

Abstract
INSIGs are proteins that underlie sterol regulation of the mammalian proteins SCAP (SREBP cleavage activating protein) and HMG-CoA reductase (HMGR). The INSIGs perform distinct tasks in the regulation of these effectors: they promote ER retention of SCAP, but ubiquitin-mediated degradation of HMGR. Two questions that arise from the discovery and study of INSIGs are: how do they perform these distinct tasks, and how general are the actions of INSIGs in biology? We now show that the yeast INSIG homologs NSG1 and NSG2 function to control the stability of yeast Hmg2p, the HMGR isozyme that undergoes regulated ubiquitination. Yeast Nsgs inhibit degradation of Hmg2p in a highly specific manner, by directly interacting with the sterol-sensing domain (SSD)-containing transmembrane region. Nsg1p functions naturally to limit degradation of Hmg2p when both proteins are at native levels, indicating a long-standing functional interplay between these two classes of proteins. One way to unify the known, disparate actions of INSIGs is to view them as known adaptations of a chaperone dedicated to SSD-containing client proteins.
Keywords: ERAD, HMG-CoA reductase, HRD genes, INSIG, sterol-sensing domain
Introduction
Mammalian cell cholesterol levels are controlled by coordinated regulation of the proteins SCAP (SREBP cleavage activating protein) and HMGR (HMG-CoA reductase) (Hampton, 2002). SCAP is an ER-resident protein that carries the membrane-anchored transcription factor SREBP from the ER to the Golgi, where it is cleaved and thus activated to transcribe genes encoding sterol pathway enzymes and the LDL receptor. HMGR is a key enzyme of the sterol pathway that undergoes sterol-regulated ubiquitination and degradation (Song and DeBose-Boyd, 2004; Song et al, 2005). When sterol levels are sufficiently high in the cell, ER exit of SCAP (and hence SREBP activation) is decreased, and HMGR degradation is increased. By these two mechanisms, abundant sterols cause coordinated decreases in both transcription of relevant genes and sterol pathway activity.
SCAP and HMGR each have multispanning membrane anchors that share a sequence motif called the sterol-sensing domain (SSD) (Kuwabara and Labouesse, 2002). The SSD is required for both cholesterol-dependent ER retention of SCAP and lanosterol-stimulated ubiquitination of HMGR (Nohturfft et al, 1999; Song et al, 2005). The SSD-domain-interacting protein INSIG1 (or its homolog INSIG2) is required for the SSD-mediated effects of sterols on each protein (Yang et al, 2002; Sever et al, 2003). How do INSIG proteins mediate these distinct actions on its two known effectors? We now show that the versions of these proteins in yeast, called NSG1 and NSG2, mediate a third variation of INSIG action, one that suggests a general view of INSIG function.
The yeast HMGR isozyme Hmg2p undergoes ER ubiquitination that is regulated by the levels of the sterol pathway molecule farnesyl pyrophosphate (FPP) or a signal derived from it (Gardner and Hampton, 1999b; Hampton, 2002). The regulation of Hmg2p ubiquitination by this specific lipid signal requires the SSD-containing transmembrane region of Hmg2p. Yeast expresses two homologs of mammalian INSIG genes called NSG1 and NSG2. We demonstrate below that these proteins directly interact with the Hmg2p transmembrane region, and participate in the regulation of Hmg2p stability at native levels by directly binding this portion of Hmg2p and promoting its folding to spare it from degradation. From these data, we propose that INSIGs can be viewed as dedicated chaperones for SSD-containing clients that have been adapted to perform the various regulatory functions so far described.
Results
Yeast Saccharomyces cerevisiae expresses two orthologs of the mammalian INSIG genes, called NSG1 and NSG2. These proteins are significantly diverged from the mammalian homologs (Figure 1), with ∼22% identity throughout. Nevertheless, our studies indicate that they have a specific function in yeast consistent with long-conserved role in controlling the processing of SSD-domain proteins.
Figure 1.

Comparison of human INSIG1 and the two yeast proteins Nsg1p and Nsg2p. Protein sequences of these three proteins were compared by T-Coffee at EMBNET. Macboxshade software was used for graphic depiction. The black sections are identities shared by all three proteins. Dark gray are identities shared by any two, and light gray are similarities.
The yeast HMGR isozyme Hmg2p undergoes regulated degradation mediated by the Hrd1p ubiquitin ligase (Hampton, 2002). We first encountered the NSG genes in a search for high-copy plasmids that stabilize Hmg2p-GFP, employing a colony fluorescence assay (Cronin et al, 2000). From that screen, we recovered a truncated version of HRD1, called ‘hemi-HRD1' that is a general dominant-negative gene for ER-associated degradation (Gardner et al, 2000), and is used as a control in the experiments below. We also identified YNL156c (NSG2), which encodes a multispanning membrane protein, and direct testing of the other homolog YHR133c (NSG1) revealed that it also stabilized Hmg2p-GFP. The seminal studies of INSIG proteins as key factors in sterol regulation allowed us to identify our stabilizing coding regions as distant INSIG homologs (Yang et al, 2002; Sever et al, 2003), and name them accordingly (NSG, pronounced ‘n-sig').
When overexpressed in yeast from the TDH3 promoter, either NSG1 or NSG2 caused significant stabilization of Hmg2p-GFP (Figure 2A). Immunoblotting of strains expressing HA-tagged NSG1 indicated that the use of the TDH3 promoter caused approximately 50- to 100-fold increase of the protein over the levels expressed from the genomic natural promoter (DNS). In the figure, flow cytometry was used to measure Hmg2p-GFP. In each panel, the three histograms are the steady-state levels of Hmg2p-GFP in untreated cells, cells treated with the drug zaragozic acid (ZA) that increases degradation rate by elevating FPP production or cells treated with lovastatin (LOVA) that slows degradation by decreasing FPP production (Gardner and Hampton, 1999b). When compared to empty vector controls (Figure 2A, top panels), the cells overexpressing NSGs showed a noticeable shift of the histograms to the right (brighter cells) and a blunting of the effects of the degradation-enhancing ZA, as expected for Hmg2p-GFP stabilization. The figure also shows the stabilizing action of the general dominant-negative hemi-Hrd1p, and 3HA-tagged NSG1 used in the interaction assays below. The stabilizing effects of all the constructs on Hmg2p-GFP have been confirmed by direct examination of time dependence of fluorescence loss after the addition of cycloheximide (CHX) (DNS). NSG1 and NSG2 also stabilized catalytically active, full-length 1myc-Hmg2p, as shown in Figure 2B, by direct CHX-chase assay followed by myc immunoblotting.
Figure 2.
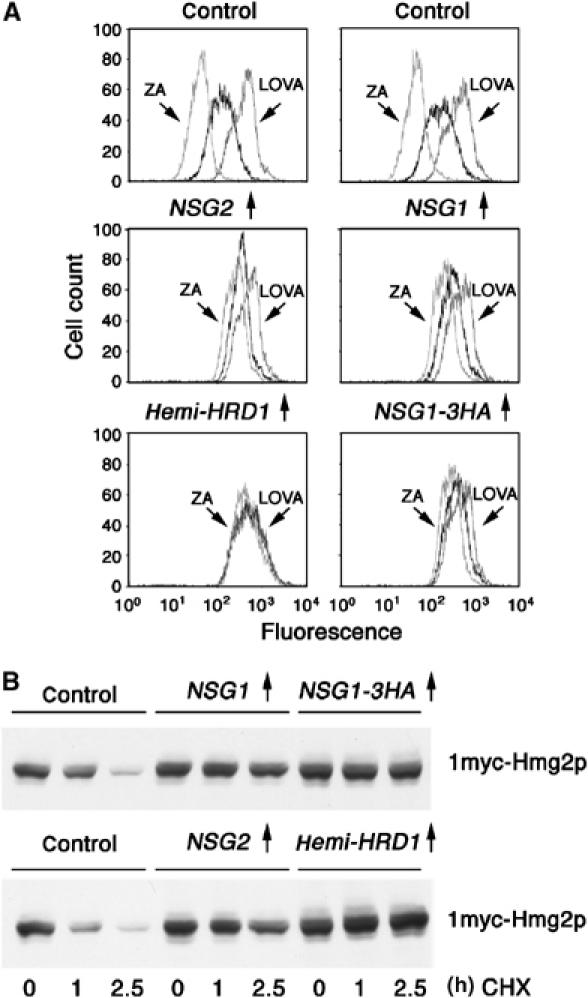
NSG overexpression stabilized Hmg2p-GFP (A) or full-length Hmg2p (B). (A) Log-phase cultures of cells expressing Hmg2p-GFP from the TDH3 promoter were subjected to flow cytometry to evaluate steady-state levels of Hmg2p-GFP fluorescence in the presence of empty vector (‘control'), TDH3-expressed Nsg1p, Nsg2 and Nsg1p-3HA, or the control dominant-negative hemi-Hrd1p. Cells were incubated with no drug, zaragozic acid (ZA) or lovastatin (LOVA) as indicated. Horizontal axis is log fluorescence in arbitrary units, and vertical axis is the number of cells per fluorescent channel. All x-axis in a column are identically aligned to facilitate visual comparison between strains. (B) Log-phase cultures of cells expressing 1myc-Hmg2p from the TDH3 promoter and the indicated proteins were treated with cycloheximide (25 μg/ml) followed by immunoblotting at the indicated times.
To determine if this stabilizing action was specific for Hmg2p, we tested the effects of NSG overexpression on three other ERAD substrates: the misfolded lumenal protein CPY* (Ng et al, 2000), the misfolded, unregulated Hmg2p variant 6myc-Hmg2p-GFP (Hampton et al, 1996; Shearer and Hampton, 2004) and the unstable mutant Sec61-2p. HA-CPY* stability was measured by CHX chase followed by immunoblotting (Figure 3A), in the presence of the indicated expression plasmids or empty vector (‘control'). Neither NSG had any effect on the degradation rate of CPY*, at levels that clearly stabilized Hmg2p included in the same experiment, while the generally acting hemi-Hrd1p did stabilize CPY*. NSG overexpression similarly had no effect on the degradation of 6myc-Hmg2p-GFP, as measured by flow cytometry after the addition of CHX (Figure 3B), in which a time-dependent shift of the histogram to the left indicates degradation of the protein. Finally, we tested the effects of NSGs on the temperature-sensitive phenotype of a sec61-2 strain as a separate, sensitive test of an ERAD defect. Inhibition of ERAD by expression of various dominant inhibitors stabilizes the mutant Sec61-2p protein, allowing growth at normally nonpermissive 35°C temperature (Sommer and Jentsch, 1993; Biederer et al, 1996). Overexpression of NSG2 (Figure 3C, top panel, dilution assay) or NSG1 (Figure 3C, bottom panel, plate assay) had no effect on the temperature sensitivity of our sec61-2 test strain, while overexpression of hemi-Hrd1p suppressed the ts phenotype, allowing robust growth at nonpermissive 35°C temperature (Figure 3C, top panel). Thus, the NSGs had no effect on the stability of three other ERAD substrates.
Figure 3.
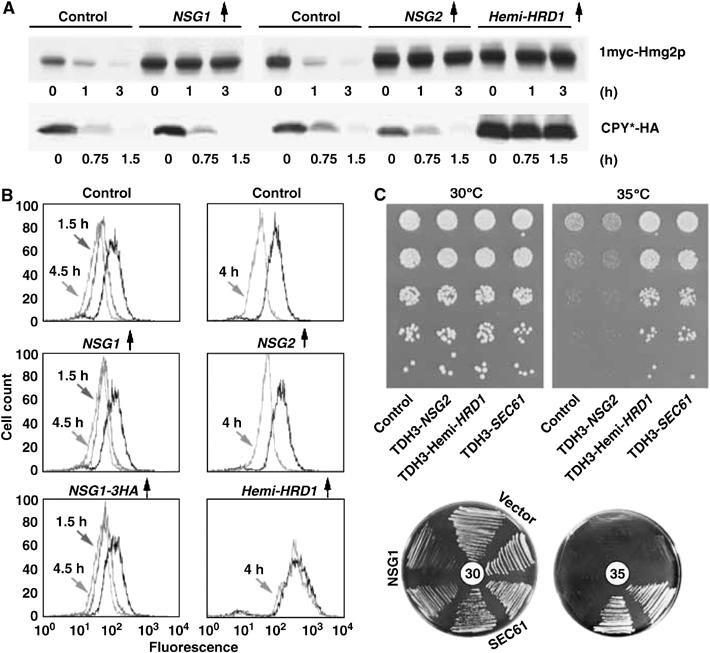
Overexpressed NSGs did not stabilize ER degradation substrates CPY* (A), 6myc-Hmg2p-GFP (B) or Sec61-2 (C). (A) Cells expressing 1myc-Hmg2p (top row) or CPY* (bottom row), along with empty vector (‘control'), TDH3-driven NSG1 and NSG2, or dominant-negative hemi-HRD1 were treated with cycloheximide for the indicated times and subjected to immunoblotting. (B) Log-phase cultures of cells coexpressing 6myc-Hmg2p-GFP with no protein (‘control'), Nsg1p, Nsg2p or hemi-Hrd1p were treated with cycloheximide and assayed for cellular fluorescence at the indicated times after treatment. A leftward shift indicates a loss of fluorescence due to HRD-dependent degradation. (C) Lack of effect of NSG2 (top rectangle) or NSG1 (bottom plates) overexpression on temperature-sensitive growth of sec61-2 mutants. Top panel: the sec61-2 strain harboring empty vector (‘control'), a plasmid expressing TDH3-driven Nsg2p, TDH3-driven hemi-Hrd1p or wild-type Sec61p were tested for growth by dilution assay on solid medium at the permissive 30°C temperature, or the nonpermissive 35°C temperature. Suspensions were serially diluted five-fold and identical samples of each dilution were spotted on agar and allowed to grow. Note the suppression by the general dominant-negative hemi-Hrd1p is as good as that provided by the complementing the SEC61 plasmid. Bottom circles: duplicate sec61-2 strains harboring vector, plasmids expressing TDH3-driven Nsg1p or wild-type Sec61p in the indicated sectors were placed on agar and allowed to grow at 30 or 35°C.
Consistent with this high specificity, neither the loss of both NSGs nor overexpression of either caused any effect on basal or stimulated UPR ER stress response (Supplementary Figure 1), as measured with the sensitive GFP reporter (Patil and Walter, 2001), by flow cytometry as in past studies (Bays et al, 2001b; Cronin et al, 2002).
Only Hmg2p or normally regulated variants such as Hmg2p-GFP are stabilized by elevated levels of Nsg proteins. One explanation for this selectivity would be that Nsg proteins somehow affect the production or action of the FPP-derived signal that controls Hmg2p degradation. To address this issue, we tested the ability of the Nsgs to stabilize Hmg2p in a cod1Δ null strain, in which Hmg2p degradation is unresponsive to the FPP-derived signal (Cronin et al, 2002). Hmg2p-GFP expressed in cod1Δ cells undergoes HRD-dependent degradation, shown here by flow cytometry after the addition of CHX (Figure 4, top panel), but degradation is completely unregulated, such that addition of LOVA has no effect on the steady-state level of the reporter. Despite the lack of regulation in the cod1Δ strain, expression of NSG1 still had a dramatic stabilizing effect on the Hmg2p-GFP degradation rate, as shown by the minimal effect of CHX on GFP fluorescence (Figure 4, middle panel). The bottom panel depicts data from a wild-type strain (as opposed to cod1Δ) that demonstrate the effect of NSG1 in the mutant or wild type are comparable. Thus, the specific action of NSG proteins on Hmg2p operated independently of FPP-mediated regulation.
Figure 4.
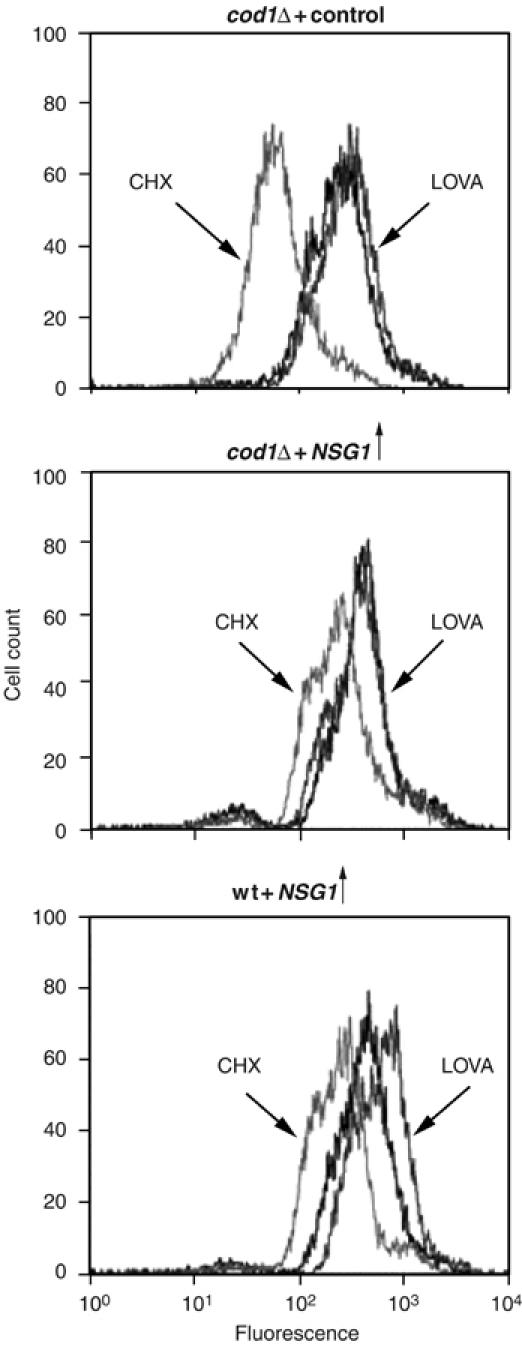
NSG1 stabilized Hmg2p in a regulation-deficient cod1Δ null. cod1Δ null mutant cells without (top panel) or with (middle panel) overexpressed NSG1 were subjected to cycloheximide treatment (50 μg/ml; light gray) or no drug (black) and incubated for 4 h to assess degradation of the Hmg2p-GFP reporter in this mutant background. Separate control cultures were incubated for 4 h with lovastatin (25 μg/ml; dark gray) to confirm the lack of Hmg2p-GFP regulation, shown by the unchanging steady-state levels. The bottom panel shows an otherwise identical wild-type (wt) strain overexpressing NSG1 subjected to the same experiment for comparison of the extent of NSG stabilization. All three panels have aligned horizontal axes to facilitate visual comparison.
As each Nsg protein stabilizes Hmg2p directly, we imagined that they would be ER-localized. Indeed, a complementing GFP-tagged version of Nsg1p showed the typical ‘concentric circle' characteristic of yeast ER, as did 3HA-tagged versions of each NSG coding region expressed from their native promoters tested by immunostaining (DNS). In addition, the newly available analysis of yeast coding regions fused with GFP also indicated that both Nsg proteins were restricted to part or all of the ER (Ghaemmaghami et al, 2003; Huh et al, 2003). Thus, by multiple criteria, both over- and normally expressed Nsg proteins appear to be denizens of the ER membrane. Although Hmg2p and Nsg are both ER proteins, we observed no alteration of Hmg2p-GFP distribution caused by overexpression or loss of NSG proteins (I Flury, S Cronin and R Hampton, personal observations).
We next investigated the action of Nsg proteins in normal yeast cells, when both Nsgs and Hmg2p are at their natural levels. Our studies on Hmg2p degradation usually employ the constitutive, strong glyceraldehyde-3-phosphate dehydrogenase promoter (pTDH3) to drive the expression of Hmg2p. Use of this heterologous promoter allows direct evaluation of Hmg2p degradation by examination of steady-state levels, since regulated degradation is the only control mode possible, facilitating various screens and flow cytometric analysis. In strains expressing high levels of Hmg2p from the TDH3 promoter, the deletion of both NSG genes had little effect, slightly accelerating FPP-regulated Hmg2p degradation. However, when Hmg2p is expressed at native levels, the normally expressed Nsg1p protein plays a central role in controlling Hmg2p stability.
To study Hmg2p at native levels, we produced an in situ gene replacement of the HMG2 locus with the coding region for 1myc-Hmg2p, allowing detection at the low native levels and in the presence of the more abundant Hmg1p. Surprisingly, we found that natively expressed Hmg2p is extremely stable even in the presence of high FPP-derived signal and a functional HRD pathway (Figure 6A). The high stability of natively expressed Hmg2p remained a conundrum until these studies on the NSG proteins were carried out. The data above indicate that THD3-driven Nsg proteins can specifically protect TDH3-driven Hmg2p from HRD-dependent degradation. We reasoned that at native levels of Hmg2p, the naturally produced Nsg proteins might be sufficient to stabilize the Hmg2p, thus causing the observed stability.
Figure 6.
Overexpression of Hmg2p transmembrane domain caused a functional loss of Nsg activity. Strains expressing the native-promoter myc-Hmg2p with the indicated overexpression plasmids (control is empty vector) were compared in cycloheximide chases as in Figures 5 and 6 for regulated stability of native-level myc-Hmg2p. Note that the only myc-tagged protein in these strains is the native promoter expressed myc-Hmg2p.
When the NSG genes are intact, natively expressed 1myc-Hmg2p is quite stable, with a half-life in excess of 4 h (Figure 5A, ‘NSG1NSG2', left group). However, in the presence of the single nsg1Δ, or the nsg1Δnsg2Δ double null, natively expressed Hmg2p was rapidly degraded, as indicated by the lower steady-state levels and its disappearance after the addition of CHX (Figure 5A). The nsg1Δ null alone allowed native Hmg2p to undergo degradation, with no added effect of the nsg1Δnsg2Δ double null. In contrast, the nsg2Δ alone did not affect the stability of native Hmg2p, despite the ability of the Nsg2p protein to stabilize Hmg2p when overexpressed. Thus, it appears that although both NSGs are capable of stabilizing Hmg2p when overexpressed, Nsg1p plays the major role in controlling Hmg2p stability at native levels. Not surprisingly, the presence of the cod1Δ null mutation did not affect the Nsg1-imposed stability of native Hmg2p (DNS), as expected from the lack of need for COD1 in the stabilizing action of NSGs shown above.
Figure 5.
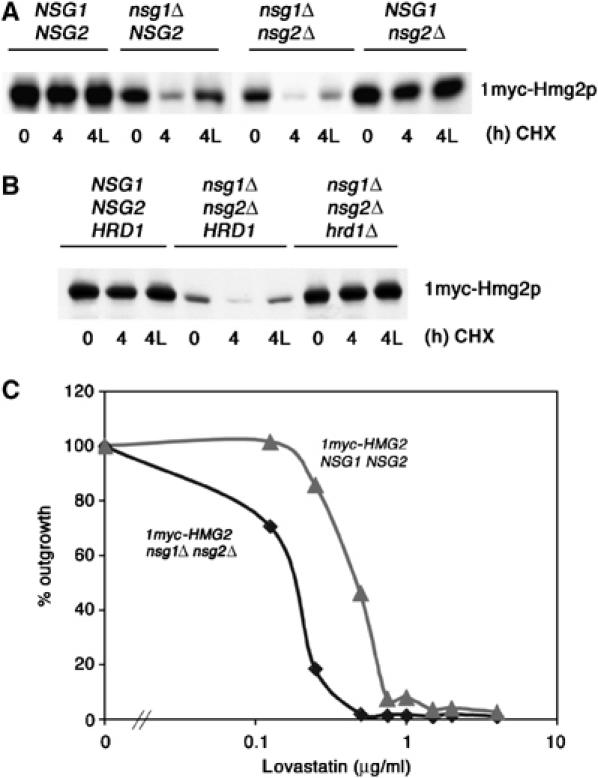
NSG1 functions naturally to limit HRD-dependent, regulated degradation of native Hmg2p. (A) Removal of NSG1 allowed regulated degradation of native Hmg2p. Log-phase cultures of strains expressing native levels of 1myc-Hmg2p from its native promoter with the indicated NSG genotypes (wt, nsg1 null, double null, nsg2 null) were treated for 0 or 4 h with cycloheximide (50 μg/ml) in the presence or absence of lovastatin (25 mg/ml; 4L), and subjected to immunoblotting for myc-Hmg2p. (B) Strains with the indicated NSG and HRD1 genotypes were subjected to the same protocol as in (A) to evaluate HRD dependence of regulated degradation that occurs in the absence of NSGs. (C) Removal of NSGs results in lower levels of native Hmg2p enzyme activity. Strains expressing only native levels of Hmg2p as the sole source of HMGR, with the indicated NSG phenotypes, were compared for sensitivity to lovastatin growth inhibition. Low-density cultures (OD∼0.02) were grown for 49 h in the presence of the indicated concentrations of lovastatin, read for OD600, and normalized to the no-drug culture for each genotype. The nsg null was approximately three times more sensitive to lovastatin.
The degradation of native-level Hmg2p observed in nsgΔ null strains was normally regulated, as indicated by the stabilizing effect of LOVA (Figure 5A, lane ‘4L'). Furthermore, native Hmg2p degradation in the nsgΔ null strain remained HRD dependent: the added presence of the hrd1Δ null in the nsg1Δ nsg2Δ completely stabilized the native-level myc-Hmg2p (Figure 5B), resulting in wild-type steady-state levels and no discernable loss after the addition of CHX.
The stabilization of native Hmg2p by Nsgs had phenotypic consequences. In an HMG2 hmg1Δ strain, Hmg2p is the only HMGR isozyme present; the levels of Hmg2p alone determine cellular levels of HMGR enzyme activity, and thus the sensitivity to the HMGR inhibitor LOVA. To examine if NSG-mediated stabilization of Hmg2p had consequences at the level of enzyme function, we compared the LOVA sensitivity of HMG2 hmg1Δ strains, with and without functional NSG genes. In strains that only expressed Hmg2p, the absence of NSG genes caused a three-fold greater sensitivity to LOVA as indicated by killing curves (Figure 5C). Thus, the presence of Nsg proteins affects the activity of Hmg2p in the cell, altering the enzyme activity levels of this isozyme by controlling stability.
The above studies showed that Nsgs proteins have a direct effect on Hmg2p stability. The simplest model is that Nsgs bind the Hmg2p transmembrane region, which is necessary and sufficient for HRD-dependent ER degradation of this substrate. We have evaluated this model with coexpression studies, in which the effects of sequestering Nsg with excess Hmg2p were evaluated, and with two direct assays of interaction: co-immunoprecipitation and blue native PAGE (BN-PAGE).
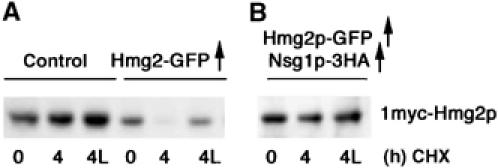
We first examined if excess expression of the Hmg2p transmembrane domain could compete for and sequester Nsg proteins, similar to sequestration studies between SCAP and INSIGs (Sever et al, 2003). Using the native-level myc-Hmg2p strain described above, we evaluated the effects of overexpressing untagged Hmg2p or Hmg2p-GFP. We observed that coexpression of high levels of untagged Hmg2p (DNS) or Hmg2p-GFP (Figure 6A) caused degradation of the small amount of normally stable, myc-tagged Hmg2p expressed from the genomic locus; the resulting degradation was regulated, as indicated by the stabilization caused by the addition of LOVA (Figure 6A, middle group, lane ‘4L'). This effect did not depend on the degradation of the overexpressed protein, since overexpression of Hmg2p or Hmg2p-GFP with stable K6R point mutations (Gardner and Hampton, 1999a) also caused myc-Hmg2p to undergo regulated degradation (DNS). This action of overexpressed Hmg2p on the stability of native levels of myc-Hmg2p could be explained by competition for the limited pool of cellular Nsg1p, effectively lowering its levels enough to allow 1myc-Hmg2p degradation. Consistent with this, addition of an Nsg-overexpressing plasmid to the strains expressing high levels of untagged Hmg2p reimparted stability to the natively expressed myc-Hmg2p (Figure 6, right panel).
We directly tested for Nsg1p–Hmg2p interactions by co-immunoprecipitation and BN-PAGE (Figure 7), using the 3HA-Nsg1p protein shown to specifically stabilize Hmg2p (Figures 2A, B and 3B). We prepared strains that coexpressed 3HA-Nsg1p and 1myc-Hmg2p, each from the strong TDH3 promoter. Detergent-solubilized microsomes were subjected to one-step immunoprecipitation with anti-HA beads, followed by SDS–PAGE and immunoblotting for Nsg1p (HA) or Hmg2p (myc) immunoreactivity. When 3HA-Nsg1p was present, 1myc-Hmg2p was co-precipitated, indicating a physical interaction with the tagged Nsg protein (Figure 7A). When the identical experiment was performed with strains coexpressing only myc-Hmg2p (Nsgs present at genomic levels, untagged), no myc-Hmg2p was detectable in the bead lysates. Similarly, use of untagged coexpressed Nsg1p showed no precipitation of Hmg2p (DNS). The co-precipitation was specific for Hmg2p: when the identical experiment was performed with the 6myc-Hmg2p that is not stabilized by the NSGs in vivo (Figure 2B), no myc immunoreactivity was captured by HA-Nsg precipitation (DNS).
Figure 7.
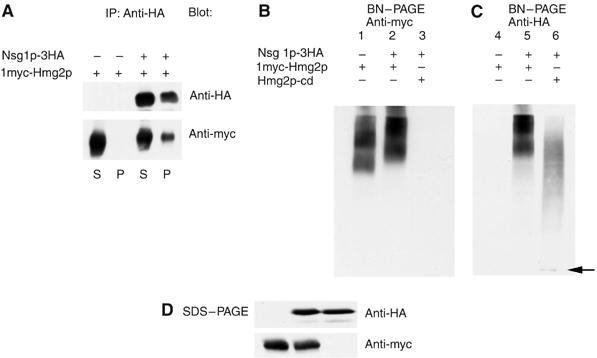
Direct assays of Nsg1p–Hmg2p interaction. (A) Co-precipitation of myc-Hmg2p by Nsg1p-3HA. Nondenaturing detergent lysates of microsomes from cells coexpressing the two tagged proteins or the individual molecules were immunoprecipitated with anti-HA beads as described in Materials and methods. Samples of the supernatants after precipitation, or protein captured by the beads were resolved by SDS–PAGE and immunoblotted for the Nsg1p-3HA or myc-Hmg2p as indicated. (B, C) Blue native PAGE (BN-PAGE) electrophoresis of Nsg1p-3HA and myc-Hmg2p. Strains expressing myc-Hmg2p alone, and strains coexpressing Nsg1p-3HA with full-length myc-Hmg2p or soluble Hmg2p catalytic domain were lysed and subjected to BN-PAGE followed by immunoblotting. Panel B (‘anti-HA') shows the mobility of Nsg1p-3HA in the presence of coexpressed myc-Hmg2p or the soluble catalytic domain. Panel C (‘anti-myc') shows the mobility of myc-Hmg2p in the presence of empty vector or Nsg1p-3HA. To generate the B and C panels, the same nitrocellulose was first probed for HA, then stripped as described in Materials and methods (‘protein degradation and immunoblotting'), and then reprobed for myc. (D) Immunoblotting of identical samples of each of the three lysates to show total levels of Nsg1p-3HA and myc-Hmg2p.
We used nondenaturing BN-PAGE to independently test for Nsg1p–Hmg2p interaction (Dekker et al, 1996). This approach has been employed to demonstrate direct INSIG interactions with SCAP (Yang et al, 2002). Detergent-solubilized microsomes were subject to BN-PAGE followed by immunoblotting for Nsg1p (HA) or Hmg2p (myc). The myc-Hmg2p protein migrated with slower mobility in lysates prepared from cells coexpressing HA-Nsg1p (Figure 7B, lanes 1 and 2). The Hmg2p forms a doublet in this gel system, and the entire doublet was shifted by Nsg1p coexpression. Immunoblotting the same blots for Nsg1p-HA revealed that in lanes with both proteins, the mobility and pattern of the HA-Nsg1p and myc-Hmg2p were identical, indicating they migrate as a complex (Figure 7B and C, lanes 2 and 5). To discern the mobility of Nsg1p in the absence of the coexpressed Hmg2p transmembrane domain, we prepared lysates from a strain expressing HA-Nsg1p and the soluble C-terminal catalytic domain of Hmg2p, called cat-Hmg2p, that provides the essential HMGR activity without the transmembrane domain. 3HA-Nsg1p coexpressed with full-length myc-Hmg2p had a significantly lower mobility when coexpressed with the truncated soluble cat-Hmg2 (Figure 7C, lanes 5 and 6). The Nsg1p immunoreactivity from the lysates coexpressing the soluble Hmg2p catalytic domain had diffuse HA immunoreactivity at lower mobilities and a major discreet band near the bottom of the blot, indicated with an arrow (Figure 7C, lane 6). Parallel SDS–PAGE immunoblots of the lysates indicated that identical amount of the tagged proteins were present in the different lysates. These results indicate that full-length Hmg2p and Nsg1p can from a stable complex that requires the transmembrane domain of Hmg2p.
We finally examined if the presence of Nsg1p had direct effects on Hmg2p in three in vitro assays of Hmg2p degradation: limited proteolysis, in vitro HRD-dependent ubiquitination and Hmg2p-Hrd1p crosslinking. Our previous work indicates that Hmg2p stability is regulated by degradation signals causing Hmg2p to adopt a less folded conformation that enhances HRD pathway recognition. Those studies used a limited proteolysis assay to follow changes in Hmg2p structure in vivo (Shearer and Hampton, 2004), and in vitro (Shearer and Hampton, 2005). Microsomes from cells expressing a normally regulated, lumenally tagged Hmg2p-GFP, called mycL-Hmg2p-GFP, are treated with trypsin for various times, followed by immunoblotting for the protected lumenal tag. High levels of FPP-derived signal cause more rapid proteolysis, whereas chemical chaperones such as glycerol, that stabilize Hmg2p in vivo, cause lower accessibility of Hmg2p and a slower limited proteolysis time course (Shearer and Hampton, 2005).
We used the limited proteolysis assay to directly evaluate the effects of Nsg proteins on Hmg2p structure (Shearer and Hampton, 2004). Microsomes from strains expressing mycL-Hmg2p-GFP with or without coexpressed Nsg1p were prepared and subject to the limited proteolysis assay. The presence of Nsg1p slowed the appearance of the trypsinolysis products without altering the digestion pattern (Figure 8A), in the same manner caused by other stabilizing conditions, such as high levels of chemical chaperones. Thus, Nsg1 changes the folding of Hmg2p in a manner very similar to these agents.
Figure 8.
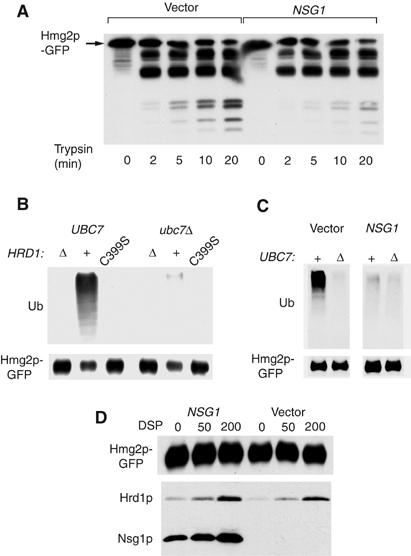
In vitro assays of Nsg1p action on Hmg2p. (A) Effect of coexpressed Nsg1p on limited protection assay of lumenally tagged mycL-Hmg2p. Microsomes of otherwise identical strains expressing the lumenally tagged mycL-Hmg2p with or without added Nsg1p were incubated with trypsin for the indicated times, followed by immunoblotting for the lumenal myc tag. (B, C) In vitro ubiquitination of Hmg2p. In vitro ubiquitination of myc-Hmg2p was evaluated by preparing microsomes of various assay strains, and incubation of the microsomes with cytosol with (UBC7) or without (Δ) Ubc7p, followed by lysis, immunoprecipitation of Hmg2p-GFP and immunoblotting for ubiquitin or GFP as indicated. (B) Hrd1p dependence of in vitro Hmg2p-GFP ubiquitination. Microsomes with Hrd1p (‘+'), no Hrd1p (‘Δ') or dominant-negative C399S Hrd1p were compared in ubiquitination assay. (C) Effect of Nsg1p on in vitro ubiquitination of Hmg2p by Hrd1p. Otherwise identical, strains with or without TDH3-expressed Nsg1p were compared. (D) Hrd1p-3HA crosslinking to Hmg2p-GFP in the presence and absence of Nsg1p. Microsomes from strain coexpressing Hrd1p-3HA and Hmg2p-GFP, with (‘NSG1') or without (‘vector') Nsg1p-3HA, were prepared as in the ubiquitination assay, treated with crosslinker DSP at the indicated concentrations for 1 h, quenched and subject to anti-GFP immunoprecitipation and immunoblotting for GFP, or HA. Note that significant Nsg1p is co-precipitated in the absence of crosslinker.
We also tested the effect of excess Nsg1p on in vitro transfer of ubiquitin to Hmg2p in isolated microsomes, using a recently developed assay characterized in detail elsewhere. In our assay, ubiquitination of Hmg2p in isolated microsomes was Hrd1p dependent, failing to occur in microsomes without Hrd1p, or with the dominant C399S version (Figure 8B). Transfer required the addition of the correct E2-conjugating enzyme Ubc7p (Figure 8B and C). When cytosol containing Ubc7p as added to Hmg2p-containing microsomes, Hrd1p-dependent transfer to Hmg2p is observed (Figure 8C, left panel, ‘+'), whereas cytosol without Ubc7p does not allow transfer (Figure 8C, left panel, ‘Δ'). In this assay, the presence of elevated levels of Nsg caused a striking decrease in HRD-dependent Hmg2p ubiquitination (Figure 8C, right panel), as expected from its in vivo effects on stability, and its in vitro effect on Hmg2p structure.
It appeared that Nsgs stabilizes Hmg2p in a manner similar to chemical chaperones or lowered degradation signal: by promoting the more tightly folded form. To more fully test this idea, we evaluated the effect of Nsg1p on Hmg2p-Hrd1p crosslinking, developed previously to directly study the ligase–substrate interaction (Gardner et al, 2001). In those studies, we discovered that stabilizing conditions such as addition of chemical chaperones or lowering degradation signals have no effect on the Hrd1p–Hmg2p interaction evaluated by this assay. In microsomes with levels of Nsg1p sufficient to block in vitro ubiquitination (Figure 8B) and alter the proteolysis profile (Figure 8A), the crosslinking of Hrd1p to Hmg2p was unaffected either in extent or concentration of DSP required (Figure 8D) Thus, Nsg1p does not inhibit Hmg2p ubiquitination by blocking the interaction of Hrd1p with its substrate. (Note that Nsg1p is readily precipitated in the no-crosslinker lane (‘NSG1', 0 DSP) when Hmg2p-GFP is immunoprecipitated.)
Taken together, these interaction studies indicated that Nsg1p's stabilizing action was analogous to a chemical chaperone: it increased Hmg2p folding by the proteolytic assay, inhibited Hrd1p-dependent ubiquitination, but did not affect the interaction of Hrd1p with its substrate.
Discussion
The above studies showed that the two yeast INSIG homologs specifically inhibit Hmg2p-regulated degradation through direct interaction with the transmembrane region of the protein. Furthermore, Nsg1p functions naturally to promote Hmg2p stability, providing an added layer of regulation to its lipid-mediated degradation. More generally, these studies indicate that the interplay between INSIG proteins and SSD-domain targets is a long-standing one, probably arising before the progenitors of yeast and mammals parted ways. Clearly, INSIG proteins have a broadly conserved role in the action of SSD proteins involving formation of complexes to alter the function of SSD-bearing proteins.
What is less clear is why this added layer of regulation for Hmg2p is present. One possibility is that FPP-mediated degradation of Hmg2p serves to remove elevated levels of Hmg2p induced by transient stimuli, with Nsg1p serving as a ‘brake' to ensure a minimum amount of this isozyme always remains. This could be particularly important when Hmg2p is the predominant form of HMGR, as it appears to be the case in anaerobiosis. Another possibility is that the Nsg–Hmg2p interaction is regulated as well. Perhaps, there are situations or conditions in which the yoke of Nsg control is lifted to allow regulated degradation of Hmg2p. This could include alteration of Nsg levels, or alteration of the Nsg–Hmg2p interaction itself.
How similar are the functions of INSIGs in yeast and mammal? In the simplest terms, the function of Nsg proteins in yeast appears to be the ‘opposite' to that of their mammalian homologs. Mammalian INSIG is required for the sterol-regulated degradation of HMGR, whereas yeast Nsgs prohibit the action of the pathway degradation signal. These disparate actions are both mediated by direct interaction of the INSIG protein with the SSD-containing transmembrane domains.
A unifying idea that comes from considering mammals and yeast is that the INSIGs have a broadly conserved role as binding partners for their SSD-domain-containing effectors, perhaps as stabilizing factors or structural chaperones. The limited proteolysis assay above indicated that the interaction of NSG on Hmg2p results in improved folding. Furthermore, the presence of stabilizing levels of Nsg1p did not affect the Hrd1p–Hmg2p interaction, also consistent with an action on the folding of Hmg2p. Thus, NSGs in yeast appear to be selective chaperones that stabilize the structure of the SSD-containing Hmg2p transmembrane domain. In the case of yeast Hmg2p, this interaction protects the Hmg2p from entry into the HRD quality control pathway by promoting the more stable structure. In mammals, INSIGs function in two other ways: they bind to SCAP in a sterol-dependent manner to cause retention in the ER, and they bind to HMGR in a sterol-dependent manner to cause ubiquitin-mediated degradation. While these three actions sound distinct, they are all familiar ways that chaperones can be adapted to perform distinct functions. In addition to their familiar roles in folding, ER-resident Hsp70 protein BiP (in mammals; Kar2p in yeast) are employed to promote retention of proteins in that organelle (Suzuki et al, 1991; Jorgensen et al, 2000), as are the chaperones calnexin and calreticulin (Zhang et al, 1997). A precedent for chaperones mediating specific ubiquitination is set from a recent work on CHIP and Hsp70 (Murata et al, 2001; Cyr et al, 2002; Xu et al, 2002). CHIP has ubiquitin ligase activity, and binds to Hsp70- and Hsp90-type chaperones, thus forming a ubiquitin ligase that targets these chaperones' client proteins for ubiquitination. In the same manner, it has recently been shown that INSIG recruits the transmembrane ER ubiquitin ligase gp78 (one of two Hrd1p homologs) to the HMGR–INSIG complex, allowing ubiquitination of HMGR in mammals (Song et al, 2005). Thus, improved folding, organelle retention and coordinating ubiquitination are all functions that have been adapted from chaperone-mediated recognition of proteins. The INSIGs have been similarly adapted for the numerous ways that they affect SSD-containing membrane proteins.
In the future, it will be important to discover the extent to which known features of the Nsg–Hmg2p interaction is conserved. The Hmg2p protein has a clear SSD domain, and we are actively performing an analysis of the role of this motif in the binding and stabilizing action of the NSG proteins. Another related question is the possible role of sterols in the interaction of yeast Nsg with its target. In both SCAP and mammalian HMGR, sterols play an integral part in their interactions with INSIGs. It may be that the abundant sterols present in aerobic yeast are required for this interaction, but that remains to be tested. It is not yet clear why the Nsgs regulate Hmg2p stability, and possibly the measurement of sterol levels is integrated into this action in yeast. Finally, the other functions or targets of the NSGs will be important to understand as well. From our competition experiments, it appears that the stable Hmg1p isozyme can also bind NSGs, although the functional significance of this interaction is not clear. Furthermore, the complex mobility of the Nsg1p in the BN-PAGE experiments implies that it may have other binding partners, whose identities may lead to new ideas about the actions of this family of proteins.
Materials and methods
LOVA and ZA were generously provided by Merck (Rahway, NJ). Anti-myc antibody was used as cell culture supernatant obtained by growing the 9E10 hybridoma cell line (CRL 1729 American Type Culture Collection) in RPMI1640 culture medium (GIBCO BRL, Grand Island, NY) with 10% fetal calf serum and supplements. All other reagents were purchased from commercial sources as described in detail in the Supplementary data.
Plasmids and DNA
Plasmids were prepared by standard techniques. A complete description of cloning details for all plasmids with pRH numbers for request is available in the Supplementary data, or by request.
Yeast culture and strains
Yeast strains were grown in minimal media (0.67% yeast nitrogen base; Difco, Detroit, MI) with glucose and the appropriate supplements, and transformed with plasmids by LiOAc as described previously (Hampton and Rine, 1994). All strains (except for those with sec61-2 mutation) were isogenic with the standard Hampton laboratory strain based on S288C, as used in other studies (e.g. Bays et al, 2001a); experimental strains differed from each other only by the addition of expression plasmids or empty vectors, and/or disruption of one or both NSG genes as indicated. The strains with the sec61-2 ts mutation (Figure 3C) were generated from a parent strain obtained from the laboratory of R Schekman (Berkeley, CA). A full table of all strain genotypes can be obtained from the Supplementary data, or our laboratory upon request.
Flow cytometry of yeast cells
Flow microfluorimetry was used to analyze GFP fluorescence and was carried out as described previously (Gardner and Hampton, 1999a, 1999b; Cronin et al, 2000). Briefly, strains were grown to early log phase in minimal media. After the addition of drugs, cultures were incubated for the indicated amount of time (usually 4 h) at 30°C before analysis on a BD Biosciences FACSCalibur™ flow cytometer with Cell Quest software. Data from 10 000 cells were used for each histogram.
Protein degradation assay and immunoblotting
CHX-chase assays to analyze regulated degradation were performed as described previously (Gardner and Hampton, 1999a). Briefly, log-phase cultures were treated with 50 μg/ml CHX and with or without LOVA (25 μg/ml), as indicated. After various incubation times, cells were lysed and protein degradation was evaluated by SDS–PAGE and immunoblotting. Myc- or HA-tagged proteins were detected using monoclonal antibodies. In the BN-PAGE experiment, the nitrocellulose blot (Figure 8C and D) was first probed with anti-HA, stripped, and probed with anti-myc, to insure that mobilities were directly comparable. Stripping was accomplished by incubating the blot in ∼30 ml stripping buffer (100 mM B-mercaptoethanol, 2% SDS, 62.5 mM Tris–HCl, pH 6.8) for 30 min at 55°C in a zip lock bag, followed by 3 × 15 min washes with 100 ml PBS+0.5% Tween 80. The blot was then subjected to the next round of immunoblotting.
Preparation of microsomal membranes
Microsomal membranes were prepared essentially as described previously (Fraering et al, 2001), with the modification that cells were grown at 30°C. Briefly, pelleted cells were resuspended in buffer TMMDP (50 mM Tris–HCl, pH 7.4, 1 mM MgCl2, 1 mM MnCl2, 1 mM DTT, 1 mM PMSF, 1 μg/ml chymostatin) and lysed by agitation with glass beads at 4°C. The homogenate was centrifuged 8 min at 3000 g to remove unbroken cells, and the supernatant was centrifuged for 40 min at 20 000 g. The membrane pellet was washed and the microsomal pellet was resuspended at 200 μl/1000 OD equivalents in membrane buffer (35% glycerol, 50 mM Tris–HCl, pH 7.4, 1 mM MgCl2, 1 mM MnCl2, 1 mM DTT, 1 mM PMSF, 1 μg/ml of each of chymostatin, pepstatin, antipain and leupeptin) and frozen at −80°C.
Solubilization of membrane proteins
After thawing the microsomal membranes, 300 μl TM buffer (0.2 M mannitol, 0.1 M NaCl, 50 mM Tris–HCl, pH 7.4, 1 mM MgCl2, 1 mM CaCl2, 1 mM MnCl2, 1 mM DTT, 1 mM PMSF plus the protease inhibitor mix described for the membrane buffer) was added to every 100 μl of membrane suspension. The samples were then incubated on a nutator at room temperature for 45 min with 0.2 mg/ml DNase I (3000 U/mg; Sigma, St Louis, MO). Glycerol was adjusted to 10%. The protein concentration was determined by Bradford and adjusted to 7 μg/ml with GTM buffer (TM buffer+10% glycerol). For solubilization of membrane proteins, digitonin (recrystallized; Sigma, St Louis, MO or Fluka AG, Buchs, Switzerland) was added at a final concentration of 1.5%, and 6-aminocaproic acid was added to a final concentration of 620 mM. Samples were incubated on a nutator at 4°C for 1 h. Insoluble material was removed by centrifugation at 100 000 g at 4°C for 45 min. The concentration of solubilized proteins was determined and samples were shock frozen in liquid nitrogen and stored at −80°C.
Blue native PAGE
Buffers and gel composition were used essentially as described (Fraering et al, 2001). Briefly, solubilized proteins were diluted in GTM buffer containing 525 mM 6-aminocaproic acid and 1.5% digitonin. A 0.15 × volume of sample buffer (100 mM Tris–HCl, pH 7.5, 500 mM 6-aminocaproic acid, 5% Serva Blue G) was added. Samples were mixed gently and loaded onto 5–18% polyacrylamide gels with a 4% stacking gel. Electrophoresis was carried out at 4°C. For immunoblotting, proteins were transferred onto PVDF membranes at 4°C in transfer buffer (25 mM Tris, 192 mM glycine, 10% methanol) using a tank blotting system, destained and probed with anti-myc or anti-HA antibodies.
Native co-immunoprecipitation
Native co-immunoprecipitation was performed using digitonin-solubilized microsomal fractions that were prepared and solubilized as for BN-PAGE. The volume of solubilized membranes corresponding to 200 μg protein from each experimental strain was diluted with two volumes of TM buffer to adjust the digitonin concentration to 0.5%. The lysate was mixed with 20 μl anti-HA beads (HA.11 Monoclonal Antibody, Affinity Matrix; Covance Inc., Berkeley, CA) and 100 μl agarose slurry. Samples were incubated on a nutator at 4°C for 2 h. The supernatant was carefully removed, and beads were harvested by centrifugation and washed three times with TM buffer. Proteins were removed from the beads by the addition of 100 μl 2 × USB and incubation at 55°C for 10 min. Samples corresponding to unbound fraction (supernatant; 10 μg protein loaded) or immunoprecipitated proteins (IP; 50 μg protein loaded) were analyzed by SDS–PAGE and immunoblotting.
Limited proteolysis assay
Limited proteolysis to evaluate Hmg2p conformation was carried out as described previously (Shearer and Hampton, 2004). Briefly, right-side out microsomes from strains expressing lumenally myc-tagged Hmg2p were prepared, resuspended in XL buffer (1.2 M sorbitol, 5 mM EDTA, 0.1 M KH2PO4/K2HPO4, pH 7.5) and incubated with trypsin for the times indicated at 30°C. Time points were then evaluated by 14% SDS–PAGE and myc immunoblotting.
In vitro ubiquitination of Hmg2p
A detailed procedure for this assay is described in the Supplementary data and will be characterized fully elsewhere. Briefly, microsomes from a ubc7Δ null yeast strain expressing Hmg2p-GFP and Hrd1p, from the TDH3 promoter, were prepared and resuspended in B88 buffer (20 mM HEPES, pH 6.8, 250 mM sorbitol 150 mM KOAc, 5 mM MgOAc, 1 mM DTT and protease inhibitors). For each experimental condition (presence of NSG1, absence of HRD1, etc.), a set of microsomes was identically prepared. Cytosol from a Ubc7p-overexpressing, hrd1Δ null strain or control ubc7Δ null cytosol was prepared by freeze–thaw lysis and ultracentrifugation.
The ubiquitination assay was initiated by the addition of microsomes, cytosol and ATP, followed by incubation at 30°C, typically for an hour. The reaction is terminated by the addition of 2 × sample buffer, followed by the addition of IP buffer, and subsequent immunoprecipitation of Hmg2p and immunblotting for Hmg2p-GFP or ubiquitin with appropriate monoclonal antibodies.
In vitro crosslinking of Hmg2p and Hrd1p
Crosslinking of Hmg2p and Hrd1p was assayed using the method described previously for cells (Gardner et al, 2001), adapted for in vitro microsomes. Microsomes from strains expressing Hmg2p-GFP and HA-Hrd1p from the TDH3 promoter, with or without TDH3-driven Nsg1-3HA, were prepared in the same manner as the in vitro ubiquitination assay, except that B88 buffer (with HEPES, instead of TRIS) was used instead of the Tris-containing MF buffer. Microsomes were resuspended in cytosol prepared as in the ubiquitination assay, DSP was added to the indicated concentrations, allowed to incubate at 30°C for 1 h, quenched with hydroxylamine, lysed, immunoprecipitated and immunoblotted as described for the cellular crosslinking assay.
Supplementary Material
Supplemental Results
Acknowledgments
We thank the members of the Hampton lab for general and specific support. We thank Randy Sheckman (UC Berkeley, CA) and Davis Ng (National University of Singapore) for strains and plasmids, and Robert Rickert (Burnham Institute, San Diego, CA) for use of the FACSCalibur™ flow cytometer. RYH wishes further to acknowledge Christopher Hager for omnidirectional curiosity. This work was supported by NIH (NIDDK) Grant #GM51996-06, an AHA Established Investigator Award, a Swiss Science Foundation Postdoctoral Fellowship to IF and NIH minority supplement (BIORGMS) to RG.
References
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, Hampton RY (2001a) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol 3: 24–29 [DOI] [PubMed] [Google Scholar]
- Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, Hampton RY (2001b) HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Mol Biol Cell 12: 4114–4128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Volkwein C, Sommer T (1996) Degradation of subunits of the Sec61p complex, an integral component of the ER membrane, by the ubiquitin proteasome pathway. EMBO J 15: 2069–2076 [PMC free article] [PubMed] [Google Scholar]
- Cronin SR, Khoury A, Ferry DK, Hampton RY (2000) Regulation of HMG-CoA reductase degradation requires the P-type ATPase Cod1p/Spf1p. J Cell Biol 148: 915–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin SR, Rao R, Hampton RY (2002) Cod1p/Spf1p is a P-type ATPase involved in ER function and Ca2+ homeostasis. J Cell Biol 157: 1017–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyr DM, Hohfeld J, Patterson C (2002) Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem Sci 27: 368–375 [DOI] [PubMed] [Google Scholar]
- Dekker PJ, Muller H, Rassow J, Pfanner N (1996) Characterization of the preprotein translocase of the outer mitochondrial membrane by blue native electrophoresis. Biol Chem 377: 535–538 [PubMed] [Google Scholar]
- Fraering P, Imhof I, Meyer U, Strub JM, van Dorsselaer A, Vionnet C, Conzelmann A (2001) The GPI transamidase complex of Saccharomyces cerevisiae contains Gaa1p, Gpi8p, and Gpi16p. Mol Biol Cell 12: 3295–3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Hampton RY (1999a) A ‘distributed degron' allows regulated entry into the ER degradation pathway. EMBO J 18: 5994–6004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Hampton RY (1999b) A highly conserved signal controls degradation of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase in eukaryotes. J Biol Chem 274: 31671–31678 [DOI] [PubMed] [Google Scholar]
- Gardner RG, Shearer AG, Hampton RY (2001) In vivo action of the hrd ubiquitin ligase complex: mechanisms of endoplasmic reticulum quality control and sterol regulation. Mol Cell Biol 21: 4276–4291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY (2000) Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol 151: 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS (2003) Global analysis of protein expression in yeast. Nature 425: 737–741 [DOI] [PubMed] [Google Scholar]
- Hampton RY (2002) Proteolysis and sterol regulation. Annu Rev Cell Dev Biol 18: 345–378 [DOI] [PubMed] [Google Scholar]
- Hampton RY, Gardner RG, Rine J (1996) Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell 7: 2029–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Rine J (1994) Regulated degradation of HMG-CoA reductase, an integral membrane protein of the endoplasmic reticulum, in yeast. J Cell Biol 125: 299–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK (2003) Global analysis of protein localization in budding yeast. Nature 425: 686–691 [DOI] [PubMed] [Google Scholar]
- Jorgensen MM, Jensen ON, Holst HU, Hansen JJ, Corydon TJ, Bross P, Bolund L, Gregersen N (2000) Grp78 is involved in retention of mutant low density lipoprotein receptor protein in the endoplasmic reticulum. J Biol Chem 275: 33861–33868 [DOI] [PubMed] [Google Scholar]
- Kuwabara PE, Labouesse M (2002) The sterol-sensing domain: multiple families, a unique role? Trends Genet 18: 193–201 [DOI] [PubMed] [Google Scholar]
- Murata S, Minami Y, Minami M, Chiba T, Tanaka K (2001) CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep 2: 1133–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng DT, Spear ED, Walter P (2000) The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J Cell Biol 150: 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohturfft A, DeBose-Boyd RA, Scheek S, Goldstein JL, Brown MS (1999) Sterols regulate cycling of SREBP cleavage-activating protein (SCAP) between endoplasmic reticulum and Golgi. Proc Natl Acad Sci USA 96: 11235–11240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil C, Walter P (2001) Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol 13: 349–355 [DOI] [PubMed] [Google Scholar]
- Sever N, Yang T, Brown MS, Goldstein JL, DeBose-Boyd RA (2003) Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol Cell 11: 25–33 [DOI] [PubMed] [Google Scholar]
- Shearer AG, Hampton RY (2004) Structural Control of Endoplasmic Reticulum-associated Degradation: effect of chemical chaperones on 3-hydroxy-3-methylglutaryl-CoA reductase. J Biol Chem 279: 188–196 [DOI] [PubMed] [Google Scholar]
- Shearer AG, Hampton RY (2005) Lipid-mediated, reversible misfolding of a sterol-sensing domain protein. EMBO J 24: 149–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer T, Jentsch S (1993) A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature 365: 176–179 [DOI] [PubMed] [Google Scholar]
- Song BL, DeBose-Boyd RA (2004) Ubiquitination of 3-hydroxy-3-methylglutaryl-CoA reductase in permeabilized cells mediated by cytosolic E1 and a putative membrane-bound ubiquitin ligase. J Biol Chem 279: 28798–28806 [DOI] [PubMed] [Google Scholar]
- Song BL, Sever N, DeBose-Boyd RA (2005) Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell 19: 829–840 [DOI] [PubMed] [Google Scholar]
- Suzuki CK, Bonifacino JS, Lin AY, Davis MM, Klausner RD (1991) Regulating the retention of T-cell receptor alpha chain variants within the endoplasmic reticulum: Ca(2+)-dependent association with BiP. J Cell Biol 114: 189–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L (2002) Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci USA 99: 12847–12852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS (2002) Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110: 489–500 [DOI] [PubMed] [Google Scholar]
- Zhang JX, Braakman I, Matlack KE, Helenius A (1997) Quality control in the secretory pathway: the role of calreticulin, calnexin and BiP in the retention of glycoproteins with C-terminal truncations. Mol Biol Cell 8: 1943–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Results