

Abstract
Centromere protein F (CENP-F) (or mitosin) accumulates to become an abundant nuclear protein in G2, assembles at kinetochores in late G2, remains kinetochore-bound until anaphase, and is degraded at the end of mitosis. Here we show that the absence of nuclear CENP-F does not affect cell cycle progression in S and G2. In a subset of CENP-F depleted cells, kinetochore assembly fails completely, thereby provoking massive chromosome mis-segregation. In contrast, the majority of CENP-F depleted cells exhibit a strong mitotic delay with reduced tension between kinetochores of aligned, bi-oriented sister chromatids and decreased stability of kinetochore microtubules. These latter kinetochores generate mitotic checkpoint signaling when unattached, recruiting maximum levels of Mad2. Use of YFP-marked Mad1 reveals that throughout the mitotic delay some aligned, CENP-F depleted kinetochores continuously recruit Mad1. Others rebind YFP-Mad1 intermittently so as to produce ‘twinkling', demonstrating cycles of mitotic checkpoint reactivation and silencing and a crucial role for CENP-F in efficient assembly of a stable microtubule–kinetochore interface.
Keywords: centromere associated protein CENP-F, kinetochore, Mad1, Mad2, microtubule
Introduction
Kinetochores are the macromolecular complexes that assemble each cell cycle at the centromere of each chromosome to ensure accurate chromosome segregation during mitosis and meiosis. The mature kinetochore and the different proteins that constitute it play a central role in the correct segregation of chromosomes by (i) establishing and maintaining the attachment to microtubules of the mitotic spindle and (ii) regulating the mitotic checkpoint (also known as the spindle assembly checkpoint), which controls cell cycle advance through mitosis. Unattached kinetochores generate a partially diffusible ‘wait anaphase' inhibitor that blocks cell cycle advance to anaphase until all kinetochores have achieved proper attachment to microtubules of the spindle (reviewed in Cleveland et al, 2003).
Kinetochore assembly is highly dynamic and organized. The amorphous electron dense material observed at prophase (Brenner et al, 1981) organizes to form a complex structure that appears as a trilaminar disk (Rieder, 1982), with each layer comprised of distinct protein compositions ensuring different roles in kinetochore function (McEwen et al, 1993). The inner plate of the vertebrate kinetochore contains the proteins CENP (centromere protein)-A, CENP-B, CENP-C, CENP-G, CENP-H, CENP-I, and Mis12. These are involved in kinetochore assembly, whereas components of the outer plate are implicated in microtubule attachment (e.g. CENP-E (Yao et al, 2000), the Ndc80 complex (DeLuca et al, 2002), and RanBP2 (Joseph et al, 2004)), microtubules dynamics (CLASP1; Maiato et al, 2003) and checkpoint signaling (including Mad1, Mad2, Bub1, BubR1 and Mps1 (see Cleveland et al, 2003; Maiato et al, 2004; Kline-Smith et al, 2005)).
The assembly of a mature three-dimensional kinetochore initiates in G2, continues into mitosis with the breakdown of the nuclear envelope and persists until the completion of cell division. One of the earliest components that assembles at the immature kinetochore is the 350 kDa CENP-F (also known as mitosin) (Rattner et al, 1993; Casiano et al, 1995; Zhu et al, 1995b), a transient kinetochore component that accumulates to become an abundant nuclear protein in G2, assembles at the nascent kinetochores in late G2, remains kinetochore-bound until anaphase, and is then degraded at the end of mitosis (Liao et al, 1995). Like CENP-E, the CENP-F has been localized at the more distal region of the outer kinetochore (Zhu et al, 1995a). Through its tail domain, CENP-F associates with the outer kinetochore earlier than any of the other known transient kinetochore proteins (Liao et al, 1995; Zhu et al, 1995a), suggesting that CENP-F may act in the initial steps of its assembly.
Several indirect data also suggest that CENP-F acts to regulate progression from the G2 phase to M phase (Zhu et al, 1995b; Ashar et al, 2000; Crespo et al, 2001; Hussein and Taylor, 2002). CENP-F is a substrate for farnesylation, and farnesyltransferase inhibitors (FTIs) prevent its farnesylation without disturbing its normal localization at the kinetochore (Ashar et al, 2000; Crespo et al, 2001). A suggestive link to CENP-F is that analyses of FTIs have demonstrated that human tumor cell lines sensitive to these agents accumulate in prometaphase. In addition, overexpression of the tail domain of CENP-F induces a delay in G2/M progression (Zhu et al, 1995b; Hussein and Taylor, 2002).
Contradictory evidence on the direct role of CENP-F (as assessed by CENP-F depletion using RNAi) has also emerged. One study focusing on the function of the Forkhead transcription factor FoxM1 during mitosis has identified the CENP-F gene as one of its targets. In total, 50% of cells depleted of CENP-F by siRNA have been reported to initiate anaphase and undergo cytokinesis in the presence of misaligned chromosomes and to be unable to sustain long-term activation of the mitotic checkpoint after spindle disruption with microtubule drugs (Laoukili et al, 2005). In direct contrast, another study also lowering CENP-F levels by siRNA reported that CENP-F depleted cells transiently arrest in pseudo-metaphase with most chromosomes aligned but then proceeded to DNA decondensation and cell death without exiting mitosis (Yang et al, 2005). These analyses leave unanswered how CENP-F absence from interphase nuclei in S or G2 and during mitosis affects cell cycle progression, mitotic checkpoint signaling and how or whether any deficit can provoke cell death from any of these cell cycle positions.
To address this, we have investigated the role of CENP-F in interphase and in mitosis using two independent methods of gene silencing and by targeting two distinct regions of CENP-F mRNA. Both methods reveal that in the absence of CENP-F, kinetochore assembly fails in a minority of cells leading to gross chromosome mis-segregation. In others, CENP-F depletion induces a strong mitotic delay with chromosomes aligned in a less tightly clustered metaphase. CENP-F depletion resembles inhibition of the Ndc80 complex, which is marked by decreased tension between sister kinetochores and a reduction of stable microtubule attachment at kinetochores. Mitotic checkpoint signaling in most CENP-F depleted cells is fully active, as demonstrated by normal levels of Mad2 recruitment at kinetochores and by the sustained mitotic arrest in the presence of microtubule depolymerizing agents. The continued presence of Mad1 at kinetochores of some aligned chromosomes and the transient reassociation (twinkling) of Mad1 at others demonstrate directly that the mitotic delay is due to continued activation of the mitotic checkpoint caused by unstable microtubule capture by CENP-F depleted kinetochores.
Results
CENP-F depletion does not affect cell cycle progression across S or G2, but provokes either premature anaphase or mitotic checkpoint mediated delay
To investigate the role of CENP-F during S, G2 and mitosis, we repressed its expression by using both transcription-mediated siRNA and siRNA duplexes (Figure 1A and B). In the first approach, a plasmid encoding the surface marker CD20 was cotransfected with the CENP-F siRNA construct. Successfully transfected cells were recovered by affinity chromatography with immobilized antibodies to CD20 (Figure 1A). For introduction of siRNA duplexes, cotransfection of a Cy3-tagged luciferase encoding gene revealed uptake by >99% of the targeted cells (Figure 1B). With both methods, CENP-F accumulation in randomly cycling cells was reduced by more than 90% by 48 h post-transfection (Figure 1C). All detectable kinetochore localization was eliminated (Figure 1D).
Figure 1.
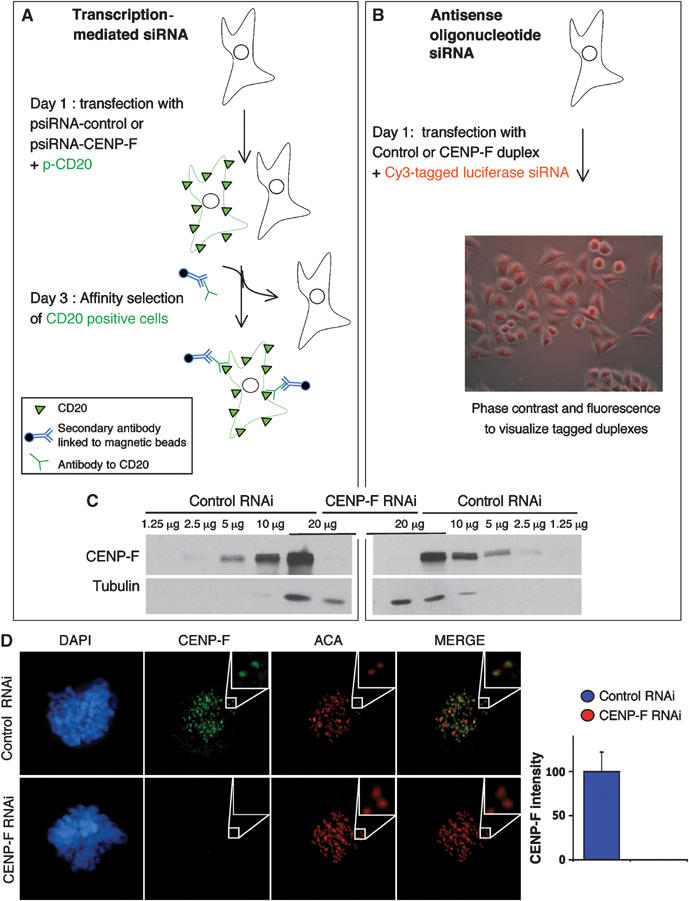
Transcription-mediated siRNA and RNA duplex siRNA both result in >90% depletion of CENP-F. (A–D) CENP-F repression was obtained by transcription-mediated siRNA and RNA duplex siRNA. HeLa cells were transfected with (A) the corresponding siRNA plasmids along with pCD20 that encodes the cell surface marker CD20 or (B) siRNA duplex RNAs and a luciferase–Cy3 RNA duplex. Accumulated CENP-F levels were visualized by (C) immunoblotting whole-cell lysates and (D) immunofluorescence 48 h after transfection.
To define if absence of CENP-F affected cell cycle progression, transcription-based siRNA was combined with subsequent CD20 selection and cell synchronization to enrich cells at the G1/S boundary following an 18 h DNA synthesis arrest caused by high levels of exogenous thymidine (Figure 2A). Detectable CENP-F was eliminated from nuclei in cells transfected with the CENP-F siRNA construct, but not those transfected with a control siRNA plasmid (Figure 2B and C). The thymidine block was released and time lapse microscopy was used to monitor the progression of the entire cell population across S and G2 using the cell rounding that occurs at late prophase/early prometaphase as a measure of mitotic entry. (This measure was validated by filming cells transfected to express histone H2B-EYFP to visualize chromosome condensation that initiates at prophase (data not shown).) Release from the S phase arrest produced indistinguishable kinetics of mitotic entry in comparing the CENP-F depleted and control cell populations (Figure 2D), with both peaking 10 h after release. Thus, the absence of nuclear CENP-F did not affect the progression across either S or G2 phases.
Figure 2.
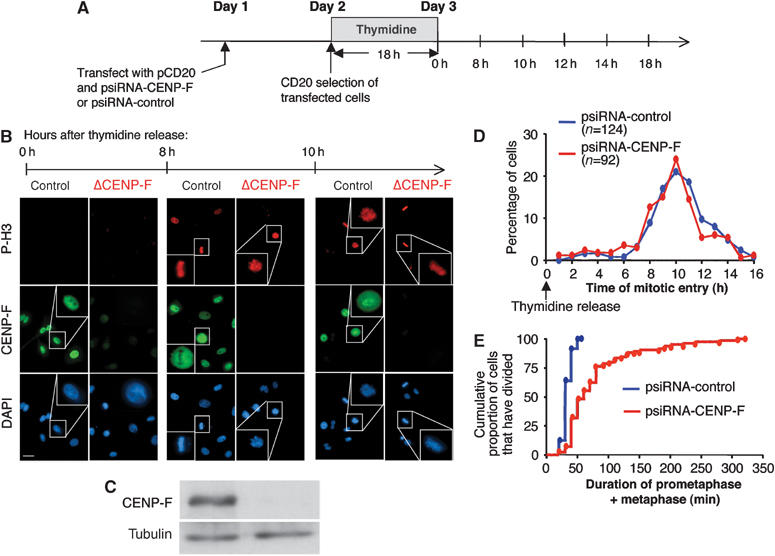
Depletion of CENP-F does not perturb cell cycle timing in S or G2, but affects the duration of mitosis. (A) HeLa cells were transfected with the corresponding siRNA plasmids along with pCD20. After 24 h, transfected cells were affinity isolated, replated and incubated with 2 mM thymidine for 18 h. At 8 h after release from the drug, CENP-F depletion was verified by (B) immunofluorescence and (C) immunoblot. (B) Thymidine-induced synchronization in S phase or at the G1–S boundary was verified by the increase in late G2/mitotic cells 8 h after release (as revealed by immunopositivity for phosphorylated histone H3) and the presence of dividing cells after 10 h (revealed by DAPI staining). (D) Determination of mitotic entry following thymidine release in control RNAi cells (blue lines) and CENP-F RNAi cells (red lines). Mitosis entry is defined by the time each cell became rounded before dividing, as observed by time lapse brightfield microscopy. (E) Determination of the duration of prometaphase/metaphase, as defined by the time each cell spent between becoming round and the beginning of cytokinesis. The cumulative proportion of the cells that have divided is represented to show that all control cells spent <1 h in mitosis, while this is extended to >5 h in CENP-F RNAi cells. Scale bar, 10 μm.
The length between mitotic entry and exit was also measured in CENP-F depleted and normal cells, as defined by the interval from mitotic rounding to the first frame in which cytokinesis furrow first appeared. Although all control cells divided within 60 min, with an average time of 38.6±7.5 min, CENP-F depleted cells showed a wide variability in the duration of mitosis that extended up to 320 min, with an average time of 84.4±57.7 min (Figure 2E). All cells ultimately underwent cell division.
To define the underlying mechanism responsible for the pre-anaphase mitotic delay in the majority of CENP-F depleted cells, HeLa cells expressing histone H2B-EYFP were monitored beginning at prophase (Figure 3). Both transcription mediated and siRNA duplexes were used to lower CENP-F synthesis. For transcription based RNAi, cells that expressed high yellow fluorescent protein (YFP) levels were analyzed. RNA duplexes were introduced into an HeLa cell line stably expressing H2B-EYFP. Both methods gave similar results in that CENP-F was undetectable in 90% of these (data not shown). Control RNAi cells were homogeneous with regard to the time spent in prophase, prometaphase and anaphase, with averages of 6.8, 27.8 and 22.7 min, respectively (Figure 3; Supplementary Movie 1). Although 17% of the control RNAi cells had somewhat extended metaphases (yielding an average of 30.5 min for all the cells), all of them divided in less than 147 min (with an average time of 87.8 min for all control cells; Figure 3A). CENP-F depleted cells were quite different. While all spent a time similar to the control RNAi cells in prophase (with an average of 8 min), most had very extended prometaphases/metaphases, with some cells strongly blocked at prometaphase (see an example in Figure 3C-1 and Supplementary Movie 2) or in metaphase (Figure 3C-2; Supplementary Movie 3), giving an average time of 87.3 min in prometaphase and 112.5 min in metaphase (Figure 3A).
Figure 3.
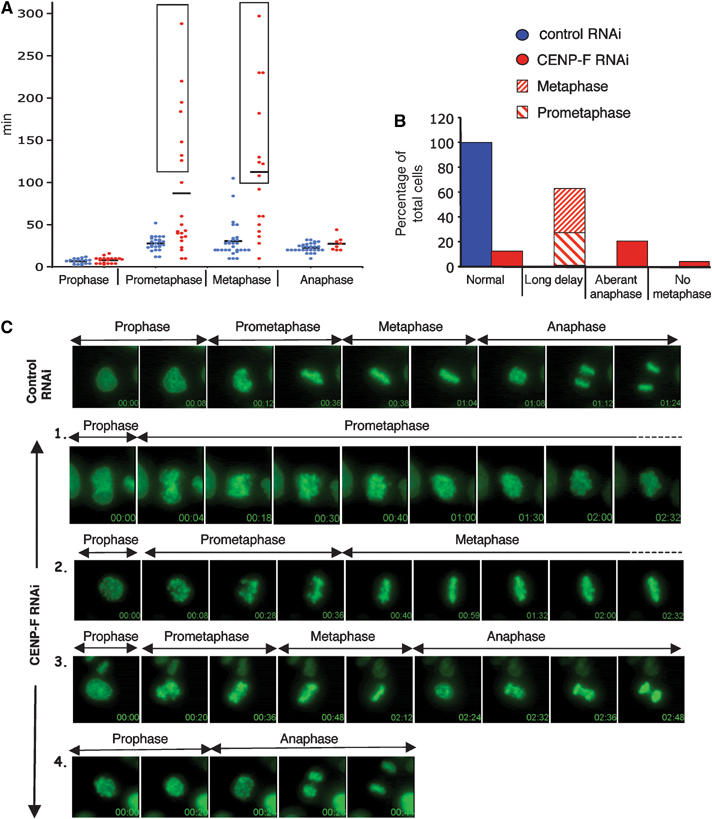
Premature anaphase and mitotic delay in the absence of CENP-F. (A–C) Time lapse imaging was performed 48 h after transfection of (i) HeLa cells with psiRNAi-control/-CENP-F with pH2B-EYFP and (ii) an HeLa cell line stably expressing H2B-EYFP with control/CENP-F antisense RNA duplexes along with a Cy3-tagged RNA duplex to luciferase. (i) Bright EYFP positive cells or the (ii) Cy3 positive cells were recorded from prophase. (A) Time spent in different stages of mitosis for control RNAi cells (in blue) and CENP-F RNAi cells (in red). The individual time for each cell is represented (in circle), as the average time (black line). Cells that were still at the corresponding phase when the filming was stopped are boxed. All other cells proceeded through mitosis and divided. (B) Relative proportions of the different phenotypes in control RNAi cells (blue) and CENP-F RNAi cells (red). (C) Time-lapse sequences of mitosis in a control RNAi cell (Supplementary Movie 1) and a prometaphase delay (1, see Supplementary Movie 2), metaphase delay (2, Supplementary Movie 3), abnormal anaphase (3, Supplementary Movie 4) and division without metaphase (4, Supplementary Movie 5) observed in CENP-F RNAi cells.
Despite initial alignment, chromosomes of CENP-F depleted cells failed to remain tightly aligned, with some arms protruding from the metaphase axis and producing ‘fat metaphase' indicative of unstable alignment (Supplementary Movie 3). Only 12.5% of the population of CENP-F depleted cells divided with normal timing, with a majority (62.5%) showing a very strong block at either prometaphase (46.7% of the arrested cells) or at metaphase (Figure 3A and B). A proportion (20.7%) of CENP-F depleted cells underwent an abnormal anaphase with many lagging chromosomes (see an example in Figure 3C-3; Supplementary Movie 4). A small proportion (4.3%) of CENP-F depleted cells proceeded directly into anaphase without detectable prometaphase or metaphase. For example, the cell in Figure 3C-4 entered anaphase only 24 min after mitotic entry and without prior alignment at metaphase, yielding gross chromosomal missegregation (see also Supplementary Movie 5). Thus, for such cells, CENP-F appears to play a crucial role in an early step of kinetochore assembly.
Reduced tension at kinetochores of bi-oriented chromosomes after assembly without CENP-F
CENP-F has been localized to the outer surface of the kinetochore (Zhu et al, 1995a), a position appropriate for affecting spindle microtubule capture or stabilization. As an initial measure for how the absence of CENP-F affected kinetochore–microtubule interactions, spacing between sister kinetochores was measured for chromatid pairs that had aligned. This revealed that tension-mediated stretching between the two sister kinetochores was reduced (inter-kinetochore spacing of 1.05±0.11 μm (Figure 4A-2) versus 1.39±0.09 μm for CENP-F containing kinetochores (Figure 4A-1)) on apparently bi-oriented sister kinetochores depleted of CENP-F. Some tension remained, however, since fully relaxed kinetochores, measured after nocodazole-mediated microtubule disassembly, showed even smaller spacing (0.87±0.04 μm (Figure 4A-3)).
Figure 4.
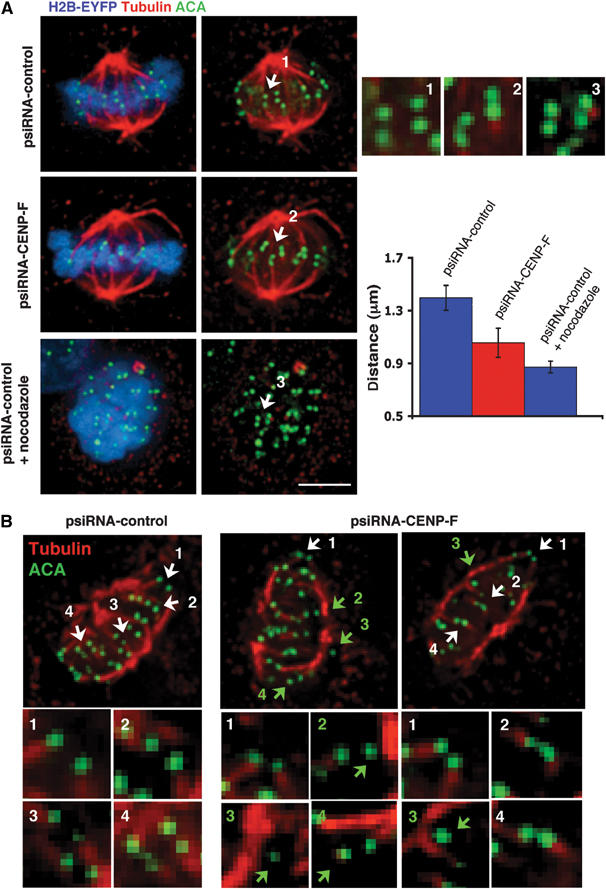
Reduced capture and/or stability of microtubules at kinetochores in the absence of CENP-F. (A) Reduced tension between sister kinetochores in the absence of CENP-F. At 48 h after cotransfection of the corresponding psiRNA with pH2B-EYFP, high expressing H2B-EYFP cells in metaphase were imaged. (Blue) H2B-EYFP; (red) tubulin (green) ACA. In total, 120 kinetochore pairs in which both sister kinetochores were in the same focal plane were analyzed for both psiRNA-CENPF and psiRNA control cells with and without nocodazole treatment. Insets 1–3 show enlargements of deconvolved planes for the psiRNA-control (1) without and with (3) 20 μM nocodazole for 3 h and (2) for psiRNA-CENP-F. Scale bar, 5 μm. The averages of 1.39±0.09, 1.05±0.11, and 0.87±0.04 μm for kinetochores in cells from (1) (2) and (3) are significantly different (using ANOVA single factor test with α values <0.05 and P-values <0.001). (B) Reduced stability of kinetochore fibers in the absence of CENP-F. psiRNA-control and psiRNA-CENP-F cells were cooled on ice for 10 min prior to extraction and fixation. High expressing H2B-EYFP were stained for (red) tubulin and (green) ACA and processed to deconvolution. Attachment of kinetochores to microtubules was determined by following >100 individual kinetochores through all the focal planes. Kinetochores in regions where fibers were not easily visualized were not taken into account. The images and enlargements correspond to the merge of selected focal planes. Kinetochores attached to microtubules (white arrows), and unattached ones (green arrows).
CENP-F depleted kinetochores bind microtubules less stably
To test whether the absence of CENP-F affected stability of microtubule capture at kinetochores, microtubule attachment was examined after briefly cooling the cells to 4°C to destabilize most nonkinetochore microtubules (Rieder, 1981). While kinetochore microtubule bundles uniformly remained attached to centromeres in control cells (Figure 4B), most cells transfected with the CENP-F encoding siRNA plasmid showed 1–3 unattached kinetochores (Figure 4B), consistent with those kinetochores either unbound initially to kinetochore bundles or that the attachment was not stable. Neither Hec1, a member of the Ndc80 complex that has been shown to be required for maintaining stable microtubule attachment (DeLuca et al, 2002), nor Clasp1, a regulator of microtubule dynamics (Maiato et al, 2003), were affected by the absence of CENP-F (Supplementary Figure 6).
Mitotic delay in CENP-F depleted cells from persistent mitotic checkpoint signaling by kinetochores of aligned chromosomes
The extended metaphases in CENP-F depleted cells, combined with apparently less stable spindle microtubule capture by those kinetochores, suggested that the mitotic delay could be due to chronic activation of the mitotic checkpoint. To test this, recruitment of Mad2, Bub1, BubR1 and CENP-E to kinetochores was examined after depleting CENP-F. In control cells, kinetochores recruited reduced levels of CENP-E, BubR1 and Bub1 following microtubule attachment, as measured by quantification of staining intensity at individual kinetochores. Specifically, at metaphase, these kinetochores recruited 44±6% (CENP-E), 62±6% (BubR1) and 66±14% (Bub1) of the corresponding intensities observed in early prometaphase (Figure 5). Meanwhile, in the absence of CENP-F, CENP-E, BubR1 and Bub1 signals were reduced to statistical significance (to 38±5, 57±10, and 48±13%, respectively) throughout prometaphase and metaphase (Figure 5A–C). Although previous reports also showed a decreased intensity of CENP-E in CENP-F absence (Johnson et al, 2004; Yang et al, 2005), BubR1 recruitment has been reported to be either unchanged (Yang et al, 2005), decreased (Laoukili et al, 2005) or increased together with Bub1 in the presence of nocodazole (Johnson et al, 2004).
Figure 5.
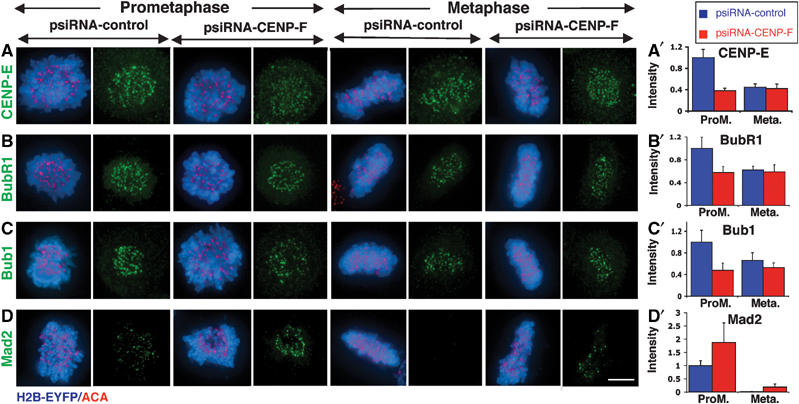
CENP-F affects the recruitment of CENP-E and checkpoint regulators to the kinetochore. Kinetochore localization in prometaphase and metaphase of (A) CENP-E, (B) BubR1, (C) Bub1, (D) Mad2 48 h after transfection of psiRNA-control/-CENP-F with pH2B-EYFP. (Blue) H2B-EYFP; (red) ACA; (green) CENP-E, BubR1, Bub1 or Mad2. Quantification of the normalized integrated intensity of (A′) CENP-E, (B′) BubR1, (C′) Bub1, and (D′) Mad2 at kinetochores in (blue) psiRNA-control and (red) psiRNA-CENP-F cells. Mad2 quantification was performed on all kinetochores, including those for which no signal was detected. The error bars represent standard errors. Heteroscedastic T-tests revealed significant differences at prometaphase between psiRNA-control and psiRNA-CENP-F: P-values of 0.0002 (CENP-E), 0.0394 (BubR1) and 0.0290 (Bub1). The same test gives a P-value of 0.0861 for Mad2 at metaphase. Aproximately 1000 kinetochores were analyzed corresponding to about 10 cells for each bar. Scale bar, 5 μm.
Mad2, whose recruitment to kinetochores is strongly correlated with generation of the mitotic checkpoint inhibitor (Chen et al, 1996; Shah et al, 2004), is lost from kinetochores in control cells after microtubule capture and alignment (Figure 5D). The overall Mad2 level at metaphase kinetochores was determined by averaging Mad2 intensity among all kinetochores (identified by anti-centromere antibodies (ACA)). Both qualitatively (Figure 5D) and quantitatively (Figure 5D′ and Figure 6A and B) such kinetochores only rarely retained detectable Mad2 in control cells. In contrast, in CENP-F depleted cells, kinetochores recruited almost twice (187±74%) the normal level of Mad2 observed at unattached prometaphase kinetochores in normal cells (Figure 5D′). Even for chromosomes that were fully aligned, an average intensity of Mad2 per CENP-F depleted kinetochore remained at 19±11% the level of a prometaphase kinetochore in CENP-F containing cells (Figure 5D′).
Figure 6.
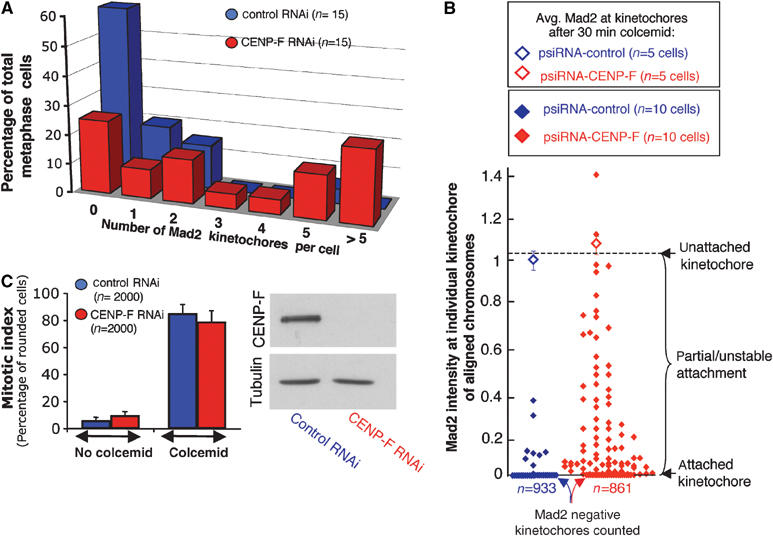
Continued recruitment of Mad2 at kinetochores of aligned chromosomes and intact checkpoint activity in CENP-F depleted cells. (A) Number of kinetochores with detectable Mad2 on aligned chromosomes in control and CENP-F depleted cells 48 h after cotransfection of the psiRNA-CENP-F/-control with pH2B-EYFP. (B) Mad2 intensity at individual kinetochores of aligned chromosomes of metaphase cells 48 h after cotransfection with the psiRNA-control/-CENP-F along with pH2B-EYFP. (Open diamonds) The average recruitment of Mad2 at kinetochores in control and CENP-F free cells after disruption of microtubule attachment (by the addition of colcemid for 30 min). (C) Mitotic indices 48 h after transfection with control or CENP-F duplexes, followed by incubation with or without 0.1 μg/ml colcemid for another 18 h. Mitotic indices of control RNAi and CENP-F RNAi are not statistically different in absence or presence of colcemid (done using Heteroscedastic T-test). In total, 20 independent fields of 100 cells were included for each siRNA. Immunoblotting confirmed the degree of CENP-F depletion.
Moreover, 50% of CENP-F depleted cells with aligned chromosomes had ⩾3 Mad2 positive kinetochores (Figures 5D and 6A). No such signal was detected on aligned chromosomes in 60% of control cells, as expected, with only one or two Mad2 positive kinetochores in the remaining control cells, presumably reflecting the last kinetochores to attach to spindle microtubules. Quantification of Mad2 at individual kinetochores showed that in addition to the increased number of Mad2 signaling kinetochores on CENP-F free aligned chromosomes, those kinetochores also recruited an increased amount of Mad2 (Figure 6B). The intensity of Mad2 at CENP-F depleted kinetochores likely reflects kinetochores that have achieved a partial or unstable attachment to spindle microtubules, since completely unattached kinetochores recruited more Mad2 on average (as assessed by brief incubation with colcemid; Figure 6B).
The fact that aligned chromosomes of CENP-F depleted cells continued to recruit Mad2 at kinetochores suggested that the pre-anaphase mitotic delay in the absence of CENP-F resulted from continued mitotic checkpoint signaling. Indeed, the maximum recruitment of Mad2 after a brief microtubule depolymerization was indistinguishable between control and CENP-F free kinetochores (Figure 6B). Mitotic checkpoint signaling was also directly assessed following an 18 h treatment with the microtubule depolymerizing drug colcemid. As in control cells, CENP-F depleted cells sustained a robust mitotic arrest. The mitotic index of CENP-F depleted cells increased (from 9.8±2.9 to 79.2±8.1%; Figure 6C). This was essentially the same as in control cells (for which the mitotic index after prolonged microtubule depolymerization increased from 6.2±2.3 to 85.5±6.3%).
CENP-F depleted kinetochores intermittently recruit Mad1, yielding sustained mitotic checkpoint activation
Activation of mitotic checkpoint signaling is initiated by recruitment of a stably bound Mad1/Mad2 complex to unattached kinetochores early in prometaphase (Shah et al, 2004). To follow checkpoint silencing and/or reactivation in real time at individual kinetochores in CENP-F depleted cells, HeLa cells stably expressing EYFP-Mad1 (Shah et al, 2004) were filmed 48 h after transfection with pH2B-mRFP and plasmids encoding CENP-F or control RNAi (Figure 7; Supplementary Movies 7 and 8). Mad1 redistributed from the nuclear envelope to kinetochores prior to nuclear envelope breakdown in both control and CENP-F RNAi cells (frames 0:00 and 0:10 for control RNAi and CENP-F RNAi, respectively). Concomitant with microtubule capture, Mad1 intensity decreased at kinetochores during progression through prometaphase (frames 0:06 and 0:14 for control RNAi and CENP-F RNAi, respectively). In control cells, it was absent after congression (frame 0:20). In most control cells, anaphase ensued within 10 min after the release of Mad1 from the last kinetochore, as expected (Rieder et al, 1994, 1995; Howell et al, 2000).
Figure 7.
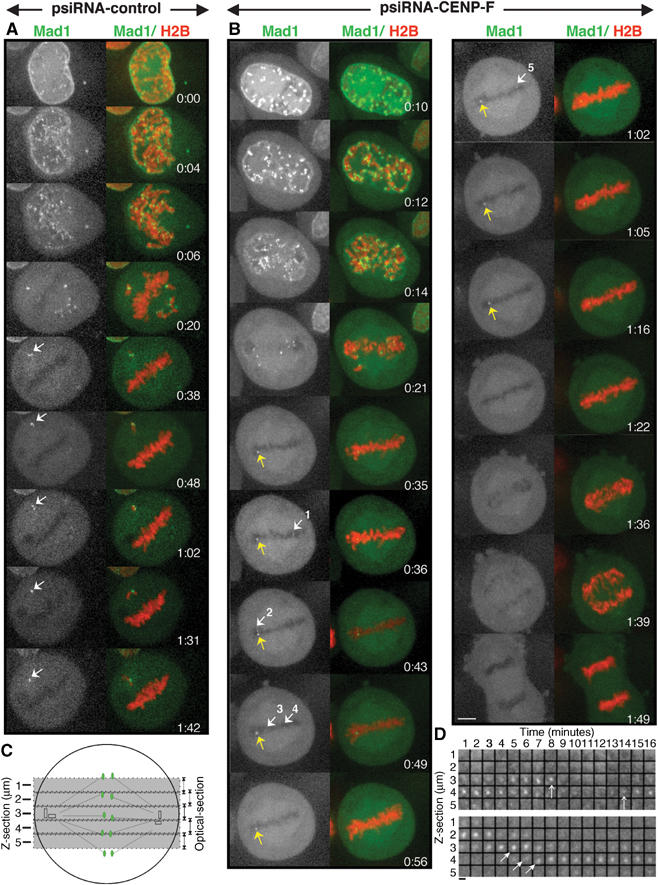
The mitotic delay of CENP-F RNAi cells is characterized by both the constitutive and transient recruitment of Mad1 at aligned chromosomes. Stably expressing EYFP-Mad1 HeLa cell lines were imaged 48 h after transfection with pH2B-mRFP along with psiRNA-control or psiRNA-CENP-F. (A) Aligned chromosomes in control cells rapidly and uniformly lose Mad1 binding coincident with alignment. In this example, one chromosome is trapped behind one spindle pole and one or both of the kinetochores of this chromatid pair chronically recruit Mad1. Selected frames of Supplementary Movie 7 of a psiRNA-control cell 48 h after transfection. (White or green) Mad1; (red) histone H2B. During the 64 min long pseudometaphase, no Mad1 is found at any aligned kinetochore. This shows that in these cells a single lagging chromosome is able to sustain mitotic checkpoint signaling to delay anaphase. (B) Images from a CENP-F depleted cell (Supplementary Movie 8). All kinetochores initially recruit Mad1 but after alignment Mad1 is released from most (see frame 0:35). However, one (yellow arrow) continued to bind Mad1 for 47 min (from frames 0:35 to 1:22). Other weaker Mad1 signals are intermittently detected (see white arrows 1–5). Finally, 14 min after Mad1 signals are lost from all kinetochores (from frames 1:22 to 1:36) anaphase initiates. (C, D). Mad1 twinkling. (C) Schematic representation of the Z-cross sections collected over time. A total of five Z-sections were collected, sampling ∼80% of the metaphase cell depth. (D) Distribution of Mad1 in every Z-section and during time in another CENP-F depleted cell. The constitutively bound Mad1 kinetochore in the lower panel moves slowly and progressively through the focal plane at 1 μm/3 min (see arrows). During the same time a kinetochore located at the center of the focal plane (first arrow in the top panel in Z-section 3) lost and regained Mad1 (second arrow in the top panel). Scale bar, 1 μm.
However, in one control cell, a chromatid pair was trapped behind one centrosome, leading to failure of one kinetochore to attach to spindle microtubules and the other to be unstably attached (the white arrow in Figure 7A; Supplementary Movie 7 frames 0:38). Mad1 remained at levels expected for an unattached kinetochore (frames 0:38 to 1:42) and anaphase was inhibited for as long as the cell was filmed (64 min). Importantly, during this metaphase delay all kinetochores of aligned chromosomes had lost Mad1 and none rebound it even transiently. While the ability of a single unattached kinetochore to generate a sustained block to anaphase had been previously demonstrated in Ptk1 cells (Rieder et al, 1994, 1995), this finding demonstrates that this is also true in this human cell line.
In CENP-F depleted cells, robust levels of EYFP-Mad1 were found not only on unaligned, unattached kinetochores (see example in frame 0:21 of Figure 7B and Supplementary Movie 8), but also on at least one kinetochore that had congressed (see the yellow arrow in the frames of Figure 7B). Other kinetochores initially aligned with no Mad1 signal, reinitiated Mad1 recruitment to produce ‘twinkling' of bound EYFP-Mad1 (see the five white arrows in frames 0:36 to 1:02 of Figure 7B). In this cell, weak Mad1 was detected for a short period of time at some kinetochores (eg, 6 min from frames 0:43 to 0:48 for the white arrow number 2) and at apparently distinct kinetochores throughout the 47 min metaphase delay (from frames 0:35 to 1:22). Both sustained and intermittent recruitment of Mad1 at kinetochores of aligned chromosomes have been observed in CENP-F depleted cells. Another example (presented in Figure 7D) shows that during the same period of time, while one aligned kinetochore constitutively recruited Mad1 (see lower panel in Figure 7D) and moved regularly and slowly over time in the z-axis at an approximate rate of 1 μm/3 min, a different kinetochore in the same spindle (represented in the top panel) clearly recruited, then lost and recruited again Mad1 within 5 min. Since all kinetochore movements tracked revealed slow transit in the z dimension, it is unlikely that this kinetochore shifts out of the focus plane since it would imply that it would oscillate orthogonally to the spindle ∼5–7 μm within 5 min. Further, our imaging included ∼80% of the complete metaphase cell depth (corresponding to five sections of 1 μm, to minimize damage to the cells; Figure 7C). While we cannot completely exclude the possibility that some kinetochores of aligned chromosomes may shift out of focus during observation, most of them (such as those tracked in Figure 7D) certainly do not. This provides evidence that in the absence of CENP-F Mad1 is recruited, then silenced, then recruited again to produce ‘twinkling' at few kinetochores of aligned chromosomes (white arrows in Figure 7B), but also repetitively at the same kinetochore (Figure 7D).
Finally, anaphase initiated 14 min after the Mad1 signal at the last kinetochore was eliminated (frames 1:22 to 1:36). Thus, mitotic delay at metaphase of CENP-F depleted cells reflects sustained mitotic checkpoint signaling in which individual few kinetochores fail to efficiently maintain capture of spindle microtubules and/or in which signaling is re-activated after initial capture and alignment.
Discussion
Depletion of nuclear CENP-F with transcription-mediated siRNA or double-stranded RNAs targeted against two different regions of the corresponding mRNA did not affect progression through S or G2 in interphase or entry into mitosis, but it did produce unstable kinetochore microtubule attachment. In a minority of cells, kinetochore assembly failed, yielding inability of any centromere to initiate or sustain mitotic checkpoint signaling. This resulted in premature anaphase with gross missegregation of chromosomes that almost certainly produces cell autonomous lethality, similar to that proven to occur after elimination of essential mitotic checkpoint components (Kops et al, 2004). This finding reveals a role for CENP-F, the earliest of the transient components to associate with kinetochores, in facilitating one or more early steps in kinetochore assembly. This aspect of function for CENP-F is most readily consistent with a scaffolding role in assembly and recruitment of other components to the outer kinetochore. Without it, a subset of kinetochores misassemble (or assemble slowly) and in the absence of an activated mitotic checkpoint to slow advance to anaphase there is insufficient time for correction of such assembly errors before a premature anaphase produces rampant chromosome missegregation.
In the majority of cells entering mitosis in the absence of CENP-F, kinetochore assembly was more successful, with at least some kinetochores assembled sufficiently well to generate a level of the mitotic checkpoint inhibitor sufficient to delay mitotic progression. This was a consequence of continued and intermittent mitotic checkpoint activation as reflected in (1) continued and transient recruitment of YFP-Mad1 to produce ‘twinkling' at kinetochores of aligned chromosomes; (2) the recruitment to unattached, CENP-F-depleted kinetochores of normal levels of several mitotic checkpoint components, including Mad2, and (3) sustained mitotic delay of CENP-F depleted cells in the presence of microtubule depolymerizing agents. Despite the two divergent outcomes for CENP-F depleted cells, both phenotypes probably reflect a common role for CENP-F in kinetochore assembly. A likely possibility is that the more severe phenotype arises from cells completely deficient in CENP-F, while the milder phenotype represents cells hypomorphic for CENP-F and in which sharply reduced levels of CENP-F uncover a CENP-F-dependency in the kinetics of kinetochore assembly.
An earlier effort using transcription based siRNA to an unspecified domain of CENP-F had claimed anaphase entry in the presence of widespread chromosome misalignment and subsequent missegregation (Laoukili et al, 2005). Our results extend this to show that continuing mitotic checkpoint signaling from unstable microtubule capture underlies a mitotic delay, but with less tightly aligned chromosomes. Anaphase initiates after capture-mediated silencing of checkpoint signaling. An additional transcription-mediated siRNA effort proposed that the absence of CENP-F produces not only mis-misaligned chromosomes, but that this is accompanied by premature chromosome decondensation before anaphase onset and subsequent death directly in mitosis without mitotic exit (Yang et al, 2005). Our evidence offers no support for either of these latter phenotypes as a direct consequent of the absence of CENP-F during mitosis. Rather, it seems likely that these severe phenotypes reflect not just the absence of CENP-F but also damage induced by repetitive, high-intensity illumination producing DNA or other damage that precludes further cycling.
The Caenorhabditis elegans proteins HCP-1 and HCP-2 have been proposed to be CENP-F relatives based upon 54% similarity to each other and a shared 20.8% identity with the carboxy terminal portion of the human CENP-F (Moore et al, 1999). Two groups have recently assessed the function of HCP-1 and HCP-2 in mitosis, by depleting simultaneously both proteins in worm embryo (Cheeseman et al, 2005; Encalada et al, 2005). Codepletion of HCP-1 and HCP-2 induces chromosome segregation defects characterized by a failure of sister chromosome bi-orientation that is dependent on Clasp2, a regulator of microtubule dynamics both in Drosophila and human cells (Maiato et al, 2002, 2003). Indeed, a direct interaction between HCP-1/2 and Clasp2 has been described including mislocalization of Clasp2 in the absence of HCP-1/2 and a similar segregation defect in embryos depleted only in Clasp2 (Cheeseman et al, 2005). We have shown that human Clasp1 is not dependent on CENP-F. The low homology, divergent consequences of the absence of CENP-F versus HCP1/2 and divergent requirement for Clasp1-2 associated with kinetochores combine to suggest that HCP1/2 and CENP-F are unlikely to be orthologues.
The delay in mitosis progression as well as the microtubule instability we observed after depletion of CENP-F is reminiscent of a similar outcome from disruption of the Ndc80 complex, comprised of the Hec1, Nuf2, Spc24 and Spc25, first identified in yeast (He et al, 2001; Wigge and Kilmartin, 2001). The crucial role of the Ndc80 complex in maintaining a stable attachment to spindle microtubules has been demonstrated in yeast and in mammals (Howe et al, 2001; DeLuca et al, 2002; Martin-Lluesma et al, 2002; Hori et al, 2003; McCleland et al, 2003, 2004). In human cells, depletion of any member of the Ndc80 complex has been shown to induce a prometaphase block of several hours (DeLuca et al, 2002; Martin-Lluesma et al, 2002; McCleland et al, 2004). Like CENP-F, Nuf2 inhibition has been shown to weaken stable microtubule capture at kinetochores, reflected in a complete absence of tension between sister kinetochores and by the complete depolymerization of kinetochore fibers after cold treatment (DeLuca et al, 2002). Similar to what happens after CENP-F and Ndc80 complex inhibition, a prometaphase delay/block has been reported when kinetochore microtubule attachment is impaired, as it is for RanBP2 and CENP-E depleted cells (Yao et al, 2000; Salina et al, 2003; Joseph et al, 2004).
While we are not aware of evidence supporting a direct role for the Ndc80 complex in microtubule capture, the evidence supports both direct and indirect roles for CENP-F. A direct contribution would be mediated by its partner CENP-E (Chan et al, 1998), whose interaction was initially proposed through a yeast two hybrid interaction (Chan et al, 1998) and was confirmed by co-immunoprecipitation (Yao et al, 2000). Indeed, we and others (Johnson et al, 2004; Yang et al, 2005) have shown CENP-E to be diminished at kinetochores depleted of CENP-F. Moreover, gene disruption (Putkey et al, 2002), antisense oligonucleotide-mediated RNA silencing (Yao et al, 2000) or antibody inhibition (McEwen et al, 2001) have all previously been used to demonstrate that the absence of kinetochore associated CENP-E reduces stable microtubule capture. An indirect role for CENP-F in microtubule capture emerges from its requirement for efficient assembly of kinetochores. Evidence for this includes cells in which kinetochore-dependent mitotic checkpoint signaling is never activated, as well as the very slow attachment and intermittent checkpoint silencing at a subset of kinetochores.
Although it is clear from our evidence that CENP-F-depleted cells can activate and maintain mitotic checkpoint signaling at unattached kinetochores, whether signaling from these kinetochores can be sustained for an extended period equal to that for CENP-F-containing kinetochores is not settled. In the HeLa cells used in our analysis, treatment for 18 h produced comparable mitotic indices from CENP-F-containing and depleted cells (Figure 6C). A similar effort with another immortalized human cell (U2OS) had reported a diminished ability of CENP-F-depleted cells to maintain microtubule drug-mediated mitotic arrest (Laoukili et al, 2005). HeLa cells robustly sustain checkpoint signaling (>80% mitotic index after 18 h). U2OS cells, on the other hand, escape that arrest much more readily producing only a 25% mitotic index after 16 h of drug induced microtubule disassembly. The divergent outcomes highlight an unresolved issue in how cells escape from chronic mitotic checkpoint activation (termed adaptation) and how that is linked to cell death either directly from mitosis or in a subsequent interphase after an abortive cytokinesis (reviewed in Weaver and Cleveland, 2005). Additional efforts are now required to identify how CENP-F, at least in some contexts, influences adaptation from chronic mitotic checkpoint signaling.
Materials and methods
RNAi
CENP-F expression was silenced in HeLa cells using antisense oligonucleotides siRNA and transcription-mediated siRNAi. A CENP-F specific RNA duplex 5′-CAAAGACCGGUGUUACCAAG-3′ (Dharmacon) and a Cy3-luciferase GL2 duplex were transfected with oligofectamine (Invitrogen) in a ratio of 10:1. The nonspecific control duplex VIII (Dharmacon), representing a RNA duplex with the same GC content as the CENP-F duplex, was used as a control. siRNA was also introduced by transfection of a polymerase-III H1-RNA promoted gene that directs expression of short hairpin RNAs containing a 19 base segment of CENP-F mRNA (5′-GAGAAGACCCCAAGTCATC-3′) (Brummelkamp et al, 2002). HeLa cells were transfected with psiRNA-CENP-F and pH2B-EYFP/-mRFP or pCMV-CD20 in a ratio 10:1, using Effectene (Qiagen).
Magnetic activated cell sorting (MACS)
HeLa cells were transfected with the psiRNA-control/-CENP-F along with a plasmid carrying a CMV promoted gene encoding the surface marker CD20 (pCMV-CD20). After transfection, cells were collected using 3 mM EDTA for 3 min. Transfected cells were selectively recovered by affinity chromoatography with immobilized antibodies to CD20. Cells were incubated with a mouse anti-CD20 antibody (DakoCytomation) for 30 min at 4°C, washed with PBS and incubated with goat anti-mouse magnetic microbeads (Miltenyl Biotech) for 30 min at 4°C. Transfected cells were loaded on a PBS-washed magnetic column (MS column, Miltenyl Biotech) in a magnetic stand, washed in PBS and eluted in PBS after the removal of the column from the magnet.
Live cell microscopy
HeLa cells were seeded onto 35-mm glass-bottom dishes (MatTek) and incubated with a CO2-independent medium (GIBCO) supplemented with 10% FBS, 0.5 μg/ml penicillin/streptomycin and 2 mM glutamine prior to recording. Dishes were placed in a heat-controlled stage set at 37°C. Time-lapse sequences were taken every 2 or 4 min. Pictures were taken on a Nikon Eclipse 300 inverted microscope with either 60 × A/1.4 or 20 × objectives. Z-stack images were collected with a Photometrics COOLSNAP HQ camera (Roper Scientific) and analyzed with Metamorph software (Universal Imaging).
EYFP-Mad1 imaging was conducted using a spinning disk confocal (McBain Instruments) attached to a Nikon TE2000e inverted microscope equipped with a 60 × /1.4NA objective lens. Fluorescence excitation as well as DIC illumination were controlled by Metamorph. Z-series images (five images at 1 μm intervals) were acquired using a Hamamatsu Orca ER camera (Bridgewater) at 1 min intervals. Z-stacks were compiled by maximum intensity projection for presentation.
Cell culture, cell lysates and immunoblots
HeLa cells were maintained in DMEM (GIBCO BRL) with 10% FCS, 0.5 μg/ml penicillin/streptomycin in a 37°C, 5% CO2 incubator. When microtubule depolymerizing agents were used, nocodazole was added to the medium at a concentration of 20 μM during 3 h, and cells were incubated during 30 min in the presence of 0.1 μg/ml colcemid. Synchronization in S and M phases were obtained after 18 h incubation in 2 mM thymidine and 0.1 μg/ml colcemid, respectively.
Cells were lysed in 50 mM Tris, 500 mM NaCl, 1% NP40 and proteinase inhibitors on ice for 20 min. Total proteins (20 μg) were run on a 6% SDS-polyacrylamide gels. Membranes were blocked in PBS containing 0.05% Tween-20 and 5% milk and blotted with antibodies diluted in the same solution: rabbit anti CENP-F antibody 1:1000 and mouse anti tubulin (DM1α) antibody 1:10 000.
Immunofluorescence, deconvolution microscopy and quantification
Immunofluorescence was performed as described (Weaver et al, 2003). Except for Hec1 and Clasp1 staining for which extraction/fixation was done in −20°C methanol during 10 min, all other stainings were performed after a 4% formaldehyde fixation for 10 min at room temperature. The dilutions of antibodies were the following: CENP-F rabbit antibody 1:1000, CENP-F sheep antibody (provided by S Taylor) 1:500, anti-human centromere proteins (Antibodies Incorporated) 1:500, phospho-histone H3 (Cell Signaling) mouse antibody 1:1000, mouse DM1α tubulin antibody 1:10 000 (Chen et al, 1996), rabbit Hec1 antibody (provided by E Nigg) 1:200, rabbit antibody to Xenopus Clasp (provided by R Heald) 1:500, rabbit Hpx antibody to CENP-E 1:200, sheep SBR1 antibody to BubR1 (provided by S Taylor) 1:1000, sheep Bub1 1:500 (S Taylor), rabbit Mad2 antibody 1:2000. Fluorophore-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) were diluted 1:200. Deconvolution images were collected and analyzed as described (Weaver et al, 2003). Kinetochore fluorescence was quantificated using the integrated intensity of three-dimentional polygons surrounding each kinetochore.
Supplementary Material
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Movie 5
Supplementary Figure 6
Supplementary Movie 7
Supplementary Movie 8
Acknowledgments
We are grateful to the members of the Cleveland group, as well as I Cheeseman and S Kline-Smith for continuous support and very enthusiastic discussions around this project. We would like to thank S Taylor, R Heald and E Nigg for their generous gift of antibodies and RY Tsien for providing the mRFP construct. PB was supported in part by a fellowship from the Fondation pour la Recherche Medicale. PSM is the Fayez Sarofim Fellow of the Damon Runyon Cancer Research Foundation. JVS was supported in part by a Postdoctoral Fellowship from the NIH. This work was supported by a grant from the NIH to DWC (GM 29513). DWC and ABD receive salary support from the Ludwig Institute for Cancer Research.
References
- Ashar HR, James L, Gray K, Carr D, Black S, Armstrong L, Bishop WR, Kirschmeier P (2000) Farnesyl transferase inhibitors block the farnesylation of CENP-E and CENP-F and alter the association of CENP-E with the microtubules. J Biol Chem 275: 30451–30457 [DOI] [PubMed] [Google Scholar]
- Brenner S, Pepper D, Berns MW, Tan E, Brinkley BR (1981) Kinetochore structure, duplication, and distribution in mammalian cells: analysis by human autoantibodies from scleroderma patients. J Cell Biol 91: 95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553 [DOI] [PubMed] [Google Scholar]
- Casiano CA, Humbel RL, Peebles C, Covini G, Tan EM (1995) Autoimmunity to the cell cycle-dependent centromere protein p330d/CENP-F in disorders associated with cell proliferation. J Autoimmun 8: 575–586 [DOI] [PubMed] [Google Scholar]
- Chan GK, Schaar BT, Yen TJ (1998) Characterization of the kinetochore binding domain of CENP-E reveals interactions with the kinetochore proteins CENP-F and hBUBR1. J Cell Biol 143: 49–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Macleod I, Yates JR III, Oegema K, Desai A (2005) The CENP-F-like proteins HCP-1 and HCP-2 target CLASP to kinetochores to mediate chromosome segregation. Curr Biol 15: 771–777 [DOI] [PubMed] [Google Scholar]
- Chen RH, Waters JC, Salmon ED, Murray AW (1996) Association of spindle assembly checkpoint component XMAD2 with unattached kinetochores. Science 274: 242–246 [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Mao Y, Sullivan KF (2003) Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 112: 407–421 [DOI] [PubMed] [Google Scholar]
- Crespo NC, Ohkanda J, Yen TJ, Hamilton AD, Sebti SM (2001) The farnesyltransferase inhibitor, FTI-2153, blocks bipolar spindle formation and chromosome alignment and causes prometaphase accumulation during mitosis of human lung cancer cells. J Biol Chem 276: 16161–16167 [DOI] [PubMed] [Google Scholar]
- DeLuca JG, Moree B, Hickey JM, Kilmartin JV, Salmon ED (2002) hNuf2 inhibition blocks stable kinetochore-microtubule attachment and induces mitotic cell death in HeLa cells. J Cell Biol 159: 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encalada SE, Willis J, Lyczak R, Bowerman B (2005) A spindle checkpoint functions during mitosis in the early Caenorhabditis elegans embryo. Mol Biol Cell 16: 1056–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Rines DR, Espelin CW, Sorger PK (2001) Molecular analysis of kinetochore–microtubule attachment in budding yeast. Cell 106: 195–206 [DOI] [PubMed] [Google Scholar]
- Hori T, Haraguchi T, Hiraoka Y, Kimura H, Fukagawa T (2003) Dynamic behavior of Nuf2-Hec1 complex that localizes to the centrosome and centromere and is essential for mitotic progression in vertebrate cells. J Cell Sci 116: 3347–3362 [DOI] [PubMed] [Google Scholar]
- Howe M, McDonald KL, Albertson DG, Meyer BJ (2001) HIM-10 is required for kinetochore structure and function on Caenorhabditis elegans holocentric chromosomes. J Cell Biol 153: 1227–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BJ, Hoffman DB, Fang G, Murray AW, Salmon ED (2000) Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J Cell Bio 150: 1233–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein D, Taylor SS (2002) Farnesylation of Cenp-F is required for G2/M progression and degradation after mitosis. J Cell Sci 115: 3403–3414 [DOI] [PubMed] [Google Scholar]
- Johnson VL, Scott MI, Holt SV, Hussein D, Taylor SS (2004) Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chromosome congression. J Cell Sci 117: 1577–1589 [DOI] [PubMed] [Google Scholar]
- Joseph J, Liu ST, Jablonski SA, Yen TJ, Dasso M (2004) The RanGAP1-RanBP2 complex is essential for microtubule-kinetochore interactions in vivo. Curr Biol 14: 611–617 [DOI] [PubMed] [Google Scholar]
- Kline-Smith SL, Sandall S, Desai A (2005) Kinetochore–spindle microtubule interactions during mitosis. Curr Opin Cell Biol 17: 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, Foltz DR, Cleveland DW (2004) Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci USA 101: 8699–8704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH (2005) FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 7: 126–136 [DOI] [PubMed] [Google Scholar]
- Liao H, Winkfein RJ, Mack G, Rattner JB, Yen TJ (1995) CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J Cell Biol 130: 507–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiato H, DeLuca J, Salmon ED, Earnshaw WC (2004) The dynamic kinetochore-microtubule interface. J Cell Sci 117: 5461–5477 [DOI] [PubMed] [Google Scholar]
- Maiato H, Fairley EA, Rieder CL, Swedlow JR, Sunkel CE, Earnshaw WC (2003) Human CLASP1 is an outer kinetochore component that regulates spindle microtubule dynamics. Cell 113: 891–904 [DOI] [PubMed] [Google Scholar]
- Maiato H, Sampaio P, Lemos CL, Findlay J, Carmena M, Earnshaw WC, Sunkel CE (2002) MAST/Orbit has a role in microtubule-kinetochore attachment and is essential for chromosome alignment and maintenance of spindle bipolarity. J Cell Biol 157: 749–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Lluesma S, Stucke VM, Nigg EA (2002) Role of Hec1 in spindle checkpoint signaling and kinetochore recruitment of Mad1/Mad2. Science 297: 2267–2270 [DOI] [PubMed] [Google Scholar]
- McCleland ML, Gardner RD, Kallio MJ, Daum JR, Gorbsky GJ, Burke DJ, Stukenberg PT (2003) The highly conserved Ndc80 complex is required for kinetochore assembly, chromosome congression, and spindle checkpoint activity. Genes Dev 17: 101–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCleland ML, Kallio MJ, Barrett-Wilt GA, Kestner CA, Shabanowitz J, Hunt DF, Gorbsky GJ, Stukenberg PT (2004) The vertebrate Ndc80 complex contains Spc24 and Spc25 homologs, which are required to establish and maintain kinetochore-microtubule attachment. Curr Biol 14: 131–137 [DOI] [PubMed] [Google Scholar]
- McEwen BF, Arena JT, Frank J, Rieder CL (1993) Structure of the colcemid-treated PtK1 kinetochore outer plate as determined by high voltage electron microscopic tomography. J Cell Biol 120: 301–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BF, Chan GK, Zubrowski B, Savoian MS, Sauer MT, Yen TJ (2001) CENP-E is essential for reliable bioriented spindle attachment, but chromosome alignment can be achieved via redundant mechanisms in mammalian cells. Mol Biol Cell 12: 2776–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore LL, Morrison M, Roth MB (1999) HCP-1, a protein involved in chromosome segregation, is localized to the centromere of mitotic chromosomes in Caenorhabditis elegans. J Cell Biol 147: 471–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putkey FR, Cramer T, Morphew MK, Silk AD, Johnson RS, McIntosh JR, Cleveland DW (2002) Unstable kinetochore–microtubule capture and chromosomal instability following deletion of CENP-E. Dev Cell 3: 351–365 [DOI] [PubMed] [Google Scholar]
- Rattner JB, Rao A, Fritzler MJ, Valencia DW, Yen TJ (1993) CENP-F is a.ca 400 kDa kinetochore protein that exhibits a cell-cycle dependent localization. Cell Motil Cytoskeleton 26: 214–226 [DOI] [PubMed] [Google Scholar]
- Rieder CL (1981) The structure of the cold-stable kinetochore fiber in metaphase PtK1 cells. Chromosoma 84: 145–158 [DOI] [PubMed] [Google Scholar]
- Rieder CL (1982) The formation, structure, and composition of the mammalian kinetochore and kinetochore fiber. Int Rev Cytol 79: 1–58 [DOI] [PubMed] [Google Scholar]
- Rieder CL, Cole RW, Khodjakov A, Sluder G (1995) The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol 130: 941–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Schultz A, Cole R, Sluder G (1994) Anaphase onset in vertebrate somatic cells is controlled by a checkpoint that monitors sister kinetochore attachment to the spindle. J Cell Biol 127: 1301–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salina D, Enarson P, Rattner JB, Burke B (2003) Nup358 integrates nuclear envelope breakdown with kinetochore assembly. J Cell Biol 162: 991–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah JV, Botvinick E, Bonday Z, Furnari F, Berns M, Cleveland DW (2004) Dynamics of centromere and kinetochore proteins; implications for checkpoint signaling and silencing. Curr Biol 14: 942–952 [DOI] [PubMed] [Google Scholar]
- Weaver BA, Bonday ZQ, Putkey FR, Kops GJ, Silk AD, Cleveland DW (2003) Centromere-associated protein-E is essential for the mammalian mitotic checkpoint to prevent aneuploidy due to single chromosome loss. J Cell Biol 162: 551–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW (2005) Decoding the links between mitosis, cancer and chemotherapy: the mitotic checkpoint, adaptation and cell death. Cancer Cell 8: 7–12 [DOI] [PubMed] [Google Scholar]
- Wigge PA, Kilmartin JV (2001) The Ndc80p complex from Saccharomyces cerevisiae contains conserved centromere components and has a function in chromosome segregation. J Cell Biol 152: 349–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Guo J, Chen Q, Ding C, Du J, Zhu X (2005) Silencing mitosin induces misaligned chromosomes, premature chromosome decondensation before anaphase onset, and mitotic cell death. Mol Cell Biol 25: 4062–4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Abrieu A, Zheng Y, Sullivan KF, Cleveland DW (2000) CENP-E forms a link between attachment of spindle microtubules to kinetochores and the mitotic checkpoint. Nat Cell Biol 2: 484–491 [DOI] [PubMed] [Google Scholar]
- Zhu X, Chang KH, He D, Mancini MA, Brinkley WR, Lee WH (1995a) The C terminus of mitosin is essential for its nuclear localization, centromere/kinetochore targeting, and dimerization. J Biol Chem 270: 19545–19550 [DOI] [PubMed] [Google Scholar]
- Zhu X, Mancini MA, Chang KH, Liu CY, Chen CF, Shan B, Jones D, Yang-Feng TL, Lee WH (1995b) Characterization of a novel 350-kilodalton nuclear phosphoprotein that is specifically involved in mitotic-phase progression. Mol Cell Biol 15: 5017–5029 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie 1
Supplementary Movie 2
Supplementary Movie 3
Supplementary Movie 4
Supplementary Movie 5
Supplementary Figure 6
Supplementary Movie 7
Supplementary Movie 8