/ΔCH1 and CBPΔCH1/ΔCH1;p300+/ΔCH1) transformed with the oncogenes 12S E1A and N61 Ras, for their tumorigenic potential following subcutaneous injection into the flanks of Scid mice. The time for the tumors to reach ∼1 cm3 was not significantly different for four independent WT transformed MEF lines (16.5±1.3 days, mean±s.d.), two CBP+/ΔCH1;p300ΔCH1/ΔCH1 lines (19.9±5.3 days), and three CBPΔCH1/ΔCH1;p300+/ΔCH1 lines (19.5±2.3 days) (ANOVA, P=0.57). Volumes (P=0.54) and weights (P=0.37) of the harvested tumors were also not significantly different between the groups. Similarly, there were no statistically significant differences in the growth rate and size of tumors in nude mice following subcutaneous injection of eight independent lines of WT and ΔCH1 MEFs transformed with retroviruses expressing c-Myc and oncogenic V12 Ras (WT, 31±23 days (N=3); CBPΔCH1/ΔCH1, 12±0 days (N=2); CBPΔCH1/ΔCH1;p300+/ΔCH1, 17±3.8 days (N=3); mean±s.d., P=0.74). Therefore, substantially reducing CH1 domain function also does not significantly affect fibroblastic transformation or tumorigenesis.
C-TAD transactivation function absolutely requires the CH1 domain
To address if residual CBP or p300 produced from the remaining WT allele in the triple-ΔCH1 cells was sufficient to support hypoxia-inducible transcription, we generated two strains of triple-ΔCH1/flox MEFs that have a Cre/LoxP conditional knockout CBPflox or p300flox allele in place of the WT gene (Kang-Decker et al, 2004) (PB, submitted). Transient expression of Cre recombinase following infection with a Cre-expressing adenovirus resulted in highly efficient recombination of CBPflox to yield a CBPΔflox null allele (i.e. CBPΔCH1/Δflox;p300ΔCH1/ΔCH1 or tri-ΔCH1/Δflox #1 MEFs; Figure 4A), or p300flox to yield a p300Δflox null allele (i.e. CBPΔCH1/ΔCH1;p300ΔCH1/Δflox or tri-ΔCH1/Δflox #2 MEFs; Figure 4B). Tri-ΔCH1/Δflox MEFs had a growth rate and morphology (not shown) comparable to Cre-adenovirus-infected control cells that lacked only a single CBP allele (CBP+/Δflox;p300+/+ or Δflox #1 MEF; Figure 4C) or p300 allele (CBP+/+;p300+/Δflox or Δflox #2 MEF; Figure 4D). Transient transfection assays revealed a dramatic loss of transactivation function for Gal-HIF-1α (>99%) and Gal-HIF-2α (>96%) in both types of tri-ΔCH1/Δflox MEFs, demonstrating that both C-TADs absolutely require the CH1 domain (Figure 4E and F). By contrast, the KIX-domain-dependent activator Gal-Myb functioned normally in tri-ΔCH1/Δflox MEFs (Figure 4E and F). Thus, other coactivators, or other domains of CBP and p300, appear to be unable to mediate C-TAD function.
Figure 4.
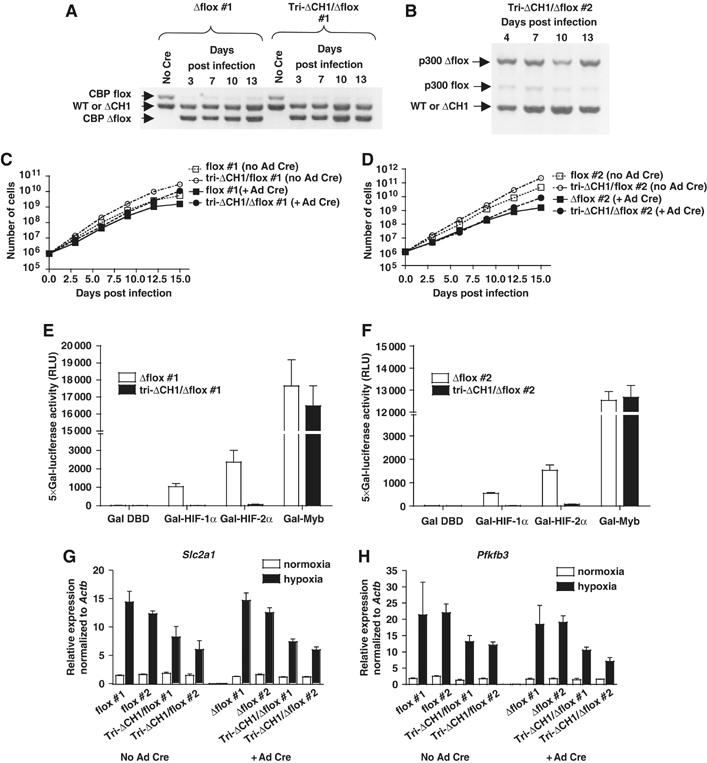
The CH1 domain is absolutely required for C-TAD transactivation function but is less essential for HIF target genes. (A, B) Deletion of CBPflox and (A) p300flox (B) in MEFs following infection with Cre-expressing adenovirus. MEF genotypes, days post infection, and allele-specific products derived from semiquantitative PCR of genomic DNA are indicated. (C, D) Comparable growth curves for tri-ΔCH1/flox and flox MEFs with or without Cre-adenovirus (Ad Cre) infection. (E, F) Normalized activity of Gal-HIF-1α, Gal-HIF-2α, and Gal-Myb in transiently transfected Δflox and tri-ΔCH1/Δflox MEFs (mean±s.e.m., N=3). (G, H) WT CBP contributes marginally to residual hypoxia-inducible gene expression in triple-ΔCH1 MEFs. qRT–PCR analysis of control flox and tri-ΔCH1/flox MEFs±Cre-adenovirus infection. Slc2a1 (G) and Pfkfb3 (H), tested under normoxia and hypoxia, normalized to β-actin mRNA (mean±s.e.m., N=2–3).
Endogenous HIF-target gene expression relies on both CH1-dependent and -independent mechanisms
We next examined the transcription of hypoxia-inducible genes in triple-ΔCH1/flox and tri-ΔCH1/Δflox MEFs by qRT–PCR. Surprisingly, there was very little difference in the expression of the HIF targets Slc2a1 and Pfkfb3 in both types of tri-ΔCH1/Δflox (infected with Cre-expressing adenovirus) and triple-ΔCH1/flox MEFs (not infected), indicating that WT CBP or p300 was not responsible for residual HIF-target gene expression in triple-ΔCH1 MEFs (Figure 4G and H). The Cre-adenovirus-infected cells were tested at 10, 13, and 16 days post infection, after they had expanded a minimum of 450-fold for tri-ΔCH1/Δflox MEFs (>250-fold for Δflox MEFs), thus greatly diluting any residual WT CBP or p300 protein and mRNA (Figure 4C and D). Comparison of 40 HIF-target genes in three independent lines each of Δflox and tri-ΔCH1/Δflox MEFs using Affymetrix microarrays revealed a 35% average decrease in gene expression (mutant to control signal ratio of 0.65±0.47, mean±s.d.) after 6 h of hypoxia (Supplementary Table S2). Thus, microarray and qRT–PCR analyses revealed that the CH1 domain is vital for a few hypoxia-responsive genes (e.g. Pgf; Figure 3C and Supplementary Table S2), moderately limiting for many (e.g. Slc2a1, Pfkfb3; Figure 4G and H), and mostly dispensable for others (e.g. Hig1; Supplementary Table S2).
The ΔCH1 mutation specifically attenuates the recruitment of CBP and p300 to HIF-target genes
We next addressed whether the ΔCH1 mutation blocked recruitment of CBP and p300 to endogenous HIF-binding sites. Quantitative real-time PCR chromatin immunoprecipitation (ChIP) assays were performed with the gene-specific signal normalized to the input DNA signal. CBPΔCH1/ΔCH1 and p300ΔCH1/ΔCH1 MEFs showed attenuated HIF-dependent recruitment of CBPΔCH1 or p300ΔCH1 to DNA sequences near the HIF-binding sites of Slc2a1, Pfkfb3, and Hig1 (Figure 5A–C). Recruitment of WT CBP or p300 in the singly homozygous mutant MEFs served as an internal control. The ΔCH1 mutation caused an 80–90% reduction in treatment-dependent CBP or p300 recruitment in CBPΔCH1/ΔCH1 and p300ΔCH1/ΔCH1 MEFs (compared to the WT CBP or p300-dependent signal in the mutant MEFs) treated for 2 h with the hypoxia mimetic dipyridyl (DP) and the proteasome inhibitors MG132 and ALLN, which together induce HIF-1α and HIF-2α (Figure 5A–C). Another independent set of MEF lines confirmed this effect of the ΔCH1 mutation following 4 h of hypoxia (Supplementary Figure S2A–C). Levels of Slc2a1, Pfkfb3, and Hig1 transcripts in tri-ΔCH1/Δflox #2 MEFs compared to Δflox #2 MEFs treated with DP, MG132, and ALLN (Figure 5D–F) were similar to results obtained with hypoxia (Figure 4G and H and Supplementary Table S2). Importantly, control experiments showed that ΔCH1 mutation did not affect recruitment to the Jun/Sp1-binding site of the non-hypoxia-regulated gene vimentin by ChIP (Wu et al, 2003), but that it strongly attenuated the interaction with HIF-1 in a co-immunoprecipitation assay (Supplementary Figure 2D and E). Although the recruitment of CBP and p300 is not completely blocked by the ΔCH1 mutation under conditions that activate HIF-1 and HIF-2, the amount of gene expression remaining in cells that only contain ΔCH1 mutant alleles suggests that a CBP/p300-independent mechanism must account for a large portion of HIF-responsive transcription.
Figure 5.
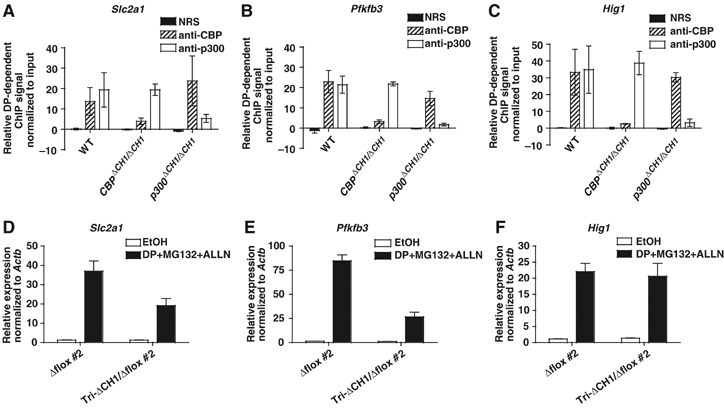
Markedly reduced recruitment of CBPΔCH1 and p300ΔCH1 to HIF-binding sites does not strongly correlate with HIF-responsive transcription. (A–C) Quantitative ChIP assays of Slc2a1, Pfkfb3, and Hig1, using WT, CBPΔCH1/ΔCH1, and p300ΔCH1/ΔCH1 MEFs treated for 2 h with ethanol vehicle (EtOH) or DP/MG132/ALLN (DP) (mean±s.e.m., N=3 independent experiments). Control (NRS) and specific (anti-CBP, anti-p300) immunoprecipitation antisera are indicated. DP-dependent ChIP signal was determined by subtracting the EtOH signal from the DP signal after normalizing to the input DNA signal. (D–F) qRT–PCR analysis of HIF-target gene expression in Δflox #2 and tri-ΔCH1/Δflox #2 MEFs after 6 h DP/MG132/ALLN, normalized to β-actin mRNA (mean±s.e.m., N=3).
A trichostatin A (TSA)-sensitive pathway cooperates with a CH1-dependent mechanism to mediate the bulk of HIF-responsive transcription
Histone deacetylases (HDACs) have been implicated in gene activation dependent on the CBP/p300-interacting transcription factors CREB and HIF (Kim et al, 2001; Brugarolas et al, 2003; Fass et al, 2003). We tested if deacetylase activity is required for the CH1-independent component of HIF-responsive transcription by pretreating MEFs with the specific HDAC inhibitor TSA for 30 min prior to inducing HIF with DP for 3 h (treatment with TSA starting 30 min after DP addition yielded similar results; LH Kasper, data not shown). TSA markedly inhibited the DP-dependent induction of Pfkfb3 and Egln3 in both Δflox controls and tri-ΔCH1/Δflox MEFs to an extent comparable to or greater than that caused by the ΔCH1 mutation alone (Figure 6A and B). TSA treatment in the absence of DP had marginal effects on these three genes over the 3.5 h of treatment, but TSA cooperated with the ΔCH1 mutation to further reduce HIF-responsive gene expression in the presence of DP. In fact, Egln3 DP-dependent expression was reduced nearly 100% by the combination of TSA and the ΔCH1 mutation (when TSA-dependent expression is subtracted from that for DP±TSA). TSA did not affect the induction, stability, nuclear entry, or DNA-binding activity of HIF-1α, nor did it have an effect on HIF-target gene mRNA stability (Supplementary Figure S3A–D). Both TSA and the ΔCH1 mutations reduced the levels of DP- and hypoxia-induced primary unspliced RNA transcripts for Pfkfb3 and Egln3 as measured by qRT–PCR, indicating that HIF transactivation functions are likely affected (Figure 6C and D and Supplementary Figure S3E).
Figure 6.
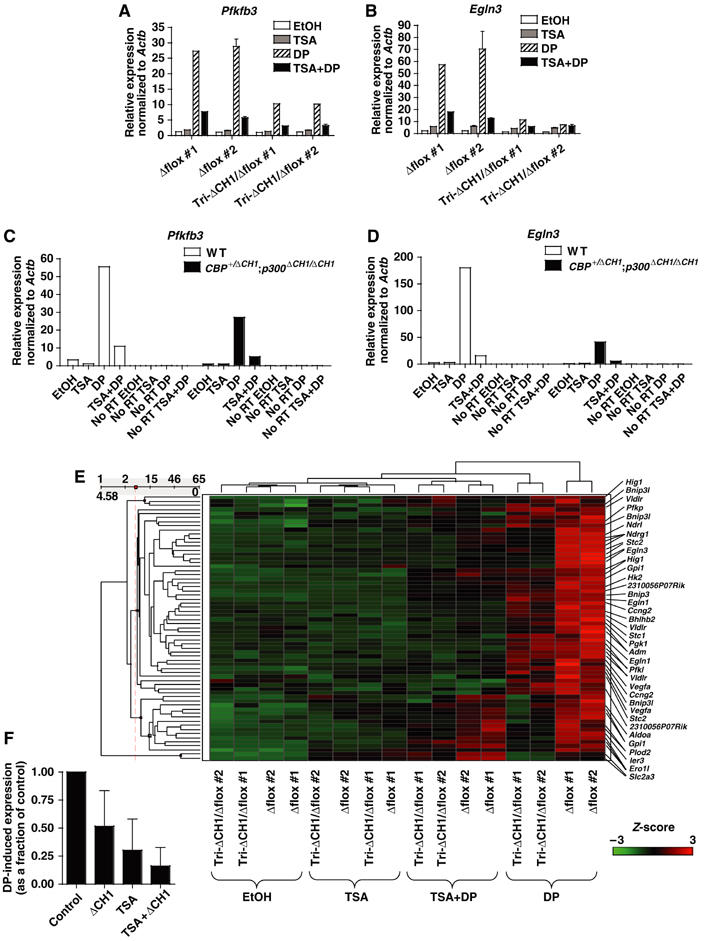
TSA and the ΔCH1 mutation have distinct but overlapping effects on the transcription of HIF-responsive genes. (A, B) qRT–PCR analysis of HIF-target gene expression in Δflox and tri-ΔCH1/Δflox MEFs treated with EtOH or TSA, followed by 3 h treatment with EtOH vehicle (EtOH) or DP, normalized to β-actin mRNA (Δflox #1 and tri-ΔCH1/Δflox #1, N=1; Δflox #2 and tri-ΔCH1/Δflox #2, mean±s.e.m., N=2). (C, D) qRT–PCR analysis of cDNA reverse transcribed from primary unspliced RNA transcripts, normalized to β-actin primary unspliced RNA, using PCR primer pairs that span an exon–intron boundary. MEFs treated with EtOH or TSA, and then for 3 h with EtOH or DP are shown. RNA was treated with DNase before reverse transcription. As a check for genomic DNA contamination, samples with no reverse transcriptase added (No RT) were analyzed. (E) Hierarchical clustering analysis of HIF-target gene probe sets in Affymetrix microarrays that showed at least a 1.5-fold induction by DP treatment and were scored as present in both Δflox MEFs treated with DP alone. MEFs of indicated genotypes were treated with EtOH or TSA, followed by a 3 h treatment with EtOH or DP. Microarray signals were normalized by Z-score transformation; red indicates induction by DP and green noninduced levels of expression. Probe sets representing genes of interest are indicated. (F) Average effect on global HIF-responsive transcription of the ΔCH1 mutation and TSA treatment alone and in combination (TSA+ΔCH1) on DP-induced signals (calculated by subtracting the appropriate control signal from the DP-treated signal). Data as in panel E (mean±s.d.).
The cooperative effects of TSA and the ΔCH1 mutation on HIF-responsive gene expression were shown using microarrays (Figure 6E). We examined 40 HIF-target genes that were induced at least 1.5-fold by DP treatment (3 h) in each of two control MEFs used (Δflox#1 and Δflox#2). Undirected hierarchical clustering of the expression signals revealed that the treatments (EtOH vehicle, TSA, TSA+DP, and DP) tended to cluster together as did the two types of control MEFs and the two types of tri-ΔCH1/Δflox mutant MEFs (Figure 6E). Expression levels were highest in the Δflox control MEFs treated with DP, whereas the tri-ΔCH1/Δflox MEFs showed reduced DP-induced expression in a gene dependent manner (Figure 6E). TSA reduced expression even more and the combination of the ΔCH1 mutation and TSA led to a profound deficit in DP-induced expression (Figure 6E). Quantitatively, the ΔCH1 mutation reduced the average global DP-inducible expression of these 40 HIF-targets to 52±32% of the Δflox controls, TSA alone reduced it to 30±28%, and the combination of the mutation and TSA reduced it to 16±16% (mean±s.d.; Table I and Figure 6F). TSA and the ΔCH1 mutation did not singly or in combination cause widespread defects in gene expression when we examined non-HIF-responsive control genes (Supplementary Figure S4A–F). Therefore, in contrast to HIF targets, the expression of non-HIF-responsive genes displayed a surprising resilience to TSA and the loss of a domain that is only found in CBP and p300.
Table 1.
Comparison of 65 Affymetrix array 430 2.0 probe sets representing 40 HIF-1α target genes as defined by Manalo et al (2005) and Greijer et al (2005)
| Probe set ID | Gene symbol | Gene title | ΔCH1 | TSA | ΔCH1+TSA |
|---|---|---|---|---|---|
| 1451385_at | 2310056P07Rik | RIKEN cDNA 2310056P07 gene | 0.63 | 0.26 | 0.17 |
| 1443339_at | 2310056P07Rik | RIKEN cDNA 2310056P07 gene | 0.95 | 0.10 | 0.22 |
| 1451678_at | 4430402O11Rik | RIKEN cDNA 4430402O11 gene | 0.19 | 0.89 | 0.00 |
| 1447839_x_at | Adm | Adrenomedullin | 0.39 | 0.26 | 0.22 |
| 1416077_at | Adm | Adrenomedullin | 0.27 | 0.24 | 0.12 |
| 1433604_x_at | Aldoa | Aldolase 1, A isoform | 0.39 | 0.29 | 0.18 |
| 1437868_at | BC023892 | CDNA sequence BC023892 | 0.35 | 0.10 | 0.00 |
| 1418025_at | Bhlhb2 | Basic helix-loop-helix domain containing, class B2 | 0.38 | 0.19 | 0.15 |
| 1422470_at | Bnip3 | BCL2/adenovirus E1B 19kDa-interacting protein 1, NIP3 | 0.78 | 0.32 | 0.33 |
| 1448525_a_at | Bnip3l | BCL2/adenovirus E1B 19kDa-interacting protein 3-like | 0.50 | 0.20 | 0.34 |
| 1416923_a_at | Bnip3l | BCL2/adenovirus E1B 19kDa-interacting protein 3-like | 0.81 | 0.01 | 0.13 |
| 1416922_a_at | Bnip3l | BCL2/adenovirus E1B 19kDa-interacting protein 3-like | 0.72 | 0.05 | 0.05 |
| 1416488_at | Ccng2 | Cyclin G2 | 0.48 | 0.07 | 0.10 |
| 1448364_at | Ccng2 | Cyclin G2 | 0.52 | 0.07 | 0.17 |
| 1428306_at | Ddit4 | DNA-damage-inducible transcript 4 | 0.43 | 0.46 | 0.20 |
| 1423785_at | Egln1 | EGL nine homolog 1 (C. elegans) | 0.83 | 0.23 | 0.26 |
| 1451110_at | Egln1 | EGL nine homolog 1 (C. elegans) | 0.63 | 0.11 | 0.01 |
| 1418648_at | Egln3 | EGL nine homolog 3 (C. elegans) | 0.14 | 0.11 | 0.00 |
| 1418649_at | Egln3 | EGL nine homolog 3 (C. elegans) | 0.14 | 0.09 | 0.03 |
| 1419029_at | Ero1l | ERO1-like (S. cerevisiae) | 0.37 | 0.69 | 0.41 |
| 1419030_at | Ero1l | ERO1-like (S. cerevisiae) | 0.36 | 0.67 | 0.66 |
| 1449324_at | Ero1l///LOC434220 | ERO1-like///hypothetical gene supported by AK009667 | 0.32 | 0.85 | 0.43 |
| 1456909_at | Gpi1 | Glucose phosphate isomerase 1 | 0.54 | 0.36 | 0.29 |
| 1450081_x_at | Gpi1 | Glucose phosphate isomerase 1 | 0.36 | 0.43 | 0.13 |
| 1420997_a_at | Gpi1 | Glucose phosphate isomerase 1 | 0.29 | 0.37 | 0.11 |
| 1450196_s_at | Gys1///Gys3 | Glycogen synthase 1, muscle///glycogen synthase 3, brain | 0.86 | 0.21 | 0.27 |
| 1448359_a_at | Hig1 | Hypoxia induced gene 1 | 0.92 | 0.46 | 0.80 |
| 1416480_a_at | Hig1 | Hypoxia induced gene 1 | 0.73 | 0.72 | 0.31 |
| 1416481_s_at | Hig1 | Hypoxia induced gene 1 | 0.74 | 0.81 | 0.56 |
| 1422018_at | Hivep2 | HIV type I enhancer binding protein 2 | 0.30 | 0.00 | 0.11 |
| 1422612_at | Hk2 | Hexokinase 2 | 0.53 | 0.32 | 0.20 |
| 1419647_a_at | Ier3 | Immediate early response 3 | 0.36 | 0.59 | 0.18 |
| 1426810_at | Jmjd1a | Jumonji domain containing 1A | 0.41 | 0.11 | 0.14 |
| 1417156_at | Krt1-19 | Keratin complex 1, acidic, gene 19 | 0.25 | 0.42 | 0.00 |
| 1418936_at | Maff | v-maf, protein F (avian) | 0.80 | 0.24 | 0.21 |
| 1450376_at | Mxi1 | Max interacting protein 1 | 0.55 | 0.17 | 0.13 |
| 1456174_x_at | Ndrg1 | N-myc downstream regulated gene 1 | 0.23 | 0.37 | 0.02 |
| 1450976_at | Ndrg1 | N-myc downstream regulated gene 1 | 0.22 | 0.29 | 0.07 |
| 1423413_at | Ndrg1 | N-myc downstream regulated gene 1 | 0.24 | 0.27 | 0.09 |
| 1420760_s_at | Ndrl | N-myc downstream regulated-like | 0.19 | 0.32 | 0.07 |
| 1426519_at | P4ha1 | Proline 4-hydroxylase, alpha 1 polypeptide | 0.51 | 0.18 | 0.57 |
| 1452094_at | P4ha1 | Proline 4-hydroxylase, alpha 1 polypeptide | 0.43 | 0.42 | 0.20 |
| 1417149_at | P4ha2 | Proline 4-hydroxylase, alpha II polypeptide | 0.41 | 0.36 | 0.15 |
| 1439148_a_at | Pfkl | Phosphofructokinase, liver, B-type | 0.79 | 0.14 | 0.06 |
| 1450269_a_at | Pfkl | Phosphofructokinase, liver, B-type | 0.67 | 0.00 | 0.02 |
| 1416069_at | Pfkp | Phosphofructokinase, platelet | 0.61 | 0.19 | 0.09 |
| 1439435_x_at | Pgk1 | Phosphoglycerate kinase 1 | 0.51 | 0.39 | 0.05 |
| 1416289_at | Plod1 | Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 | 0.50 | 0.80 | 0.17 |
| 1416687_at | Plod2 | Procollagen lysine, 2-oxoglutarate 5-dioxygenase 2 | 0.46 | 0.76 | 0.36 |
| 1416686_at | Plod2 | Procollagen lysine, 2-oxoglutarate 5-dioxygenase 2 | 0.40 | 0.83 | 0.34 |
| 1429206_at | Rhobtb1 | Rho-related BTB domain containing 1 | 1.32 | 0.09 | 0.39 |
| 1455898_x_at | Slc2a3 | solute carrier family 2 (facilitated glucose transporter), 3 | 0.28 | 0.65 | 0.27 |
| 1437052_s_at | Slc2a3 | Solute carrier family 2 (facilitated glucose transporter), 3 | 0.25 | 1.18 | 0.22 |
| 1450448_at | Stc1 | Stanniocalcin 1 | 0.45 | 0.07 | 0.05 |
| 1445186_at | Stc2 | Stanniocalcin 2 | 0.35 | 0.10 | 0.01 |
| 1449484_at | Stc2 | Stanniocalcin 2 | 0.18 | 0.09 | 0.04 |
| 1419503_at | Stc2 | Stanniocalcin 2 | 0.62 | 0.16 | 0.00 |
| 1433699_at | Tnfaip3 | Tumor necrosis factor, alpha-induced protein 3 | 0.67 | 0.02 | 0.04 |
| 1420909_at | Vegfa | Vascular endothelial growth factor A | 0.58 | 0.28 | 0.08 |
| 1451959_a_at | Vegfa | Vascular endothelial growth factor A | 0.71 | 0.03 | 0.08 |
| 1435893_at | Vldlr | Very low density lipoprotein receptor | 1.70 | 0.79 | 0.22 |
| 1417900_a_at | Vldlr | Very low density lipoprotein receptor | 0.59 | 0.09 | 0.25 |
| 1434465_x_at | Vldlr | Very low density lipoprotein receptor | 0.88 | 0.09 | 0.11 |
| 1438258_at | Vldlr | Very low density lipoprotein receptor | 0.78 | 0.09 | 0.00 |
| 1419574_at | Zfp292 | Zinc finger protein 292 | 0.61 | 0.00 | 0.05 |
| |
|
Ratio of mutation and treatment average signal to control average signal, grand mean±s.d. |
0.52±0.32 |
0.30±0.28 |
0.16±0.16 |
| Two lines of each Δflox and tri-ΔCH1/Δflox MEFs were pretreated with TSA or EtOH vehicle for 30 min followed by 3 h with EtOH or DP. Inclusion of HIF-1α target gene probe sets required induction by DP of ⩾1.5-fold and probe sets scored as present in both DP-alone-treated control MEF lines. Presented as the ratio of DP-dependent expression signal (subtracting out the appropriate control expression signal without DP) under the indicated conditions. Ratios shown are for the effect of the ΔCH1 mutation alone (ΔCH1), TSA-treated control cells (TSA), or the combination of ΔCH1 mutation and TSA treatment (ΔCH1+TSA). Data for multiple probe sets representing a single gene were averaged for the grand mean. | |||||
The microarray results confirmed that HIF-target genes do not act homogenously and that they rely to various degrees on the CH1 domain, the TSAS mechanism, and a pathway(s) that appears independent of both. A number of genes showed almost complete loss of HIF-responsive transcription by the combination of the ΔCH1 mutation and TSA treatment (e.g. Egln3, Ndrg1, Pfkl, Stc2, Vegfa; Table I and Figure 6E). None of the genes tested were completely dependent on the CH1 domain (Pgf and Egln3 were very sensitive to the mutation), although a few seemed to be more affected by the mutation than by TSA (e.g. Ero1l, Ier3, Plod2, Slc2a3; Table I and Figure 6E). Some genes were quite sensitive to TSA and less so to the ΔCH1 mutation (e.g. Bnip3l, Egln1, Pfkl, Vldlr). Yet other genes such as Bnip3, Ero1l, Hig1, P4ha1, and Plod2 retained at least a third of their DP-inducible expression in the presence of TSA and the ΔCH1 mutation, indicating an important role for a pathway independent of TSA and the CH1 domain.
Discussion
This study identifies a novel interaction between the CBP/p300 family of coactivators and a TSA-sensitive pathway that together cooperate to mediate the bulk of HIF-responsive gene transcription. Remarkably, HIF-target genes do not respond to hypoxia as a monolithic block that is wholly dependent on the CH1 domain of CBP and p300. Instead, at least three mechanisms (CH1-dependent, TSA-sensitive (TSAS), and CH1-independent/TSA-insensitive (CH1Indep/TSAI)) are utilized and the importance of each depends on the particular HIF-target gene.
The CH1-dependent mechanism was less crucial for HIF-responsive gene expression than would have been predicted from numerous in vitro studies. We found that the CH1 domain is necessary for an average of 48±32% of global HIF-responsive gene expression in MEFs treated with DP, and about 35±47% in response to hypoxia. Results from the two regimens may differ slightly because DP was applied for 3 h while the hypoxia treatment was for 6 h in our microarray studies. The somewhat greater effect of the mutation in DP-treated cells also probably reflects the strength and kinetics of HIF-target gene induction, which tends to be stronger and more rapid with DP than with hypoxia (i.e. cofactors could be more limiting). The relatively modest effect of the ΔCH1 mutation on endogenous gene expression was all the more surprising because CH1 is required for ∼80–90% of CBP and p300 recruitment to HIF-binding sites as measured by ChIP assay of DP-treated cells, and it is absolutely essential for HIF-1α and HIF-2α C-TAD transactivation function. It is unclear if the ∼10–20% residual recruitment of mutant CBP and p300 detected by ChIP reflects an alternative mechanism used by HIF to bind CBP/300, recruitment through other transcription factors, or nonspecific background levels inherent to the method. It is also unclear if the residual recruitment of ΔCH1 mutant CBP and p300 is important for CH1-independent expression of HIF targets. In this regard, the disparity between Hig1 expression and CBP/p300 recruitment in the presence of the ΔCH1 mutation implies that the CH1-independent mechanism is also independent of CBP and p300.
An important new finding is the identification of a TSA-sensitive mechanism that contributes more to overall HIF-responsive gene expression than the CH1 domain. This further suggests that HDACs have a much larger role in gene activation than typically appreciated, contrasting with their conventional involvement in repression (Yang and Gregoire, 2005). Indeed, our work significantly extends previous studies showing that TSA can inhibit the expression of the HIF-target gene Vegfa (Kim et al, 2001; Brugarolas et al, 2003), although we did not observe the TSA-dependent decrease in HIF-1α protein as reported by Kim et al (2001). Perhaps, cells exposed to TSA for periods longer than the 3.5–6.5 h used in our study would have reduced levels of HIF-1α. Intriguingly, TSA can inhibit the expression of two genes (NOR-1 and ICER) that respond to the cAMP-responsive factor CREB, suggesting that unrelated transcription factors (i.e. HIF and CREB) that interact with different domains of CBP/p300 (i.e. CH1 and KIX) share a requirement for HDAC activity (Fass et al, 2003). It is unknown whether the effect of TSA on HIF targets is direct or indirect. For example, TSA may inhibit the function of an HDAC at the affected gene promoter (i.e. a direct effect), or it may induce the expression of a repressor protein that acts on HIF targets (i.e. indirectly). However, we did not find evidence that a putative TSA-inducible repressor competes for HIF-binding sites. It is also possible that TSA upregulates non-HIF-target genes, which leads to a sequestration of a cofactor that is limiting for HIF targets (i.e. indirect squelching). Arguing against this model, we observed relatively modest effects of TSA (both repressive and inductive) on non-HIF-responsive global gene expression for the short treatment times used here (3.5 h). Regardless of the molecular mechanism, the TSAS pathway may broadly be considered as an obligate partner for signal-dependent transcription factors that rely upon CBP and p300.
The requirement for the CH1-dependent and TSAS-sensitive mechanisms varied between different HIF-responsive genes. This gene-to-gene variability suggests that cis- and trans-acting regulatory elements dictate the dependence of HIF on a specific pathway. For a number of genes (e.g. Egln3, Pfkfb3), the level of HIF-responsive transcription when both mechanisms were functioning (i.e. in WT cells) was more than additive of the levels found when each was acting alone (i.e. in TSA-treated or ΔCH1 mutant cells), thereby indicating that the two mechanisms are not independent (Herschlag and Johnson, 1993).
The importance of a CH1Indep/TSAI pathway(s) was significant for a number of HIF targets, including Bnip3, Ero1l, Hig1, P4ha1, and Plod2. Hig1 encodes a gene of unknown function and was fairly resistant to both the ΔCH1 mutation and TSA, yet ChIP analysis revealed a strong deficit in the recruitment of ΔCH1 CBP and p300, indicating that the CH1Indep/TSAI mechanism is independent of CBP/p300. This suggests that coactivators (or HDACs) of different families (i.e. not highly related to CBP and p300, or TSA-insensitive HDACs) may deliver similar functionalities to HIF. Alternatively, transcription factors other than HIF that do not bind CBP/p300, or do not require TSA-sensitive cofactors, may mediate a greater proportion of hypoxia-inducible gene expression than previously thought.
The identities of the transcriptional cofactors besides CBP and p300, and possibly HDACs, involved in these three pathways are unclear. In this regard, the coactivator SRC-1 has been implicated in HIF function, although it has been reported that this interaction is mediated by, and requires, CBP (Ruas et al, 2005). Redundant coactivation functions for HIF may also be supplied through ARNT and the HIF-1α N-terminal activation domain, which have both been reported to interact with CBP and p300 (Kobayashi et al, 1997; Ema et al, 1999). HDAC7 has been reported to bind to HIF-1α and potentiate its activity, and HDAC1 binds to p300, suggestive of a physical link between the CH1-dependent and TSAS mechanisms (Kato et al, 2004; Simone et al, 2004).
The ΔCH1 mutant mice also revealed three other important insights into the biological and transcriptional roles of this domain. First, the CH1 domains of CBP and p300 are genetically non-redundant in mice. In contrast to CBP−/− and p300−/− animals (Goodman and Smolik, 2000), the lack of a catastrophic early embryonic phenotype in CBPΔCH1/ΔCH1 and p300ΔCH1/ΔCH1 mice suggests that not all functions of CBP and p300 are limiting, or that other coactivators can function redundantly for CH1 functions. Furthermore, the notion that CBP and p300 are not necessarily interchangeable or indistinguishable in vivo is also underscored here, suggesting possible functional differences at the domain level (CH1 or other domains), or that the relative levels of CBP and p300 protein in critical target cells are important.
Second, we observed that cell growth and global gene expression were remarkably resilient to loss of the CH1 domain, despite the fact that it is unique to CBP and p300 in the genome, and the CH1 region interacts with 37 different transcriptional regulators in vitro. Indeed, these 37 proteins probably represent only a fraction of CH1 interactors, since most of the ∼2000 mammalian transcriptional regulators have not been tested in this way. It could be argued that some of the described interactions do not occur in vivo, and there is evidence to support this notion (Matt et al, 2004), but data for HIF-1α and -2α indicate that in vitro evidence can reflect true in vivo interactions. Redundancy supplied by the TAZ2 (CH3) domain of CBP and p300 also appears unlikely in most cases because its ligand-binding surface differs substantially from CH1 (Dames et al, 2002; Freedman et al, 2002). This implies that seemingly dissimilar coactivator families, or other CBP/p300 domains, provide redundancy for a wide variety of transcription factors, not just HIF.
Finally, a reduction in functional CH1 domain levels did not significantly reduce tumorigenesis. Ironically, our findings further suggest that drugs designed to specifically disrupt CH1 domain function may not always produce catastrophic side effects, despite the fact the domain binds so many different transcriptional regulators. By the same token, the efficacious reduction of hypoxia-inducible gene expression needed to treat disease may be challenging because of the two main pathways used by HIF to activate target genes. Thus, antitumor therapies that solely target the HIF C-TAD or CH1 domains may be ineffectual. There is considerable interest in HDAC inhibitors as cancer therapeutics, however, and our studies indicate that their efficacy may be enhanced by co-administering compounds that interfere with the HIF:CH1 interaction.
Materials and methods
Mice
Deletion mutations were introduced into the CH1 domains of CBP and p300 in ES cells by homologous recombination. Chimeric mice were generated from homologously targeted ES cells by standard methods. ΔCH1 mutant mice used in initial analyses were on a mixed 129 and C57BL/6 genetic background. Congenic C57BL/6 and 129 lines used to generate F1 hybrids were backcrossed at least five times. For histology, E18.5 embryos were removed from the uterus under ice-cold PBS, decapitated, and placed in 10% buffered formalin. Animal procedures were approved by the SJCRH Institutional Animal Care and Use Committee and performed in accordance with IACUC guidelines.
Plasmids and antibodies
Gal-HIF-1α contained HIF-1α residues 736–836 encoding the C-TAD fused to the Gal4 DNA-binding domain of plasmid pM2 (from I Sadowski). Gal-HIF-2α (Gal/HLF-TD 774–874) was a gift of Dan Peet. Gal-Myb contained residues 186–325 of c-Myb, Gal-Ets contained residues 2–210 of Ets-1, and Gal-CREBΔbzip contained residues 1–283 of CREB lacking the bzip domain (Kasper et al, 2002). The ΔCH1 mutation was introduced into the CBP expression vector pRC/RSV-mCBP-HA-RK (from R Goodman). A 1 μl portion of normal rabbit serum (NRS) per immunoprecipitation was used as a ChIP negative control. Specific antibodies (2 μg each) were combined for ChIP assays (CBP-A22 plus CBP-C20; p300-N15 plus p300-C20; Santa Cruz).
Cell culture and transient assays
MEFs were generated from E14.5 embryos; growth rates and morphology were comparable for WT and all mutant cells. For endogenous gene expression studies, primary MEFs were grown for the times indicated in 0.1% O2 (hypoxia) or with 100 μM DP (with or without 5 μM MG132 and 25 μM ALLN as indicated) and then immediately placed in Trizol reagent (Invitrogen) for RNA extraction. TSA (100 ng/ml) was added 30 min before hypoxia or DP treatment where noted. Transient transfection assays were performed as described; test gene luciferase activity was normalized to Renilla luciferase derived from cotransfected pRL-SV40 (Promega) (Kasper et al, 2002). For Cre-expressing adenovirus infections, MEFs were incubated overnight at 37°C, 3% O2 with adenovirus at an MOI of 100.
qRT–PCR, primary transcript qRT–PCR, and microarrays
cDNA was generated from 100 ng of total RNA in a 20 μl reaction using Superscript II reverse transcriptase (RT, Invitrogen). For primary transcript qRT–PCR to detect primary unspliced RNA transcripts, 45 μg of total RNA was treated with 6 U of RNase-free DNase (Promega) for 45 min at 37°C, inactivated by phenol/chloroform extraction, and precipitated; 200 ng of total RNA was used per 20 μl reverse transcriptase reaction; qPCR primer pairs corresponding to exonic and intronic sequences were used to distinguish cDNAs derived from primary transcripts. qRT–PCR was performed on an Opticon DNA Engine (MJ Research) using 1 μl of cDNA per 25 μl PCR reaction with SYBR Green dye. qPCR primers were designed using Primer Express software (Applied Biosystems) and confirmed to yield a single product by melt-curve analysis. Samples were normalized to β-actin (Actb) mRNA. Affymetrix microarray data were generated by the Hartwell Center (SJCRH) using Affymetrix mouse genome arrays 430A and 430 version 2.0. Spotfire software was used for analysis; signal data were normalized by Z-score transformation for the hierarchical clustering analysis. Hypoxia-inducible and DP-inducible gene probe sets were defined using control MEF data; probe sets scored present by Affymetrix software in hypoxia- or DP-treated samples from control MEFs and induced beyond a defined hypoxia/normoxia or DP/EtOH signal ratio. The 40 HIF-responsive genes were defined as meeting the above criteria (in this case, ⩾1.5-fold induction above control by hypoxia or DP) and were genes that were shown by Manalo et al (2005) to be induced by a constitutively active form of HIF-1α or by Greijer et al (2005) to have attenuated expression under hypoxic conditions in HIF-1α null MEFs as compared to control MEFs. Hypoxia- and DP-dependent signals were calculated by subtracting the normoxia signal from the hypoxia signal or the EtOH signal from the DP signal. The effect of TSA on DP-dependent expression was calculated by subtracting the DP plus TSA signal from the TSA alone signal. Array data were deposited with Gene Expression Omnibus (GEO) (GSE3318, GSE3195, GSE3196, GSE3296).
Nuclear extracts, Western blots, HAT assays, and ChIP assays
Nuclear extract preparation, Western blots, and HAT assays were performed as described (Kasper et al, 2002). ChIP assays followed a modified Upstate Biotechnology Inc. protocol. Briefly, MEFs were treated at 37°C for either 2 h with 100 μM DP, 5 μM MG132, and 25 μM ALLN or for 4 h with 0.1% O2, and immediately treated for 20 min with 3% paraformaldehyde in PBS. Cells were washed in PBS and collected by scraping. Whole-cell extracts were sonicated for 5 × 10 s at 15 μm amplitude (Sanyo Soniprep 150), precleared using NRS and salmon sperm DNA, and then incubated with specific antibodies overnight at 4°C. After washing the immunoprecipitates, DNA/antibody complexes were eluted, crosslinks reversed, and DNA was purified by phenol extraction and ethanol precipitation. Quantitative real-time PCR analysis used 8% (2 μl) of ChIP sample per 25 μl reaction and was normalized to input DNA. qPCR primers were verified to be quantitative using 10-fold serially diluted DNA.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Acknowledgments
We thank D Warburton for advice on the lung phenotype, H Stunnenberg and S Denissov for advice on ChIP assays, S Lerach and C Wang for excellent technical assistance, M Chong for help with histology and primary cell culture, D Bedford for comments on the manuscript, and D Peet for Gal-HIF-2α. We also thank the Vector Development and Production, and Transgenic core facilities at SJCRH. The Hartwell Center at SJCRH provided oligonucleotides and DNA sequencing, and performed the Affymetrix experiments. This work was supported by ALSAC and NIH grants CA076385 (PB), CA076379 (JC), and DK058199 (PB), the Cancer Center (CORE) support grant P30 CA021765, and the American Lebanese Syrian Associated Charities of St Jude Children's Research Hospital.
References
- Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A (2004) Chromatin acetylation, memory, and LTP are impaired in CBP(+/−) mice; a model for the cognitive deficit in Rubinstein–Taybi syndrome and its amelioration. Neuron 42: 947–959 [DOI] [PubMed] [Google Scholar]
- Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG Jr (2003) TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 4: 147–158 [DOI] [PubMed] [Google Scholar]
- Bruick RK (2003) Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia-inducible transcription factor. Genes Dev 17: 2614–2623 [DOI] [PubMed] [Google Scholar]
- Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, Nemery B, Dewerchin M, Van Veldhoven P, Plate K, Moons L, Collen D, Carmeliet P (2002) Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med 8: 702–710 [DOI] [PubMed] [Google Scholar]
- Dames SA, Martinez-Yamout M, De Guzman RN, Dyson HJ, Wright PE (2002) Structural basis for Hif-1 alpha/CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci USA 99: 5271–5276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ema M, Hirota K, Mimura J, Abe H, Yodoi J, Sogawa K, Poellinger L, Fujii-Kuriyama Y (1999) Molecular mechanisms of transcription activation by HLF and HIF1alpha in response to hypoxia: their stabilization and redox signal-induced interaction with CBP/p300. EMBO J 18: 1905–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fass DM, Butler JE, Goodman RH (2003) Deacetylase activity is required for cAMP activation of a subset of CREB target genes. J Biol Chem 278: 43014–43019 [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW (1996) Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380: 439–442 [DOI] [PubMed] [Google Scholar]
- Freedman SJ, Sun ZY, Poy F, Kung AL, Livingston DM, Wagner G, Eck MJ (2002) Structural basis for recruitment of CBP/p300 by hypoxia-inducible factor-1 alpha. Proc Natl Acad Sci USA 99: 5367–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccia A, Siim BG, Johnson RS (2003) HIF-1 as a target for drug development. Nat Rev Drug Discov 2: 803–811 [DOI] [PubMed] [Google Scholar]
- Giaccia AJ, Simon MC, Johnson R (2004) The biology of hypoxia: the role of oxygen sensing in development, normal function, and disease. Genes Dev 18: 2183–2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman RH, Smolik S (2000) CBP/p300 in cell growth, transformation, and development. Genes Dev 14: 1553–1577 [PubMed] [Google Scholar]
- Greijer A, van der Groep P, Kemming D, Shvarts A, Semenza G, Meijer G, van de Wiel M, Belien J, van Diest P, van der Wall E (2005) Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1). J Pathol 206: 291–304 [DOI] [PubMed] [Google Scholar]
- Gu J, Milligan J, Huang LE (2001) Molecular mechanism of hypoxia-inducible factor 1alpha -p300 interaction. A leucine-rich interface regulated by a single cysteine. J Biol Chem 276: 3550–3554 [DOI] [PubMed] [Google Scholar]
- Herschlag D, Johnson FB (1993) Synergism in transcriptional activation: a kinetic view. Genes Dev 7: 173–179 [DOI] [PubMed] [Google Scholar]
- Kalkhoven E (2004) CBP and p300: HATs for different occasions. Biochem Pharmacol 68: 1145–1155 [DOI] [PubMed] [Google Scholar]
- Kang-Decker N, Tong C, Boussouar F, Baker DJ, Xu W, Leontovich AA, Taylor WR, Brindle PK, Van Deursen JM (2004) Loss of CBP causes T cell lymphomagenesis in synergy with p27(Kip1) insufficiency. Cancer Cell 5: 177–189 [DOI] [PubMed] [Google Scholar]
- Kasper LH, Boussouar F, Ney PA, Jackson CW, Rehg J, van Deursen JM, Brindle PK (2002) A transcription-factor-binding surface of coactivator p300 is required for haematopoiesis. Nature 419: 738–743 [DOI] [PubMed] [Google Scholar]
- Kato H, Tamamizu-Kato S, Shibasaki F (2004) Histone deacetylase 7 associates with hypoxia-inducible factor 1alpha and increases transcriptional activity. J Biol Chem 279: 41966–41974 [DOI] [PubMed] [Google Scholar]
- Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, Moon EJ, Kim HS, Lee SK, Chung HY, Kim CW, Kim KW (2001) Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med 7: 437–443 [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Numayama-Tsuruta K, Sogawa K, Fujii-Kuriyama Y (1997) CBP/p300 functions as a possible transcriptional coactivator of Ah receptor nuclear translocator (Arnt). J Biochem (Tokyo) 122: 703–710 [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M (2004) CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42: 961–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung AL, Wang S, Klco JM, Kaelin WG, Livingston DM (2000) Suppression of tumor growth through disruption of hypoxia-inducible transcription. Nat Med 6: 1335–1340 [DOI] [PubMed] [Google Scholar]
- Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, Memmert K, Naegeli HU, Petersen F, Eck MJ, Bair KW, Wood AW, Livingston DM (2004) Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell 6: 33–43 [DOI] [PubMed] [Google Scholar]
- Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL (2005) Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105: 659–669 [DOI] [PubMed] [Google Scholar]
- Matt T, Martinez-Yamout MA, Dyson HJ, Wright PE (2004) The CBP/p300 TAZ1 domain in its native state is not a binding partner of MDM2. Biochem J 381: 685–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina DN, Glasscock J, Gish W, Lovett M (2004) An ORFeome-based analysis of human transcription factor genes and the construction of a microarray to interrogate their expression. Genome Res 14: 2041–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naltner A, Wert S, Whitsett JA, Yan C (2000) Temporal/spatial expression of nuclear receptor coactivators in the mouse lung. Am J Physiol Lung Cell Mol Physiol 279: L1066–L1074 [DOI] [PubMed] [Google Scholar]
- Newton AL, Sharpe BK, Kwan A, Mackay JP, Crossley M (2000) The transactivation domain within cysteine/histidine-rich region 1 of CBP comprises two novel zinc-binding modules. J Biol Chem 275: 15128–15134 [DOI] [PubMed] [Google Scholar]
- Poellinger L, Johnson RS (2004) HIF-1 and hypoxic response: the plot thickens. Curr Opin Genet Dev 14: 81–85 [DOI] [PubMed] [Google Scholar]
- Roelfsema JH, White SJ, Ariyurek Y, Bartholdi D, Niedrist D, Papadia F, Bacino CA, den Dunnen JT, van Ommen GJ, Breuning MH, Hennekam RC, Peters DJ (2005) Genetic heterogeneity in Rubinstein–Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet 76: 572–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruas JL, Poellinger L, Pereira T (2005) Role of CBP in regulating HIF-1-mediated activation of transcription. J Cell Sci 118: 301–311 [DOI] [PubMed] [Google Scholar]
- Semenza GL (2002) Physiology meets biophysics: visualizing the interaction of hypoxia-inducible factor 1 alpha with p300 and CBP. Proc Natl Acad Sci USA 99: 11570–11572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3: 721–732 [DOI] [PubMed] [Google Scholar]
- Simone C, Stiegler P, Forcales SV, Bagella L, De Luca A, Sartorelli V, Giordano A, Puri PL (2004) Deacetylase recruitment by the C/H3 domain of the acetyltransferase p300. Oncogene 23: 2177–2187 [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Naruse I, Hongo T, Xu M, Nakahata T, Maekawa T, Ishii S (2000) Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB-binding protein. Mech Dev 95: 133–145 [DOI] [PubMed] [Google Scholar]
- Warner DR, Pisano MM, Greene RM (2002) Expression of the nuclear coactivators CBP and p300 in developing craniofacial tissue. In vitro Cell Dev Biol Anim 38: 48–53 [DOI] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, Abel T (2005) Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem 12: 111–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zhang X, Zehner ZE (2003) c-Jun and the dominant-negative mutant, TAM67, induce vimentin gene expression by interacting with the activator Sp1. Oncogene 22: 8891–8901 [DOI] [PubMed] [Google Scholar]
- Yang XJ, Gregoire S (2005) Class II histone deacetylases: from sequence to function, regulation, and clinical implication. Mol Cell Biol 25: 2873–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanger K, Radovick S, Wondisford FE (2001) CREB binding protein recruitment to the transcription complex requires growth factor-dependent phosphorylation of its GF box. Mol Cell 7: 551–558 [DOI] [PubMed] [Google Scholar]
- Zhou XY, Shibusawa N, Naik K, Porras D, Temple K, Ou H, Kaihara K, Roe MW, Brady MJ, Wondisford FE (2004) Insulin regulation of hepatic gluconeogenesis through phosphorylation of CREB-binding protein. Nat Med 10: 633–637 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2