

Abstract
Cullin-4 forms a scaffold for multiple ubiquitin ligases. In Schizosaccharomyces pombe, the Cullin-4 homologue (Pcu4) physically associates with Ddb1 and the COP9 signalosome (CSN). One target of this complex is Spd1. Spd1 regulates ribonucleotide reductase (RNR) activity. Spd1 degradation during S phase, or following DNA damage of G2 cells, results in the nuclear export of the small RNR subunit. We demonstrate that Cdt2, an unstable WD40 protein, is a regulatory subunit of Pcu4–Ddb1–CSN ubiquitin ligase. cdt2 deletion stabilises Spd1 and prevents relocalisation of the small RNR subunit from the nucleus to the cytoplasm. cdt2+ is periodically transcribed by the Cdc10/DSC1 transcription factor during S phase and transiently transcribed following DNA damage of G2 cells, corresponding to Spd1 degradation profiles. Cdt2 co-precipitates with Spd1, and Cdt2 overexpression results in constitutive Spd1 degradation. We propose that Cdt2 incorporation into the Pcu4–Ddb1–CSN complex prompts Spd1 targeting and subsequent degradation and that Cdt2 is a WD40 repeat adaptor protein for Cullin-4-based ubiquitin ligase.
Keywords: Cct complex, Cullin, damage checkpoint, ribonucleotide reductase, signalosome
Introduction
Genes required for DNA synthesis are periodically transcribed during the mitotic cell cycle, peaking at G1–S. The Schizosaccharomyces pombe G1/S transition is coordinated by the DSC1 (DNA Synthesis Control or MBF factor) transcription complex. DSC1 contains the cdc10, rep1, rep2, res1 and res2 gene products and recognises the MCB (MluI cell cycle box) DNA sequence (White et al, 2001). cdc10 loss-of-function mutants have profound effects on MCB-dependent transcription (Hofmann and Beach, 1994). Most known Cdc10-dependent target genes are directly involved in DNA replication. These include genes encoding the replication initiation factors Cdc18 and Cdt1 and enzymatic functions required for DNA synthesis such as Cdc22, the large subunit of ribonucleotide reductase (RNR).
RNR catalyses the rate-limiting step for dNTP production and is required for replicative DNA synthesis and DNA repair (Elledge et al, 1992). RNR is a heterotetramer formed by two large subunits (R1 subunit; encoded by cdc22R1 in S. pombe) and two small subunits (R2 subunit; encoded by suc22R2). RNR activity is tightly regulated to maintain the balanced dNTP pools necessary for high-fidelity DNA synthesis. In Saccharomyces cerevisiae, RNR gene expression is under tight cell cycle control and separate genes encoding stress-specific subunits are induced after DNA damage via the MEC1/RAD53 pathway (Elledge et al, 1993). An additional level of control over RNR activity in S. cerevisiae is exerted by Sml1, which binds to and inhibits the function of the R1 subunit (Chabes et al, 1999). In order to allow replication, Sml1 is degraded in S phase in a MEC1/RAD53-dependent manner (Zhao et al, 1998). MEC1/RAD53-dependent Sml1 degradation accounts for the essential function of the S. cerevisiae checkpoint pathway. In fission yeast, the MEC1 homologue rad3+ is not essential and there is currently no evidence for an R1 subunit inhibitor.
A novel mode of RNR regulation by subcellular compartmentalisation was recently uncovered and is conserved between S. pombe (Liu et al, 2003) and S. cerevisiae (Yao et al, 2003). In S. pombe, when RNR activity is not required (e.g. G2 cells), the small subunit (Suc22R2) is found in the nucleus whereas the large subunit (Cdc22R1) is largely cytoplasmic. When RNR activity is required (e.g. DNA replication or DNA repair synthesis), Suc22R2 translocates to the cytoplasm where it is presumed to engage with Cdc22R1 to form active RNR (Liu et al, 2003). Suc22R2 localisation to the nucleus is dependent on the presence of the 124 aa protein Spd1, a potential Suc22R2 nuclear anchor. At the start of each S phase, Spd1 is degraded by the ubiquitin–proteasome system (UPS). Periodic degradation of Spd1 in S phase does not require rad3+ (MEC1 homologue) activity and is checkpoint pathway independent. In contrast, Spd1 degradation in G2 after DNA damage requires the rad3/chk1 DNA damage checkpoint (Liu et al, 2003). Both cell cycle and damage-induced Spd1 degradation are mediated by Spd1 ubiquitylation, which requires components of the COP9 signalosome complex (CSN), the S. pombe Cullin-4 homologue (Pcu4) and the Ddb1 protein (Liu et al, 2003; Holmberg et al, 2005).
The UPS is the primary mechanism for regulated degradation of cellular proteins (Hershko and Ciechanover, 1998). Targeting of specific substrates to UPS is mediated by E3 ubiquitin ligases. Multiple ubiquitin ligase families exist that can be broadly categorised into three classes: HECT domain E3's, single subunit RING E3's and multi-subunit RING E3's (Pickart, 2001). Multi-subunit RING E3's are often exemplified by SCF (Skp1–Cullin–F box) complexes, a collection of RING ubiquitin ligases assembled on a common cullin-1 scaffold. CUL1-based SCF complexes have a modular design; one end of cullin binds Skp1, which in turn recruits one of many different F-box proteins to act as a substrate adaptor. The other end of cullin binds Rbx1, a RING finger protein that recruits an E2 enzyme (Deshaies, 1999). Unlike CUL1, the CUL4A and B proteins do not have Skp1-binding motifs. However, CUL4A associates with DDB1 (Damaged DNA-binding protein) as well as WD40 repeat proteins and has been proposed to scaffold a subfamily of SCF-like E3 complexes (Groisman et al, 2003; Wertz et al, 2004).
CUL4A–DDB1 ubiquitin ligases play roles in gene expression and DNA replication by selectively degrading target proteins (including c-JUN and CDT1; Higa et al, 2003; Wertz et al, 2004) and were recently found tightly associated with CSN (Groisman et al, 2003; Liu et al, 2003). CSN consists of eight subunits (Csn1–8) that share one-to-one similarity to the 19S regulatory lid complex of the 26S proteasome (Wei and Deng, 2003). CSN functions are pleiotropic, which can be attributed to interactions with multiple cullins and thus multiple cullin-based ubiquitin ligases. CSN recruits several enzymatic activities to cullin-based E3's including Csn5/Jab1-dependent deneddylase and the Ubp12 deubiquitinase (Cope et al, 2002; Zhou et al, 2003). The phenotypic consequences of CSN loss reflect this pleiotropism and include cellular and developmental defects (Chamovitz et al, 1996; Schwechheimer et al, 2001) as well as DNA damage response defects (Mundt et al, 1999; Groisman et al, 2003).
We previously identified S. pombe csn1 and csn2 null mutants as DNA damage sensitive and delayed in DNA replication (Mundt et al, 1999). Null mutants corresponding to other CSN subunits do not share these phenotypes, indicating a novel function for these two components (Mundt et al, 2002). Purification of fission yeast CSN identified Ddb1 and Pcu4, but not other cullins, as strongly associated proteins (Liu et al, 2003). pcu4 and ddb1 null mutants share the DNA damage sensitivity and S-phase defects of csn1 and csn2 mutants. These phenotypes correlate with defective Spd1 degradation (Liu et al, 2003; Holmberg et al, 2005). A Pcu4–Ddb1–CSN ubiquitin ligase thus catalyses Spd1 polyubiquitylation in S phase and in response to DNA damage. Here we report that the WD40 repeat protein Cdt2 (Cdc10-dependent transcript 2) functions as the adaptor protein and key activator to target Spd1 to the Pcu4–Ddb1–CSN ubiquitin ligase. We show that Pcu4–Ddb1–CSN activity against Spd1 is largely dependent on the regulation of cdt2+ gene expression.
Results
Cdt2 copurified with Pcu4–CSN protein complex
Csn2-TAP affinity purification identified six CSN subunits and Pcu4–Ddb1 at ∼stoichiometric concentrations (Liu et al, 2003). Analysis of several repeat mass spectrometry data sets revealed one minor band generated ⩽12 peptides with masses matching the predicted fragmentation of Cdt2 (a WD repeat protein of 55.058 kDa) (32.4% coverage). These data suggested Cdt2 as a potential Csn2-TAP interactor. Concomitantly, a cdt2 mutant was found to display DNA replication and repair defects reminiscent of phenotypes associated with csn1, csn2, ddb1 and pcu4 deletion (Yoshida et al, 2003).
As judged from Spd1 degradation profiles, the Pcu4–Ddb1–CSN ubiquitin ligase is activated during unperturbed S phase in a checkpoint-independent manner and can be activated by the DNA damage checkpoint when G2 cells suffer DNA damage. No link has been established between the Pcu4–Ddb1–CSN complex and potential activating signals. Because the cdt2 transcript and gene product accumulate specifically in S phase (Yoshida et al, 2003), we investigated if Cdt2 regulates Spd1 degradation by activating Pcu4–Ddb1–CSN.
First, we confirmed the Cdt2 interaction with Pcu4–Ddb1–CSN using immunoprecipitation (IP). cdt2-HA csn1-Myc cells were arrested in S phase (4 h, 20 mM hydroxyurea (HU)) to induce Cdt2 and extracts were prepared. Cdt2-HA co-immunoprecipitated (co-IPed) specifically with Csn1-Myc (Figure 1A, lane 3). This interaction was csn2 and ddb1 dependent (lanes 4 and 6, respectively). The csn5 null mutant does not share the replication and repair phenotypes associated with loss of csn1 and csn2 (Mundt et al, 2002) and, consistent with this, the interaction was not csn5 dependent (lane 5). We next examined the interaction between Cdt2-HA and Ddb1-Myc (Figure 1B). Cdt2-HA co-IPed specifically with Ddb1-Myc. This was not dependent on any CSN subunits or pcu4. Thus, Pcu4 does not bridge the Cdt2-HA–Ddb1-Myc interaction. The data suggest either that Ddb1 and Cdt2 bind directly to each other or that their interaction is mediated by unidentified bridging factor(s). We also find that the Ddb1-Myc Csn1-TAP interaction is independent of Cdt2 (Figure 1C). Together, these results are compatible with Cdt2 interacting with Ddb1 and assembling with Pcu4 and CSN to form a multi-subunit ubiquitin ligase.
Figure 1.
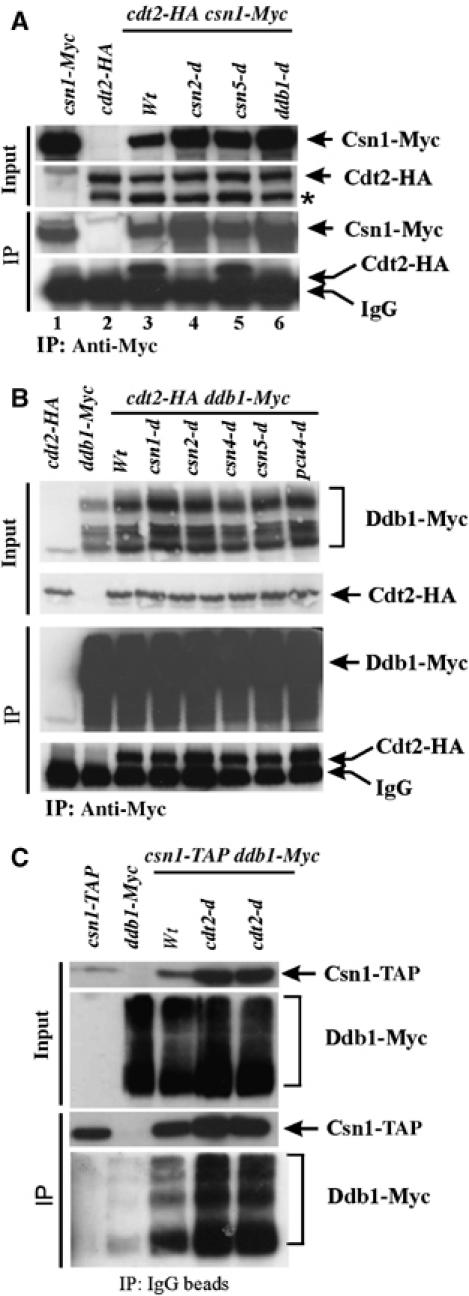
Identification of Cdt2 as an interacting protein with the Pcu4–CSN complex. (A) Co-IP of Cdt2-HA and Csn1-Myc. Csn1-Myc IP (α-Myc): In a wild-type background (Wt, lane 3), co-precipitated Cdt2-HA was detected with α-HA. The interaction is lost in csn2-d and ddb1-d (lanes 4 and 6) but not csn5-d (lane 5) extracts. *Indicates a second Cdt2-HA-dependent band we assume to be a degradation product. The intensity of this band varies between experiments. (B) Co-IP of Cdt2-HA and Ddb1-Myc. Ddb1-Myc was IPed with α-Myc. Cdt2-HA was detected with α-HA. The interaction was not affected by loss of other Pcu4–CSN subunits. (C) Co-precipitation of Ddb1-Myc and Csn1-TAP. Csn1-TAP was precipitated with IgG-coated Dynabeads. Ddb1-Myc was detected with α-Myc. Loss of cdt2 did not prevent the interaction between Csn1-TAP and Ddb1-Myc.
Cdt2 co-precipitates with the T complex
Size-exclusion chromatography (Figure 2A) indicated that Cdt2-HA fractionates as two peaks: ∼400–800 kDa (I) and ∼200 kDa (II). Peak I overlapped with Ddb1 and Csn2 proteins, consistent with a Cdt2–Ddb1–CSN complex. Peak II overlapped with Ddb1, but did not coelute with Csn2. Deletion of ddb1 disrupted peak II and deletion of pcu4 reduced its intensity. Loss of csn2 did not affect peak II integrity. This may indicate the presence of Cdt2 complex containing Ddb1 and possibly Pcu4, but not containing CSN. In all the deletion backgrounds tested, peak I, although sometimes reduced in intensity, was not lost. This could be explained if Cdt2 associates with additional protein complexes.
Figure 2.
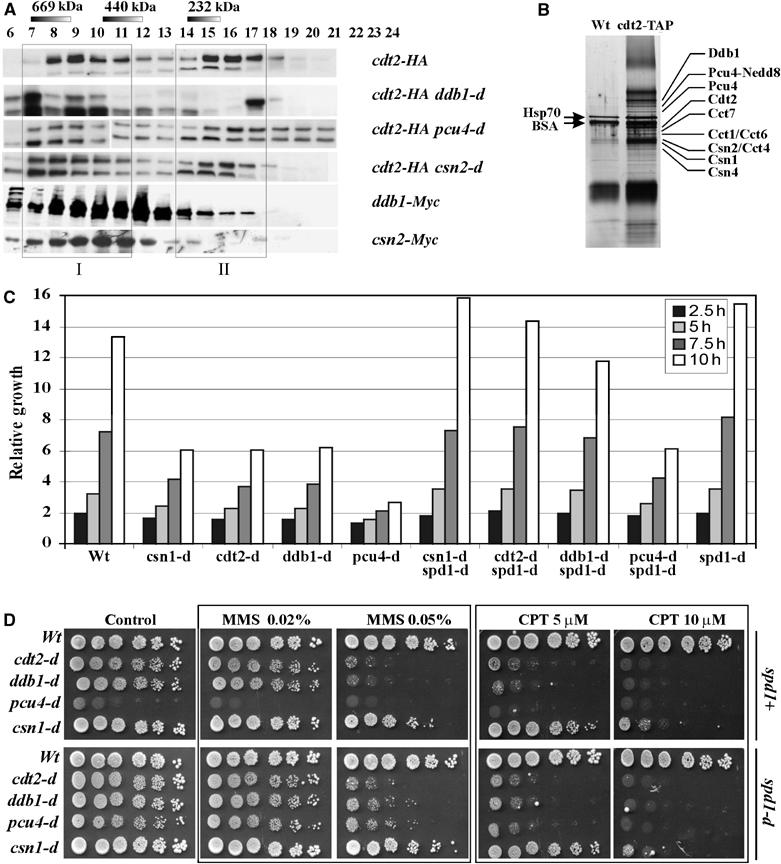
Cdt2 exists in two different protein complexes. (A) Gel filtration shows Cdt2-HA coeluting at 500–800 kDa (fraction I: FI) and 200 kDa (fraction II: FII). FI coelutes with Ddb1-Myc and Csn2-Myc. Deletion of the ddb1, pcu4 or csn2 did not result in complete Cdt2-FI loss. ddb1 deletion resulted in complete loss of Cdt2-FII. pcu4 deletion resulted in partial Cdt2-FII loss. csn2 deletion did not affect FII. (B) Silver-stained SDS–PAGE gradient gel following Cdt2-TAP purification. The four copurifying subunits (56.6–60.6 kDa) of the T chaperonin complex are indicated. The band above Ddb1 could not be unambiguously identified. Bands below Csn4 were assumed to represent other CSN subunits identified by Liu et al (2003). (C) Relative growth (cell number doublings) of strains indicated. (D) Serial dilutions of log-phase csn1, cdt2, ddb1 and pcu4 null mutant cells with (bottom) or without (top) concomitant spd1 deletion were spotted onto rich media containing indicated dosages of MMS or CPT and incubated for 4 days.
To identify further Cdt2-interacting proteins, we purified Cdt2-TAP. HU-arrested (4 h, 20 mM) cells were used to increase Cdt2 levels in the extract. Following TAP purification, Coomassie staining bands from SDS–PAGE (Figure 2B) were identified by MALDI-TOF mass spectrometry. Cdt2-TAP copurified with Pcu4–Ddb1–CSN components, as expected. No other cullin or cullin-associated proteins were identified. However, four T complex subunits (Cct proteins: Chaperonin containing TCP-1) were identified. The T complex is a group II eukaryotic cytosolic chaperonin composed of eight homologous subunits of similar size (Spiess et al, 2004). Cct proteins are essential because they function to correctly fold tubulin and actin from nascent polypeptides. They are also required for correct folding of other proteins known to form stable complexes with additional subunits (Leroux and Hartl, 2000). Thus, it is possible that peak I contains mixed complexes of Cdt2–Pcu4–Ddb1–CSN and Cdt2 associated with the T complex. If this were the case, this could explain the gel filtration data since it would reduce the impact of disrupting ddb1-d and pcu4-d on the profiles. However, we cannot formally exclude the existence of further Cdt2-containing complexes on the basis of these data.
Pcu4–Ddb1–Cdt2 has functions in addition to CSN-dependent Spd1 degradation
pcu4, ddb1 and cdt2 null mutants grow slowly and exhibit a greater number of more elongated cells when compared to csn1 or csn2 null mutants (Figure 2C). Because pcu4-d, ddb1-d and cdt2-d cells have the same Spd1 degradation defect as the more moderately growth-impaired csn1-d cells (Liu et al, 2003), it is likely that the Pcu4–Ddb1–Cdt2 complex has functions that are CSN independent.
Concomitant deletion of spd1 in csn1-d cells reverses both the growth and UV sensitivity defects (Figure 2C; Liu et al, 2003). The growth defect of cdt2-d and ddb1-d single mutants was also rescued to approximately that of control strains when spd1 was concomitantly deleted. However, the growth rate of pcu4-d cells was only partially suppressed, and thus not restored to wild-type levels by spd1 deletion. Thus, Pcu4 has Ddb1-independent functions during cell growth, possibly via association with additional adaptor proteins.
We also assayed the sensitivities of the mutants to methylmethane sulphonate (MMS) and camptothecin (CPT) (Figure 2D). In response to MMS and CPT, csn1-d colony formation was moderately impaired. This effect was more pronounced for ddb1-d and cdt2-d mutants and most pronounced for pcu4-d (upper panel). Pcu4 cells grow very slowly, so the increased cell death in CPT and MMS was verified by microscopy (data not shown). Upon concomitant spd1 deletion (lower panel), the sensitivity of the csn1-d mutant to MMS and CPT was not reversed to that seen in wild-type cells. This contrasts with the response to UV, where wild-type levels of resistance were restored to csn1-d mutants by concomitant deletion of spd1 (Liu et al, 2003; Holmberg et al, 2005) and thus indicates that csn1 has functions in the DNA damage response independent of Spd1 degradation. Similarly, the resistance to MMS and CPT of cdt2-d, ddb1-d and pcu4-d was only modestly restored when spd1-d was concomitantly deleted.
Together, these data suggest that the Cdt2–Pcu4–Ddb1–CSN complex has additional targets to Spd1 that affect sensitivity to agents that introduce damage during S phase and that Pcu4 has additional Ddb1-independent functions in cell growth.
Degradation of Spd1 and nuclear export of Suc22R2 require functional Cdt2
In budding and fission yeast, RNR activation is preceded by nuclear export of the small subunit (Liu et al, 2003; Yao et al, 2003). In S. pombe, Suc22R2 nuclear export is suppressed by Spd1, which may anchor Suc22R2 in the nucleus. Spd1 must be degraded via Pcu4–Ddb1–CSN polyubiquitylation and loss of components of this ubiquitin ligase causes Spd1 stabilisation, constitutive nuclear localisation of Suc22R2 and slow S phase. To explore if Cdt2 is required to regulate RNR localisation, Suc22R2 nuclear export was monitored by indirect immunofluorescence (Figure 3A). In wild-type cells, Suc22R2 localised in the nuclei of log-phase G2 cells and disappeared 4 h after HU treatment, a sign of its nuclear export (Liu et al, 2003). We observed that the nuclear Suc22R2 signal remained visible after HU treatment in cdt2 null cells. This indicates a failure to translocate Suc22R2 to the cytoplasm. Consistent with this, concomitant deletion of spd1 resulted in constitutive loss of Suc22R2 from the nucleus (Figure 3A). The implied loss of Suc22R2 translocation in the cdt2 mutant correlated with Spd1 stabilisation in cdt2 null mutant cells when we assayed Spd1-TAP levels in spd1-TAP cdt2+ and spd1-TAP cdt2-d extracts before and after HU treatment (Figure 3B, top). In extracts from a cdt2-HA spd1-TAP double-tagged strain (Figure 3B, bottom), Spd1-TAP degradation correlated with increased Cdt2-HA levels during HU arrest. By indirect immunofluorescence, Cdt2-HA was seen in the nucleus of HU-treated cells and in unperturbed cells during S phase (Figure 3C). In synchronised cells (Figure 3D), the nuclear Suc22R2 staining declined as cells traversed unperturbed S phase, and this coincided with an increase in Cdt2-HA nuclear staining. These data suggest that increased Cdt2 levels stimulate Pcu4–Ddb1–CSN ubiquitin ligase activity against Spd1. We noted that the nuclear localisation of Cdt2 is not csn1 dependent (Figure 3C, right).
Figure 3.
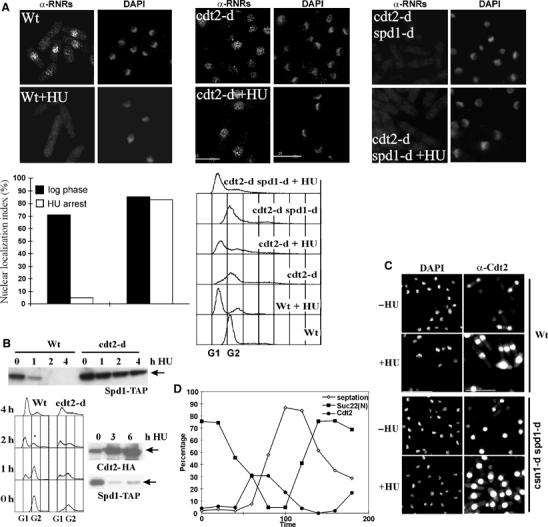
Subcellular localisation of Suc22 and Spd1 degradation in cdt2− mutants. (A) α-Suc22 (RNRs) peptide antibody indirect immunofluorescence in log-phase and HU-treated cells (4 h). Micrographs: α-Suc22 and 4′,6-diamidino-2-phenylindole (DAPI). Nuclear Suc22R2 signal disappearance indicates Suc22 translocation. In contrast to wild–type cells (top left), in cdt2 null cells (middle), Suc22R2 remained nuclear after HU treatment (4 h). Concomitant deletion of spd1 (top right) causes constitutive loss of nuclear signal. Bottom left: quantification of an equivalent experiment. Percentage of cells with nuclear Suc22R2 is shown. Bottom right: FACS analysis confirming early S-phase arrest. (B) Top: 50 μg of cdt2+ or cdt2-d extract was analysed for Spd1-TAP by Western blot before or after HU treatment. Bottom left: FACS analysis of the same strains. Bottom right: parallel detection of Cdt2-HA and Spd1-TAP in otherwise wild-type cells 3 and 6 h post HU treatment. Spd1 protein level decreased when Cdt2 protein increased. (C) Cdt2-Myc (α-Myc) indirect immunofluorescence in log-phase or HU-treated cells. Without treatment, Cdt2-Myc is only detectable in the nuclei of binuclear cells (S phase). After treatment (+HU), the nuclear signal intensified in all cells. csn1 disruption does not affect Cdt2-Myc localisation. (D) Quantification of nuclear signals of Cdt2-Myc and Suc22R2 in a G2 synchronised culture. Septation index indicates cell cycle progression. S phase occurs commensurate with the peak in septation.
Cdt2 protein is unstable and degraded by UPS
Many adaptor subunits of cullin-scaffolded ubiquitin ligases, such as F-box proteins and DDB2, are rapidly degraded upon assembly into E3 complexes (Deshaies, 1999). To examine if Cdt2 exhibits UPS-dependent instability, we prepared extracts from cdt2-HA mts3-1 cells. Mts3 is a subunit of the 19S regulatory particle of the proteasome. The mts3-1ts mutant significantly diminishes its proteolytic activity (Liu et al, 2003). Cdt2-HA protein levels increased at 35°C in the mts3-1 background (Figure 4A). Slow-mobility bands became evident, indicating the accumulation of polyubiquitylated Cdt2-HA. IP of Cdt2-HA from mts3-1 cells at the restrictive temperature (Figure 4B) results in detection of high molecular weight ubiquitylated species, further indicating that Cdt2 is polyubiquitylated.
Figure 4.
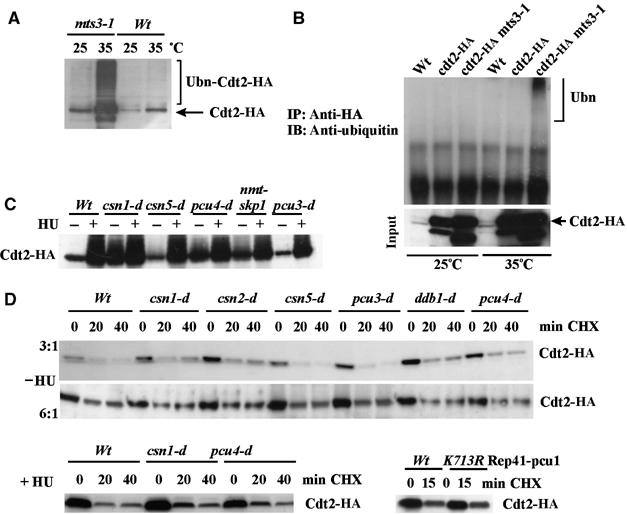
Ubiquitylation and degradation of Cdt2. (A) Cdt2-HA levels compared by Western blot (α-HA) in mts3-1 and mts3+ cell extracts at the permissive (25°C) and restrictive (35°C) temperature. Higher molecular weight species are presumed to represent ubiquitylated Cdt2-HA. (B) The strains indicated were incubated at the permissive (25°C) and restrictive (35°C) temperature and extracts were prepared and Cdt2-HA IPed, separated by SDS–PAGE and probed with α-Ub antibody. (C) Cdt2-HA levels were assayed in extracts derived from csn1-d, csn5-d, pcu4-d, nmt-skp1shut-off (nmtP3-3ha-skp1+) and pcu3-d either untreated (−) or following incubation with 10 mM HU for 4 h (+). Induced Cdt2 levels are comparable and basal levels in csn1-d, pcu4-d and nmt-skp1 are higher due to the extended S phase. Note: skp1 expression was suppressed before analysis by growing the culture in YEA media supplemented with thiamine. (D) The degradation of Cdt2-HA either without treatment (−HU) or following 4 h incubation with 10 mM HU (+HU). Protein synthesis was inhibited with 100 μg/ml cyclohexamide (CHX) at time 0. Top: two different loadings (wild type:mutant total extract ratio of 3:1 and 6:1) are shown. Cdt2-HA half-life was estimated to be 10–15 min in all strains. Bottom right: before CHX treatment, Pcu1 function was ablated by induction of the dominant-negative form Pcu1-K713R for 24 h (Osaka et al, 2000).
Analysis of Cdt2-HA protein levels in csn1-d, pcu4-d and skp1-nmt (shut-off) cells showed increased levels of Cdt2 in log-phase cells that increased further upon HU treatment (Figure 4C). Each of these strains exhibits a prolonged S phase. Because Cdt2 is present at high levels during S phase, this complicates interpretation. We thus monitored the stability of Cdt2-HA in various genetic backgrounds (Figure 4D). Cdt2-HA expression was ‘shut off' by addition of cyclohexamide to log-phase cells (Figure 4D, top) or to cells treated for 4 h with HU (Figure 4D, bottom). Cdt2-HA levels rapidly decayed when protein synthesis was terminated (estimated half-life: 10–15 min) in all strains tested, suggesting that none of the cullin-scaffolded ubiquitin ligases were necessary for Cdt2 degradation. Moreover, Cdt2 was not stabilised in cyclohexamide-treated cells arrested in S phase by HU. We conclude that Cdt2 protein can be degraded at all stages of the cell cycle and that its degradation is independent of its association with the Pcu4–Ddb1–CSN complex.
Accumulation of Cdt2 protein in S phase and upon DNA damage
Spd1 is degraded during S phase and upon checkpoint activation in G2 (Liu et al, 2003). We reasoned that Cdt2 protein levels in S phase and following irradiation of G2 cells might trigger Pcu4–Ddb1–CSN-dependent polyubiquitylation of Spd1.
To establish if Cdt2 protein levels correlated with the profile of Spd1 degradation during unperturbed S phase, cdt2-Myc strains were synchronised by elutriation and allowed to progress from the second G2 phase (time 0; see Materials and methods) through the cell cycle. Cdt2-Myc and septation index were monitored (Figure 5A). Cdt2-Myc accumulated as cells traversed S phase in wild-type, rad3-d or cds1-d backgrounds. Thus, Cdt2 levels fluctuate periodically in normal mitotic cycle in a checkpoint-independent manner. This mirrors the profile of Spd1 degradation (Liu et al, 2003).
Figure 5.
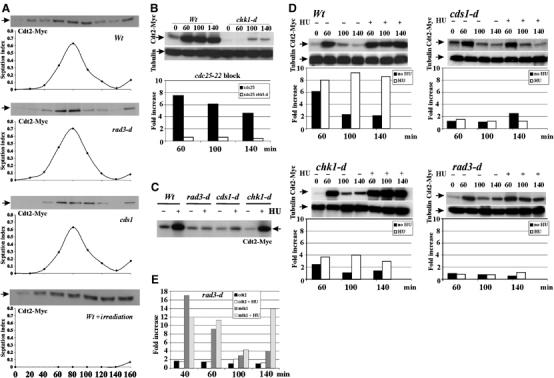
Expression of Cdt2 in normal cell cycle and upon checkpoint activation. (A) Wild-type, rad3-d or cds1-d cells (harbouring cdt2-Myc) were G2 synchronised by elutriation. Time 0: cells in the second G2 phase. First three panels: 20 min samples were taken through subsequent mitosis and S phase for extract preparation and septation index. Bottom panel: wild-type cells were irradiated (500 Gy γ-irradiation) at time 0. A 50 μg portion of total protein was separated by SDS–PAGE and probed with α-Myc. Cdt2 peaks in S phase. (B) Cdt2-Myc and cdt2 mRNA levels were analysed following γ-irradiation. cdt2-Myc cdc25-22 cells either with or without chk1 deletion were synchronised in G2 (35°C, 4 h) and irradiated (500 Gy). Total Cdt2-Myc protein and mRNA levels (relative to G2 unirradiated cells, assayed by QPCR) are shown for indicated times. chk1-d cells were defective for Cdt2 protein and mRNA accumulation. (C–E) Cdt2 protein and mRNA levels after replication stress were measured by Western blotting (α-Myc) and QPCR. α-Tubulin is used as a loading control. (C) Cdt2-Myc in unsynchronised rad3-d, cds1-d and chk1-d cells treated or not with 10 mM HU for 4 h. (D) Cdt2-Myc and cdt2 mRNA levels in synchronised wild-type, chk1-d, cds1-d and rad3-d cells. Half the sample was treated with 10 mM HU (+) during the first cell cycle to activate replication checkpoint. (E) mik1 and cdt2 mRNA in synchronised rad3-d cells either with (+) or without HU treatment.
Cells synchronised in G2 and γ-irradiated activate the Rad3–Chk1 DNA damage checkpoint and arrest in G2 until the DNA lesions are repaired (Carr, 2002). Concurrently, Spd1 is degraded, Suc22R2 is translocated from the nucleus to the cytoplasm and an increase in dNTP synthesis is postulated to facilitate DNA repair synthesis in G2 (Liu et al, 2003). To establish if Cdt2 levels increase in response to DNA damage, and thus correlated with Spd1 degradation profile after DNA damage, synchronised G2 cells were γ-irradiated (500 Gy). Cell extracts were prepared at 20-min intervals from 60 min post-irradiation. Cdt2-Myc levels increased after treatment and were maintained until cells re-entered the cell cycle (Figure 5A, bottom panel). This precisely mimicked the kinetics of Spd1 degradation (Liu et al, 2003). Thus, Cdt2 levels are elevated during S phase and following DNA damage in G2, inversely correlating with Spd1 protein levels.
Induction of cdt2+ transcription upon checkpoint activation
Cdt2 accumulation during S phase correlates with Cdc10/DSC1-dependent transcription (Yoshida et al, 2003). To characterise cdt2 RNA and protein expression after DNA damage, cdt2-HA cdc25-22 cells were arrested for 4 h at 35°C to accumulate G2 cells before γ-irradiation (500 Gy). cdt2+ mRNA expression was enhanced (∼7-fold; Figure 5B) and Cdt2-HA levels significantly elevated. Both responses were significantly reduced in a chk1-d background. Thus, chk1-dependent cdt2+ expression correlates with Spd1 degradation and activation of Pcu4–Ddb1–CSN after DNA damage in G2.
To characterise Cdt2 protein profiles following replication checkpoint activation, we next analysed Cdt2-Myc protein levels following replication stress by HU (Figure 5C). Both wild-type and chk1-d cells accumulated Cdt2-Myc when treated with HU. Protein levels remained low in rad3-d and cds1-d cells that lack a functional replication checkpoint. This profile is identical to that observed for the mitotic inhibitor Mik1 (Christensen et al, 2000).
To explore the relationship between cdt2+ transcription and Cdt2 protein levels in detail, we synchronised wild-type, chk1-d, cds1-d and rad3-d cultures by centrifugal elutriation and monitored both cdt2+ transcript and Cdt2-Myc protein levels following S-phase arrest (Figure 5D). G2 cultures were divided in half and HU added to one aliquot. At time 0 (G2), 60 (peak of S phase), 100 (untreated cells return to G2) and 140 (G2) cells were collected for protein and mRNA extraction. As expected, cdt2 mRNA and protein levels peaked during normal S phase of wild-type cells, but continued to accumulate in the presence of HU. A similar profile was observed for chk1-d, although the level of transcriptional induction was significantly decreased.
In contrast, for rad3-d and cds1-d cultures, Cdt2 protein peaked during S phase in untreated cells, but did not accumulate following HU treatment (Figure 5D). This behaviour is similar to the profile we previously reported for Mik1 protein levels through the cell cycle in similar experiments (Christensen et al, 2000). However, we observed a significant difference between the cdt2+ transcript profile and the mik1+ transcript profile: previous analysis of mik1+ transcription (Christensen et al, 2000) demonstrated that cell cycle-dependent transcription in unperturbed cells was independent of the rad3 and cds1 replication checkpoint genes. In contrast, these data (Figure 5D) and several repeats (not shown) demonstrate that cdt2+ transcription in unperturbed S phase is rad3 and cds1 dependent.
To verify this difference, we compared the transcription profiles of cdt2+ and mik1+ in the same synchronous rad3-d culture (Figure 5E). In contrast with the steady expression level of cdt2+, mik1+ mRNA level displayed a dramatic increase upon S-phase entry in rad3-d cells (Figure 5E). These results suggest that the rad3+-dependent checkpoint pathway plays an additional role in controlling the expression of cdt2+ during unperturbed cell cycle progression as well as during checkpoint activation. Because the S-phase profile of cdt2+ transcription and Cdt2 protein levels are uncoupled in unperturbed cell cycles in the rad3 mutant background (cf. Figure 5A and E), we also infer that an unknown post-transcriptional mechanism coexists with the transcriptional control of cdt2+ to ensure that the Cdt2 protein levels oscillate during the normal cell cycle.
Cdt2 is the adaptor protein for Spd1
cdt2 is required for Spd1 degradation and Cdt2 protein accumulation correlates with Spd1 degradation. However, the role of Cdt2 as a Pcu4–Ddb1–CSN activator remains elusive. To determine if the simple presence of Cdt2 results in Spd1 degradation, cdt2 was transcriptionally induced from the nmt promoter. Rep41(HA-Cdt2) was introduced into spd1-TAP cdt2-d and spd1-TAP csn1-d cells and cdt2 transcript was induced by removal of thiamine from the media, both with and without HU treatment. Spd1-TAP protein levels were monitored by Western blot. The presence of the cdt2-expresing plasmid resulted in lower levels of Spd1-TAP protein, irrespective of HU treatment (Figure 6A, lanes 3 and 4). Even basal level of Cdt2 expression from the plasmid (in the presence of thiamine) was sufficient to promote Spd1 degradation (data not shown). Overproduction of Cdt2 mimicked the situation seen in HU-treated cells (cf. lanes 2 and 3) but did not significantly affect cell cycle progression profiles (Figure 6A, bottom right): Spd1-TAP levels were already low and were not further decreased by HU treatment (lane 4). However, Cdt2 expression did not overcome the Spd1 degradation defect of csn1-d cells (lanes 5 and 6). As expected, overexpression of Cdt2 reduced nuclear localisation of Suc22R2 (Figure 6A, bottom left). Thus, the abundance of Cdt2 protein determines, at least in part, the activation of Pcu4–Ddb1–CSN ubiquitin ligase against Spd1.
Figure 6.
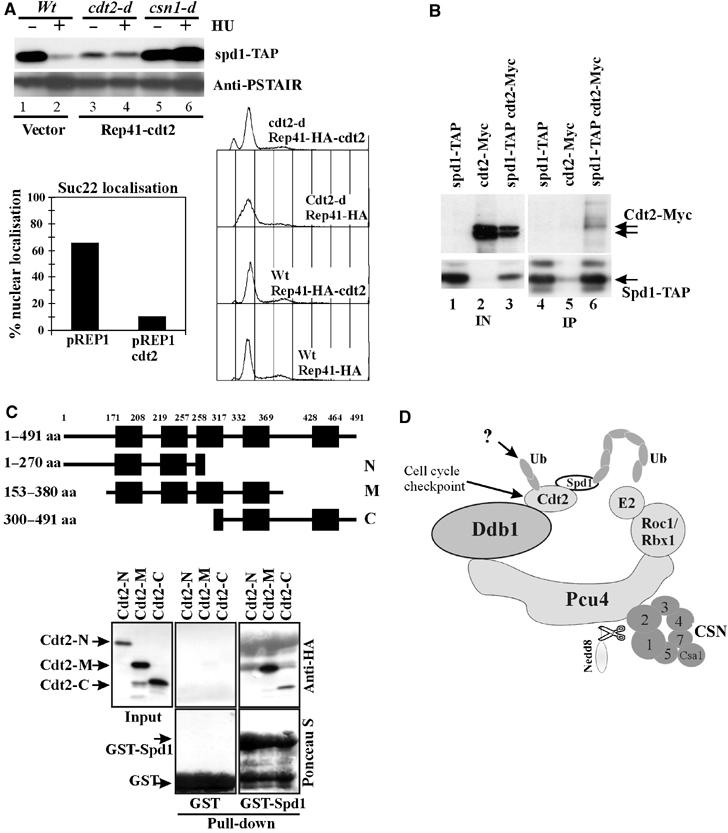
Cdt2 is an adaptor protein for Spd1. (A) Top: Rep41(HA-Cdt2) plasmid or vector control was induced by the absence of thiamine (22 h) in spd1-TAP strains shown. Half of each culture was treated (20 mM HU for 4 h). Spd1 levels were monitored by SDS–PAGE and IgG binding to TAP. α-PSTAIR (Cdc2 peptide antibody) was used as a loading control. Overexpressed Cdt2-HA (α-HA, top) caused Spd1 degradation. Overproduction of Cdt2 did not rescue the csn1-d defect for Spd1 degradation (lanes 5 and 6). Bottom left: nuclear signal of Suc22R2 was quantified in the presence of overexpressed Cdt2 (Rep1 plasmid, 22 h without thiamine). Bottom right: Cdt2-HA overexpression rescues the slow S-phase phenotype of cdt2-d cells but does not affect cell cycle profiles of wild-type cells. (B) Co-precipitation from extracts of indicated strains of Cdt2-Myc with Spd1-TAP. Strains contained the csn1-d mutation to prevent Spd1-TAP degradation. We estimated <5% of Cdt2-Myc co-precipitates. (C) Truncated HA-tagged Cdt2 constructs containing different WD40 repeat domains expressed in S. pombe from Rep41-HA. Cdt2 constructs were pulled down from soluble extracts using purified Spd1-GST (∼38 kDa) or GST alone (25 kDa). Only Cdt2-M and Cdt2-C fragments were efficiently pulled down by Spd1-GST. (D) A model of Pcu4–CSN complex containing Cdt2. Cdt2 protein is regulated by a combination of constant UPS-dependent degradation, cell cycle and checkpoint-driven transcription and an uncharacterised cell cycle-dependent post-transcriptional mechanism. Together with Ddb1, Cdt2 adapts Spd1 to the Pcu4-scaffolded RING-E2 enzyme for ubiquitylation. CSN is obligatory for Pcu4–Ddb1–Cdt2 function, possibly via regulation of neddylation and E2 loading.
To demonstrate that Cdt2 can interact with Spd1, Cdt2-Myc was tested for co-precipitation with Spd1-TAP. This experiment was performed in csn1-d background to ensure that Spd1 remained stable in the presence of HU-induced Cdt2. A significant amount of Cdt2-Myc protein co-precipitated with Spd1-TAP (Figure 6B, lane 6). No signal was detected in control strains (lanes 4 and 5). To define an interaction domain of Cdt2 for Spd1, extracts from cells expressing truncated HA-Cdt2 fragments were probed with recombinant GST-Spd1 or GST protein as control. Only HA-Cdt2 middle and C-terminal fragments containing the third and fourth WD40 repeats specifically bound to GST-Spd1 beads (Figure 6C).
Discussion
In this work, we have identified the WD repeat protein Cdt2 as an adaptor protein for a Pcu4–Ddb1 ubiquitin ligase that directs the degradation of the RNR inhibitor Spd1. We provide evidence that Cdt2 protein level determines (at least in part) Spd1 degradation. Cdt2 protein levels fluctuate during unperturbed S phase in a manner independent of the DNA structure checkpoint pathway. In contrast, DNA damage induces Cdt2 levels in G2 cells in a manner that is dependent on the DNA damage checkpoint.
The Pcu4–Ddb1–CSN–Cdt2 protein complex
Pcu4 is the closest S. pombe protein homologous to human CUL4A. All characterised CUL4A-scaffolded ubiquitin ligases are associated with DDB1 and one or more WD40 repeat proteins such as DDB2, CSA or COP1 (Groisman et al, 2003; Liu et al, 2003; Wertz et al, 2004). The best-characterised CUL4A-scaffolded ubiquitin ligase is the DCXDET1−COP1 complex (DDB1–CUL4A–X-box). Its proposed architecture conforms to the SCF paradigm, with an unknown motif in DET1 (termed the ‘X-box') permitting association of a dimeric substrate adaptor (DET1–COP1) with DDB1. The C-terminal WD40 repeats of COP1 then function to recruit substrates including c-JUN and other bZIP transcription factors (Wertz et al, 2004). The architecture of the Spd1-specific Pcu4-scaffolded E3 is consistent with the DCX model: Ddb1 is required for the interaction between the Cdt2 ‘substrate adaptor' and the cullin (Pcu4). Cdt2 recruits the target protein Spd1 for ubiquitylation, possibly via WD40 repeats (Figure 6C). However, no S. pombe DET1 analogue copurified with Cdt2 and we believe it unlikely (but it cannot be formally excluded) that Cdt2 associates with a secondary adaptor (i.e. a DET1-like protein). This is consistent with characterisation of the human DCXCSA and DCXDDB2 complexes (Groisman et al, 2003), each of which contained DDB1, CUL4A, a putative WD repeat substrate adaptor and CSN, but not DET1. Possibly, the ‘X-box' is contained within CSA and DDB2 proteins or the WD40 repeats may bind both substrates and DDB1. Indeed, multiple mechanisms of adaptor protein recruitment may be available to DDB1.
The role of Pcu4–Ddb1–CSN ubiquitin ligase
The CSN complex interacts with several cullins in human cells and is required to regulate their deneddylation. However, CSN plays a novel role in association with cullin-4-scaffolded E3's. In S. pombe, Pcu4 and CSN specifically copurify and CSN is essential for Pcu4-dependent ubiquitylation of Spd1, implying a positive regulatory function. Conversely, biochemical data suggest a negative regulatory role for human CSN in CUL4A-dependent ubiquitylation (Groisman et al, 2003). Resolution of this conundrum awaits further experimentation.
Current models, based on the interaction with other cullins (Wu et al, 2002), suggest that CSN attaches to the Pcu4 C-terminus and that this is important for regulating Pcu4 neddylation status, which in turn facilitates E2 loading onto Rbx1/Pip1 within the Pcu4 complex. This suggests that the substrate recognition by the Pcu4–Ddb1–CSN complex is mediated by Cdt2 protein association with Ddb1. We can exclude that CSN regulates the association of Cdt2 with Pcu4–Ddb1–CSN complex because Cdt2 levels, subcellular localisation and association with Ddb1 do not require csn1 or csn2.
Substrate degradation by CUL1-scaffolded ubiquitin ligases (SCF complexes) is regulated by substrate phosphorylation, which promotes substrate binding to the F-box substrate adaptor subunits. Despite extensive analysis (unpublished data), we have found no evidence for Spd1 phosphorylation and since simple overexpression of Cdt2 can mimic HU-induced Spd1 degradation (Figure 6A), we conclude that Cdt2 protein abundance is the key step in activating Pcu4–Ddb1–CSN ubiquitin ligase against Spd1.
Cdt2 protein abundance is regulated, in part, through cdt2+ transcription and in part through an unknown post-transcriptional mechanism. This mechanism may dictate the translation efficiency of the cdt2 message. The Cdt2 protein itself is unstable and this instability is not cell cycle regulated. Uncovering what factors are required for Cdt2 degradation (we have shown here that it does not require any cullin-based ubiquitin ligases or CSN) and what mechanism underlies the increase in Cdt2 levels during S phase and after DNA damage will be informative.
The interaction with T complex
Besides actin and tubulin folding, the Cct chaperonin has been implicated in the correct assembly of several multisubunit E3 ligases. The CDC20 and CDH1 subunits of the anaphase-promoting complex (APC) require Cct proteins for binding and activation of APC (Camasses et al, 2003). Cct proteins also mediate the formation of the VHL-elongin BC ubiquitin ligases and unassembled VHL protein remains associated with the T complex (Feldman et al, 1999; Melville et al, 2003). Some tumour-causing mutations interfere with chaperonin association of VHL and thus its incorporation into the complex (Feldman et al, 2003). Furthermore, it has been demonstrated that a group of WD40 beta-propeller proteins transiently interact with Cct and require the chaperonin to reach their native folded state (Siegers et al, 2003). Thus, it is perhaps not surprising that this chaperonin associates with Cdt2 in S. pombe. Most likely, Cct proteins facilitate Cdt2 folding and/or assembly with the Pcu4–Ddb1–CSN scaffold. This additional association of Cdt2 protein with Cct proteins may explain why, during gel filtration, a large Cdt2-containing protein complex remains (Figure 2, peak I) in the absence of ddb1 or pcu4. However, we cannot formally exclude the presence of other Cdt2-interacting proteins in peak I.
rad3+-dependent checkpoints and cdt2 regulation
We demonstrate that increased Cdt2 abundance in S phase and after DNA damage drives Spd1 degradation, subsequent Suc22R2 cytoplasmic relocalisation and RNR activation. cdt2+ is a Cdc10/DSC target during S phase and transcription is also under the control of the chk1+-dependent checkpoint pathways following DNA damage in G2. The rad3+–chk1+ pathway is absolutely required for the cdt2+ messenger and protein accumulation after DNA damage of G2 cells. The simplest explanation is that Chk1 phosphorylates and activates a transcription factor that promotes G2 transcription of cdt2+ and potentially of other genes. Previously, we identified several genes that are transcriptionally induced after DNA damage of G2 cells (Watson et al, 2004). A subset of these were DNA damage checkpoint dependent. Intriguingly, this subset included genes previously identified as DSC/Cdc10 transcription targets at the G1–S transition, including Cdc22R1, Cdc18 and Cdt2 itself. It is thus possible that Chk1 specifically activates Cdc10/DSC-dependent transcription when DNA damage is detected outside of S phase in order to ensure a supply of enzymes and dNTPs for DNA repair synthesis.
In response to replication arrest by HU, we observed that the rad3–cds1-dependent replication checkpoint was required to maintain cdt2+ transcript and Cdt2 protein levels. We have previously observed that the activation of the Rad3–Cds1-dependent replication checkpoint resulted in similar accumulation of mik1+ transcript and Mik1 protein (Christensen et al, 2000). However, we did not find evidence that checkpoint directly targeted mik1 expression or Mik1 protein stability. Instead, we proposed that the replication checkpoint indirectly led to mik1+ transcript and Mik1 protein accumulation because cells are blocked within S phase, where mik1+ is transcribed and Mik1 protein is predetermined to be resistant to degradation. Thus, our observations concerning Cdt2 do not require an interpretation in which the replication checkpoint directly controls cdt2+ transcription or Cdt2 protein abundance.
Our analysis of Cdt2 protein stability in S- and G2-phase-arrested cells suggests that degradation rates do not dramatically vary through the cells cycle. We thus propose that transcription and/or translation determine Cdt2 protein levels. It is possible that Cdt2 accumulates in S-phase-arrested cells because the S-phase checkpoint directly targets cdt2+ transcription and/or translation factors. However, the most parsimonious interpretation of the data is that checkpoint activity maintains cells within the S-phase compartment of the cell cycle where cdt2+ is predetermined to be transcribed and translated.
Unlike our previous findings concerning the lack of dependence of mik1+ transcription on the rad3 and checkpoint pathway in unperturbed S phase, we identified a requirement for both rad3+ and cds1+ function for periodic cdt2+ transcription in unperturbed S phase. However, loss of periodic S-phase transcription did not prevent periodic Cdt2 accumulation. Thus, while transcriptional profiles clearly contribute to Cdt2 protein profiles, mechanisms additional to transcription must act to regulate Cdt2 protein level. Why cdt2+ transcription and mik1+ transcription differ with respect to rad3–cds1 dependence remains to be established. Both are Cdc10/DSC dependent, so it must reflect a subtle aspect of transcriptional control that has not yet been addressed in S. pombe.
Conclusion
RNR regulation by checkpoint pathways is conserved through evolution, reflecting the importance of controlling dNTP pools. Disrupting relative and total dNTP concentrations is mutagenic (Chabes et al, 2003) and destabilises the genome. In addition, other metabolic products depend on the correct nucleotide pool balance. A recent example is the identification in S. pombe of interactions between RNR and phthalate metabolism (Stolz et al, 2004). In both S. pombe and S. cerevisiae, subcellular compartmentalisation of RNR subunits is one of several mechanisms contributing to RNR regulation. Understanding how this conserved pathway operates and is regulated will inform our understanding of an aspect of DNA metabolism potentially important in maintaining genomic stability and which is a target of medically important pharmaceuticals.
Materials and methods
Plasmid construction and protein expression
spd1 ORF was amplified by PCR from an S. pombe cDNA library and cloned into pGEX-KG. A sequenced clone was transformed into BL21 (Escherichia coli) and induced with 1 mM IPTG (OD600=0.5; 4 h, 37°C). GST-Spd1 was extracted, enriched and purified at 4°C in NETN buffer (0.5% NP-40; 20 mM Tris, pH 8.0; 100 mM NaCl; 1 mM EDTA; 1 mM PMSF) on glutathione beads.
Genetics, cell biology and centrifugal elutriation
Strain nmtP3-3ha-skp1+ was a gift from Dr T Toda. Double mutants were isolated by genetics, except cdt2 ddb1 as the two genes are 9.736 kb apart. Sequential tagging/deletion was used in these cases. Elutriation (JE-5.0, Beckman Coulter): Mid-log cultures were loaded and small G2 cells were collected, harvested and resuspended in rich medium. Cell cycle progression and septation were followed by staining cells with DAPI and Calcofluor, respectively. In all elutriation experiments, time zero (time 0) was defined as cells that had been through one complete cycle and entered the second G2 after elutriation.
Protein extracts, gel filtration and Western blots
Cells were disrupted with glass beads in either HB buffer (25 mM Tris–HCl, pH 7.5; 15 mM EGTA, pH 7.5; 15 mM MgCl2; 0.1% NP-40; 1 mM DTT) or IP buffer (20 mM Tris–HCl, pH 7.4; 150 mM NaCl; 0.5% Triton X-100; 1 mM DTT), supplemented with protease inhibitor (Complete EDTA-free, Roche) and 1 mM PMSF. Extracts were precleared (14 000 r.p.m., microfuge). Protein concentration was quantified by Bradford assay. For size exclusion, 1 mg of protein was passed through a Sephadex 200HR 10/30 column (Pharmacia) and 500 μl fractions were collected. To ensure detectable levels of Cdt2 protein, cells were first incubated in 10 mM HU for 4 h.
Immunoprecipitation
Precleared supernatants were incubated with anti-HA/Myc antibodies (1 h, 4°C). Immunocomplexes were absorbed onto 20 μl protein G beads (1 h). Beads were pelleted, washed 3 × (HB or IP buffer) and 50 μl SDS sample buffer was added before SDS–PAGE. For TAP purification, the protocol was as described (Liu et al, 2003). Elutes were concentrated and resolved in 4–20% gradient SDS–PAGE. Coomassie-stained bands were identified by MALDI-TOF (Yates, 1998).
Immunofluorescent microscopy
Briefly, cells were fixed in 3.75% paraformaldehyde in PEM (100 mM PIPES; 1 mM EGTA, pH 6.9; 1 mM MgCl2), permeabilised with 1.25 mg/ml zymolase T20 (ICN Biomedicals, Cleveland, OH) and 1% Triton X-100 in PEM+1.2 M sorbitol. Proteins were visualised using TRITC or FITC-conjugated secondary antibodies in PEM+1% BSA, 0.1% NaN3 and 100 mM lysine hydrochloride.
Quantitative PCR
Total RNA (∼20 μg) was RQ1 RNAse free DNAse (Promega, Madison, WI) treated and reverse transcribed (Stratascript from Stratagene). Quantitative PCR (QPCR) used BrilQuantiTect SYBR Green PCR mix (Qiagen). Duplicate samples were run on an MX4000 (Stratagene). Relative transcript quantities were determined using the 2−ΔΔCt formula: Ct=cycle at which fluorescence is statistically above background; ΔCt=difference in Ct of the gene of interest and Ct of the normaliser gene (cdc2); ΔΔCt=difference in ΔCt at time of sampling after synchronisation (time=t) and ΔCt at time=0. Reaction efficiencies for the primers were determined as equivalent using serial S. pombe genomic DNA dilutions.
Acknowledgments
We thank Dr Stead (COGEME proteomics facility, Aberdeen) for invaluable help with mass spectrometry and Dr Kanji Furuya and Dr Jo Murray for critical reading of the manuscript. CL was supported by Cancer Research UK grant C5514. AW and MP were supported by MRC grant G0001129.
References
- Camasses A, Bogdanova A, Shevchenko A, Zachariae W (2003) The CCT chaperonin promotes activation of the anaphase-promoting complex through the generation of functional Cdc20. Mol Cell 12: 87–100 [DOI] [PubMed] [Google Scholar]
- Carr AM (2002) DNA structure dependent checkpoints as regulators of DNA repair. DNA Repair 1: 983–994 [DOI] [PubMed] [Google Scholar]
- Chabes A, Domkin V, Thelander L (1999) Yeast Sml1, a protein inhibitor of ribonucleotide reductase. J Biol Chem 274: 36679–36683 [DOI] [PubMed] [Google Scholar]
- Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L (2003) Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 112: 391–401 [DOI] [PubMed] [Google Scholar]
- Chamovitz DA, Wei N, Osterlund MT, von Arnim AG, Staub JM, Matsui M, Deng XW (1996) The COP9 complex, a novel multisubunit nuclear regulator involved in light control of a plant developmental switch. Cell 86: 115–121 [DOI] [PubMed] [Google Scholar]
- Christensen PU, Bentley NJ, Martinho RG, Nielsen O, Carr AM (2000) Mik1 levels accumulate in S phase and may mediate an intrinsic link between S phase and mitosis. Proc Natl Acad Sci USA 97: 2579–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope GA, Suh GS, Aravind L, Schwarz SE, Zipursky SL, Koonin EV, Deshaies RJ (2002) Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science 298: 608–611 [DOI] [PubMed] [Google Scholar]
- Deshaies RJ (1999) SCF and Cullin/Ring H2-based ubiquitin ligases. Annu Rev Cell Dev Biol 15: 435–467 [DOI] [PubMed] [Google Scholar]
- Elledge SJ, Zhou Z, Allen JB (1992) Ribonucleotide reductase: regulation, regulation, regulation. Trends Biochem Sci 17: 119–123 [DOI] [PubMed] [Google Scholar]
- Elledge SJ, Zhou Z, Allen JB, Navas TA (1993) DNA damage and cell cycle regulation of ribonucleotide reductase. BioEssays 15: 333–339 [DOI] [PubMed] [Google Scholar]
- Feldman DE, Spiess C, Howard DE, Frydman J (2003) Tumorigenic mutations in VHL disrupt folding in vivo by interfering with chaperonin binding. Mol Cell 12: 1213–1224 [DOI] [PubMed] [Google Scholar]
- Feldman DE, Thulasiraman V, Ferreyra RG, Frydman J (1999) Formation of the VHL–elongin BC tumor suppressor complex is mediated by the chaperonin TRiC. Mol Cell 4: 1051–1061 [DOI] [PubMed] [Google Scholar]
- Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y (2003) The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 113: 357–367 [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67: 425–479 [DOI] [PubMed] [Google Scholar]
- Higa LA, Mihaylov IS, Banks DP, Zheng J, Zhang H (2003) Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat Cell Biol 5: 1008–1015 [DOI] [PubMed] [Google Scholar]
- Hofmann JF, Beach D (1994) cdt1 is an essential target of the Cdc10/Sct1 transcription factor: requirement for DNA replication and inhibition of mitosis. EMBO J 13: 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg C, Fleck O, Hansen HA, Liu C, Slaaby R, Carr AM, Nielsen O (2005) Ddb1 controls genome stability and meiosis in fission yeast. Genes Dev 19: 853–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux MR, Hartl FU (2000) Protein folding: versatility of the cytosolic chaperonin TRiC/CCT. Curr Biol 10: R260–R264 [DOI] [PubMed] [Google Scholar]
- Liu C, Powell KA, Mundt K, Wu L, Carr AM, Caspari T (2003) Cop9/signalosome subunits and Pcu4 regulate ribonucleotide reductase by both checkpoint-dependent and -independent mechanisms. Genes Dev 17: 1130–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melville MW, McClellan AJ, Meyer AS, Darveau A, Frydman J (2003) The Hsp70 and TRiC/CCT chaperone systems cooperate in vivo to assemble the von Hippel–Lindau tumor suppressor complex. Mol Cell Biol 23: 3141–3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundt KE, Liu C, Carr AM (2002) Deletion mutants in COP9/signalosome subunits in fission yeast Schizosaccharomyces pombe display distinct phenotypes. Mol Biol Cell 13: 493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundt KE, Porte J, Murray JM, Brikos C, Christensen PU, Caspari T, Hagan IM, Millar JBA, Simanis V, Hofmann K, Carr AM (1999) The COP9/signalosome is conserved in fission yeast and has a role in S-phase. Curr Biol 9: 1427–1430 [DOI] [PubMed] [Google Scholar]
- Osaka F, Saeki M, Katayama S, Aida N, Toh EA, Kominami K, Toda T, Suzuki T, Chiba T, Tanaka K, Kato S (2000) Covalent modifier NEDD8 is essential for SCF ubiquitin-ligase in fission yeast. EMBO J 19: 3475–3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM (2001) Mechanisms underlying ubiquitination. Annu Rev Biochem 70: 503–533 [DOI] [PubMed] [Google Scholar]
- Schwechheimer C, Serino G, Callis J, Crosby WL, Lyapina S, Deshaies RJ, Gray WM, Estelle M, Deng XW (2001) Interactions of the COP9 signalosome with the E3 ubiquitin ligase SCFTIRI in mediating auxin response. Science 292: 1379–1382 [DOI] [PubMed] [Google Scholar]
- Siegers K, Bolter B, Schwarz JP, Bottcher UM, Guha S, Hartl FU (2003) TRiC/CCT cooperates with different upstream chaperones in the folding of distinct protein classes. EMBO J 22: 5230–5240 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Spiess C, Meyer AS, Reissmann S, Frydman J (2004) Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets. Trends Cell Biol 14: 598–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz J, Caspari T, Carr AM, Sauer N (2004) Cell division defects of Schizosaccharomyces pombe liz1− mutants are caused by defects in pantothenate uptake. Eukaryot Cell 3: 406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson A, Mata J, Bahler J, Carr A, Humphrey T (2004) Global gene expression responses of fission yeast to ionizing radiation. Mol Biol Cell 15: 851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei N, Deng XW (2003) The COP9 signalosome. Annu Rev Cell Dev Biol 19: 261–286 [DOI] [PubMed] [Google Scholar]
- Wertz IE, O'Rourke KM, Zhang Z, Dornan D, Arnott D, Deshaies RJ, Dixit VM (2004) Human De-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase. Science 303: 1371–1374 [DOI] [PubMed] [Google Scholar]
- White S, Khaliq F, Sotiriou S, McInerny CJ (2001) The role of DSC1 components cdc10+, rep1+ and rep2+ in MCB gene transcription at the mitotic G1–S boundary in fission yeast. Curr Genet 40: 251–259 [DOI] [PubMed] [Google Scholar]
- Wu K, Chen A, Tan P, Pan ZQ (2002) The Nedd8-conjugated ROC1-CUL1 core ubiquitin ligase utilizes Nedd8 charged surface residues for efficient polyubiquitin chain assembly catalyzed by Cdc34. J Biol Chem 277: 516–527 [DOI] [PubMed] [Google Scholar]
- Yao R, Zhang Z, An X, Bucci B, Perlstein DL, Stubbe J, Huang M (2003) Subcellular localization of yeast ribonucleotide reductase regulated by the DNA replication and damage checkpoint pathways. Proc Natl Acad Sci USA 100: 6628–6633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JR III (1998) Mass spectrometry and the age of the proteome. J Mass Spectrom 33: 1–19 [DOI] [PubMed] [Google Scholar]
- Yoshida SH, Al-Amodi H, Nakamura T, McInerny CJ, Shimoda C (2003) The Schizosaccharomyces pombe cdt2(+) gene, a target of G1–S phase-specific transcription factor complex DSC1, is required for mitotic and premeiotic DNA replication. Genetics 164: 881–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Muller EG, Rothstein R (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol Cell 2: 329–340 [DOI] [PubMed] [Google Scholar]
- Zhou C, Wee S, Rhee E, Naumann M, Dubiel W, Wolf DA (2003) Fission yeast COP9/signalosome suppresses cullin activity through recruitment of the deubiquitylating enzyme Ubp12p. Mol Cell 11: 927–938 [DOI] [PubMed] [Google Scholar]