

Abstract
The crystal structure of subunit F of vacuole-type ATPase/synthase (prokaryotic V-ATPase) was determined to of 2.2 Å resolution. The subunit reveals unexpected structural similarity to the response regulator proteins that include the Escherichia coli chemotaxis response regulator CheY. The structure was successfully placed into the low-resolution EM structure of the prokaryotic holo-V-ATPase at a location indicated by the results of crosslinking experiments. The crystal structure, together with the single-molecule analysis using fluorescence resonance energy transfer, showed that the subunit F exhibits two conformations, a ‘retracted' form in the absence and an ‘extended' form in the presence of ATP. Our results postulated that the subunit F is a regulatory subunit in the V-ATPase.
Keywords: ATP synthase, CheY, crystal structure, rotary motor, V-ATPase
Introduction
The vacuole-type ATPase/synthases (V-ATPases) are commonly found in many organisms involved in a variety of physiological processes (Nishi and Forgac, 2002). V-ATPases in eukaryotic cells (eukaryotic V-ATPases) translocate protons across the membrane consuming ATP. They reside within intracellular compartments, including endosomes, lysosomes, and secretory vesicles, and within plasma membranes of certain cells including renal intercalated cells, osteoclasts, and macrophages. V-ATPase and F-type ATPase/synthase (F-ATPase) are evolutionarily related and share a similar rotary mechanism, coupling ATP synthesis/hydrolysis and proton translocation across the membrane (Boyer, 1993; Forgac, 2000; Yoshida et al, 2001; Imamura et al, 2003; Yokoyama et al, 2003b). However, these two types of ATPases also show significant differences. For example, reversible association/dissociation of the V1 domain (soluble) and the V0 domain (membrane bound) in eukaryotic V-ATPase is a unique regulatory mechanism not found in the F-ATPase (Parra and Kane, 1998; Kane and Parra, 2000; Nishi and Forgac, 2002). The subunit composition and structure in the stalk region of V-ATPase, which connects the V0 and V1 domains, is suggested to be significantly different from that of the F-ATPase (Iwata et al, 2004) (Figure 1A). Therefore, this region is possibly responsible for the association/dissociation of the complex.
Figure 1.
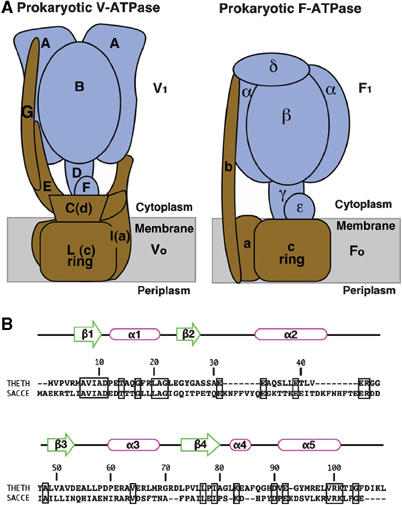
(A) Schematic models of the prokaryotic V-ATPase from T. thermophilus and the prokaryotic F-ATPase from E. coli. The V1 and F1 domains are colored in blue, and the F0 and V0 domains are in brown. (B) Sequence alignment of subunit F from the prokaryote T. thermophilus and the eukaryotic yeast Saccharomyces cerevisiae, with secondary structure from our T. thermophilus structure. The sequence alignment was performed using program water of EMBOSS (Rice et al, 2000). Identical residues in this alignment are enclosed by thick rectangles. Identity 23.5%, similarity 40.9%, and gaps 18.3% were shown in this alignment. Sequence numbering is for T. thermophilus. THETH, T. thermophilus; SACCE, S. cerevisiae.
Eukaryotic V-ATPase-like proteins are found in some prokaryotic cell membranes and are here referred to as prokaryotic V-ATPases (Yokoyama et al, 1990, 1994; Murata et al, 1997). Bacterial V-ATPases, particularly those from archaea, are sometimes also called A-type ATPases (A-ATPase) (Müller and Grüber, 2003). The prokaryotic V-ATPase from Thermus thermophilus, which lacks an F-ATPase, is solely responsible for aerobic ATP synthesis (Yokoyama et al, 1998). The T. thermophilus prokaryotic V-ATPase is composed of nine different subunits that are arranged within the atp operon in the order G–I–L–E–C–F–A–B–D, with gene products of 13, 71, 8, 20, 35, 12, 64, 54, and 25 kDa molecular weight, respectively (Yokoyama et al, 2000, 2003a). The ATPase-active V1 domain is composed of four subunits with a stoichiometry of A3B3D1F1. The V0 domain, which is involved in proton translocation across the membrane, is an assembly consisting of subunits G, I, L, E, and C (Yokoyama et al, 2003a). Each T. thermophilus prokaryotic V-ATPase subunit shows a sequence similarity to its eukaryotic counterpart. Subunit C, an essential component, shows rather low (18%) but significant sequence similarity to subunit D of the yeast enzyme (Yokoyama et al, 2003a; Iwata et al, 2004). This subunit is part of the V-ATPase central stalk (Iwata et al, 2004), and has no counterpart in F-ATPases (Figure 1A). Subunit F, which is also known as Vma7p after the name of the yeast protein, is important not only for the ATPase activity of the V-type enzyme (Imamura et al, 2004) but also for the association of the V1 domain with the V0 domain (Graham et al, 1994). Subunit F of the T. thermophilus prokaryotic V-ATPase also shows an apparent sequence similarity to its eukaryotic counterpart (Yokoyama et al, 2003a) (Figure 1B). For instance, the T. thermophilus subunit F shows 23.5% identity and 40.9% similarity to its yeast counterpart (Figure 1B). Although subunit F had been proposed to have a similar function and structure as that of the ɛ subunit, a regulatory subunit of F-type ATPase, the structure and function of subunit F had not been well understood up to now.
Here, we present the 2.2 Å resolution structure of protein F, a subunit from the T. thermophilus prokaryotic V-ATPase, and discuss its possible function based on the crosslinking experiments and single-molecule analysis using fluorescence resonance energy transfer (FRET).
Results and discussion
Subunit F structure
The structure of subunit F was solved by multiple anomalous dispersion (MAD) using a Se-Met-containing protein, and was refined to 2.2 Å resolution (see Materials and methods). There are six subunit F molecules with 109 amino-acid residues each in the asymmetric unit. Data collection and refinement statistics are summarized in Table I. An example of typical electron density found in the map is shown in Figure 2A. In the crystals, the molecules exist as a dodecamer that is composed of two asymmetric-unit hexamers related by a crystallographic two-fold axis. The dodecamer can be split into six dimers that are almost identical. These are domain-swapped dimers of monomers in the extended form, which are related by a noncrystallographic two-fold axis (Figure 2B and C). This dimer has an elongated shape with dimensions of 30 × 35 × 70 Å. The N-terminal domain of the monomer (residues 1–70) is composed of an α/β fold with three β strands (β1–3) and three α-helices (α1–3). It is connected by a flexible loop (residues 71–74) (Figure 2B) to the C-terminal domain (residues 75–109), composed of one β strand (β4) and one short (α4) and one long (α5) α helix. The density for the flexible loop (residues 71–74) is more disordered than in other regions. The first three strands (residues 5–11 (β1), 24–28 (β2), and 48–53 (β3)) from one monomer and one strand (residues 74–79 (β4)) from another monomer form a β sheet with a parallel conformation. The N-terminal domain (residues 1–70) of one monomer in the dimer forms a globular fold together with the C-terminal domain (residues 75–109) of another monomer. It has been suggested that the subunit stoichiometry of V1 complex is A3B3D1F1 (Murata et al, 1999; Xu et al, 1999). Single-pair FRET experiments also strongly support this idea as described later. Thus, it is probable that the dimer in the crystal is a crystallization artifact. However, this still leaves the possibility that the monomeric subunit F in the prokaryotic V-ATPase complex either exists in the extended form (extended subunit F; left in Figure 2D) found in the crystal or in the retracted form (retracted subunit F; right in Figure 2D), where the complex is formed by the N-terminal domain (residues 1–70, α1–3 and β1–3) from one monomer (colored in green in Figure 2B and C) and the C-terminal domain (residues 75–109, α4–5 and β4) from the other monomer (colored in magenta in Figure 2B and C). These conformations could be interchangeable depending on the conformation of the flexible loop (residues 71–74). As will be discussed below, the single pair FRET study shows that subunit F exists mainly in the retracted form in the complex, but can undergo a conformational change to the extended form during ATP hydrolysis.
Table 1.
Summary of data collection and phase determination
| Native | Se-Met (peak) | Se-Met (inflection) | Se-Met (remote) | ||||
|---|---|---|---|---|---|---|---|
| Resolution range (Å) | 26–2.2 | 30–3.0 | 30–3.0 | 30–3.0 | |||
| Wavelength (Å) | 0.9790 | 0.9794 | 0.9796 | 0.9724 | |||
| Number of reflections | |||||||
| Overall | 147 450 | 110 466 | 110 096 | 104 224 | |||
| Unique | 38 603 | 15 719 | 15 675 | 15 518 | |||
| Completenessa (%) | 99.6 (99.9) | 100 (100) | 100 (99.9) | 100 (100) | |||
| Mean I/σ(I)a | 25.3 (2.4) | 28.8 (5.5) | 29.8 (2.9) | 28.4 (5.3) | |||
| Rmergea,b (%) | 6.5 (47.4) | 6.3 (46.8) | 7.0 (89.7) | 6.8 (40.7) | |||
| Heavy atom sites | 12 | ||||||
| Rcullisc | 0.79 | 0.78 | |||||
| X-ray source | X06SA/SLS | ID14-EH4/ESRF | ID14-EH4/ESRF | ID14-EH4/ESRF | |||
| Phasing statistics | |||||||
| Resolution (Å) | 14.55 | 8.11 | 5.62 | 4.30 | 3.48 | 2.93 | Total |
| Reflections | 85 | 503 | 1306 | 2486 | 4047 | 4859 | 13 286 |
| Figure of merit | 0.67 | 0.62 | 0.65 | 0.58 | 0.48 | 0.32 | 0.47 |
| aValues for the highest resolution shell are given in parentheses. | |||||||
| bRmerge=∑h∑f∣Ii(h)−〈I(h)〉∣/∑h∑i〈I(h)〉. | |||||||
| cRcullis=∑∣∣FPH–FP∣–∣FH(calc)∣∣/∑∣FPH–FP∣. FPH and FP are defined above, and FH(calc) is the calculated heavy atom structure factor. Summation was carried out by using centric reflections only. | |||||||
Figure 2.
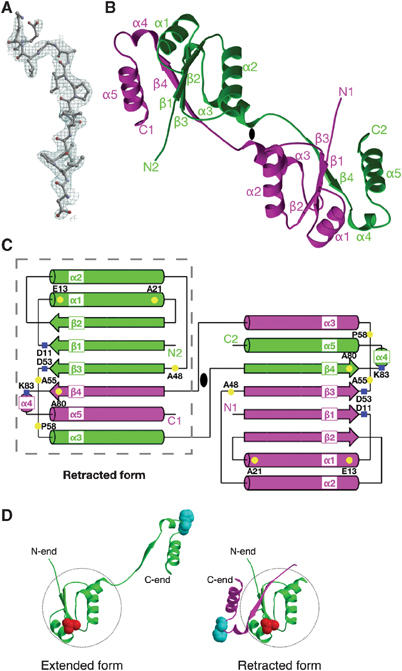
Structure of subunit F from the T. thermophilus V-ATPase. (A) 2Fo–Fc electron density map from Asp-72 to Glu-84 is shown at a contour level of 1.5σ. (B) Ribbon diagram of subunit F related by a two-fold axis. One monomer is colored in magenta, and the other is colored in green. The two-fold axis is represented as a black ellipse. (C) Schematic representation of subunit F colored as in (B). The residues that were mutated in the crosslinking experiments (Figure 5) are highlighted in yellow. Residues Asp-11, Asp-53 and Lys-83 are highlighted in blue. The structure of the retracted form is enclosed by the gray dashed line. This schematic representation was made using TopDraw (Bond, 2003). (D) The structures of the extended (left) and retracted (right) forms of the subunit F monomer. The N-terminal domain of the monomer (residues 3–70) is enclosed by the gray dashed circle. The extended form (left) is colored green. The retracted form (right) is colored green (residues 1–69) and magenta (residues 70–109 from another monomer). The residues A55 (colored red) and H89 (colored cyan) for FRET experiments (Figure 4) are represented by spheres. All figures with molecules and densities were prepared with Molscript (Kraulis, 1991), Bobscript (Esnouf, 1997), and Raster3D (Merritt and Murphy, 1994) unless noted otherwise.
Comparison of structures of subunit F and CheY
The retracted form of subunit F has a flat spherical shape with dimensions of 30 × 35 × 40 Å. It has a four-stranded parallel β-sheet (β1–4) as a core that is surrounded by four long and one short α helices (α1–α5). The last β strand (β4) and the last two helices (α4 and 5) are detached from the rest of the structure in the extended subunit F structure as discussed above (Figure 2C).
The structure of the retracted subunit F was shown to have a striking similarity to the structure of CheY (Stock et al, 1989; Lee et al, 2001a, 2001b) and related regulator proteins (for example, Spo0A) (Lewis et al, 2000) using the Dali database (Holm and Sander, 1996), although there is no apparent sequence similarity between subunit F and these regulator proteins. CheY is the chemotaxis regulator protein from Escherichia coli that is activated through the phosphorylation of an Asp-57 residue by the CheA protein (Falke et al, 1997; Lee et al, 2001a). It is then inactivated through self-dephosphorylation or by the CheZ protein (Silversmith et al, 2003). In its activated state, phospho-CheY interacts with the flagella motor subunit FliM, causing a reversal in the direction of rotation from counter-clockwise to clockwise, which in turn causes the cell to tumble. Figure 3A and B compares the structures of CheY and the retracted subunit F. The two structures can be superimposed with a root-mean-square (r.m.s.) deviation of 2.0 Å for 49 Cα atoms. In the structure of CheY, strand β5 and helix α4, which form the binding site for FliM, are inserted after strand β4 (Figures 2C and 3A).
Figure 3.
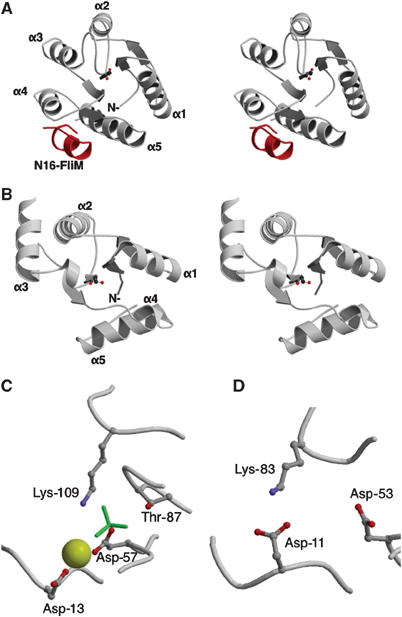
Structural comparison between subunit F and CheY. (A) Stereo view of the structure of CheY with the first 16 residues of FliM (N16-FliM, colored in red). The CheY (PDB ID 1F4V; Lee et al, 2001b) structure is shown with the active site directed towards the reader. The active site residue Asp-57 is shown by a ball-and-stick representation. (B) Stereo view of the subunit F structure (retracted subunit F) is shown with the equivalent site Asp-53, which is shown by a ball-and-stick representation. (C) Close-up of CheY active site. The active site of CheY consists of four residues (Asp-13, Asp-57, Thr-87, and Lys-109) shown by a ball-and-stick representation. The BeF3− and Mg2+ molecules are colored in green and yellow, respectively. (D) Close-up of the equivalent active site of subunit F. The three equivalent residues (Asp-11, Asp-53, and Lys-83) are shown by a ball-and-stick representation.
Another interesting similarity is observed between the phosphorylation site of CheY (Asp-57 and surroundings) and the equivalent site on subunit F. The structure of the CheY phosphorylation site has been studied using a BeF3− activated protein (Lee et al, 2001a, 2001b). In this structure, Asp-57 binds BeF3− and an Mg2+ ion, which are surrounded by residues Asp-13, Thr-87, and Lys-109 (Figure 3C). The activation signal induced by the phosphorylation of Asp-57 can be transmitted to the FliM-binding site through residues Thr-87 and Lys-109. In the structure of the subunit F, residues Asp-53, Asp-11, and Lys-83, which are at the equivalent positions to Asp-57, Asp-13, and Lys-109 of CheY, form a site (‘Active site') similar to the phosphorylation site of CheY (Figure 3D). Residue Lys-83 of subunit F is located on helix α4 within the C-terminal domain, which is detached from the N-terminal domain in the extended subunit F structure (Figure 2C). Within the six subunit F molecules in the asymmetric unit, which are in slightly different environments, Lys-83 on two of the subunits is hydrogen bonded to Asp-13, while the other four subunits do not have this hydrogen bond. This suggests that this hydrogen bond is not permanent and its formation could be regulated by a ligand binding to the ‘active site'. The hydrogen bond could be the key for the conformational change between the extended and retracted forms of the subunit F. The equivalent residues, Lys-83 and Asp-11 in subunit F of T. thermophilus, are well conserved among subunit F of eukaryotic V-ATPase according to the Pfam database (Bateman et al, 2004) (Supplementary Figure 3). This strongly suggests that eukaryotic subunit F forms a similar ‘active site', which might play an important role in the regulatory mechanism of V-ATPase due to phosphorylation of the carboxyl residue or binding of small molecules. The ligand could be a small molecule or a protein subunit of the V-ATPase. This is the case in the equivalent CheY site, which serves as the docking site to CheA and CheZ (Zhao et al, 2002). We have tried soaking and cocrystallization experiments with the following compounds, but no binding was observed in the crystal: AMP-PNP-Mg2+, BeF3−-Mg2+, or BeF3−-Mn2+ (data not shown). Some prokaryotic subunit F lacks the equivalent lysine residue (Supplementary Figure 3). These prokaryotic V-ATPases might have lost the regulatory mechanism at this ‘active site' of the subunit F during evolution.
Single-pair FRET analysis of subunit F conformation
FRET is a physical phenomena that occurs as a result of the long-range dipole–dipole interactions between fluorescent dyes (Lakowicz, 1999). The efficiency of FRET depends on the extent of spectral overlap between the emission and absorption spectra of the donor and acceptor dye, respectively, the quantum yield of the donor, relative orientation of the donor and acceptor transition dipoles, and the distance between the donor and the acceptor. Among these parameters, the distance which corresponds to 50% FRET efficiency is called the Förester (1948) distance. Typical Förester distances are around 50 Å, and FRET measurement is useful as a ruler that can be applied to the biological macromolecules which have the dimension comparable to the Förester distance (Stryer, 1978). This technique has been successfully applied to probe the structural organization and the conformational states of biological molecular machines, such as RNA polymerase (Mekler et al, 2002), myosin (Suzuki et al, 1998; Shih et al, 2000), and kinesin (Rice et al, 1999). Furthermore, recent technical advances in optical microscopy made it possible to measure FRET at the single-molecule level (single-pair FRET) in biologically relevant environments (Deniz et al, 2001; Ha, 2001). Single-pair FRET potentially gives more detailed information about the conformations of biological macromolecules, since the heterogeneity in the population, which may exist, can be directly examined. Single-pair FRET has been successfully applied to the ribozyme (Zhuang et al, 2000), SNARE complex (Weninger et al, 2003), F-ATPase/synthase (Yasuda et al, 2003; Diez et al, 2004), and so on. In this study, we applied this technique to probe the conformational states of the subunit F in the V1 complex.
A mutant subunit F (A55C, H89C) was randomly labeled with Cy3 and Cy5 and reconstituted into the V1 complex (A3B3DF). FRET between single pairs of Cy3 (a donor) and Cy5 (an acceptor) was then measured. The distance between the residues Ala-55 and His-89 in the crystal structure of the retracted and the extended subunit F molecules is 24 and 61 Å, respectively. Since the typical Förester distance (the distance which exhibits 50% FRET efficiency) is around 50 Å (Ha, 2001), a large difference in the FRET efficiency between the retracted and the extended conformations was expected. When the donor was selectively excited at 532 nm, roughly 30–40% of the total spots appeared in the acceptor channel. Recovery of the donor fluorescence concomitant with photo-bleaching of the acceptor, or simultaneous disappearance of the donor and acceptor spots due to photo-bleaching of the donor, indicated single-pair FRET (data not shown). This result also strongly supports the idea that only a single copy of subunit F exists in one V1 complex. As illustrated in Figure 4, most pairs exhibited high FRET efficiency (around 95%) in the absence of nucleotide. The Förester distance of the FRET pair in our study was calculated to be in the range of 40–72 Å (see Material and methods), where 95% efficiency corresponds to the distance of 24–44 Å. These results support the idea that residue Ala-55 is in the proximity of residue His-89 in the V1 complex. The addition of ADP or AMP-PNP does not affect the FRET efficiency (Figure 4B and C). These results clearly indicate that, in the complex, subunit F is found in the retracted conformation both in the absence of nucleotide or in the presence of a nonhydrolysable nucleotide. However, in the presence of ATP, a significant population of the pairs shows much lower FRET efficiency, indicating that subunit F molecules in some of the complexes are in the extended conformation (Figure 4A). Since this is not observed with ADP or AMP-PNP, it is likely that this conformational change is coupled to hydrolysis of ATP. When the same experiment was performed using another double-labeled mutant (V2C, A55C), where the distance between two residues is constant (32 Å) for both retracted and extended forms, the addition of ATP did not cause any change in fluorescence transfer efficiency (Figure 4E and F). This result further supports the idea that observed change represents the transition between the retracted and extended forms of subunit F in the complex.
Figure 4.
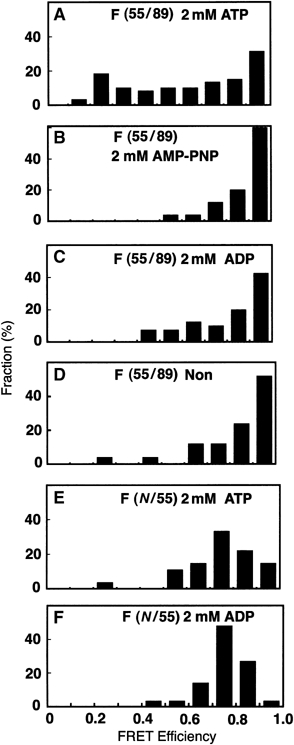
Effect of nucleotides on the distribution of single-pair FRET efficiency of subunit F. (A–D) Distribution of the single-pair FRET efficiency of the double-labeled subunit F (A55C, H89C) in the presence or absence of various nucleotides. (A) ATP (2 mM) in the presence of an ATP regeneration system (n=72). (B) AMP-PNP (2 mM) (n=25), (C) 2 mM ADP (n=40), (D) in the absence of nucleotide (n=25). (E, F) Distribution of the single-pair FRET efficiency of the double-labeled subunit F (V2C, A55C) in the V1 complex. (E) ATP (2 mM) in the presence of ATP, a regeneration system (n=27). (F) ADP (2 mM) (n=30). Measurement of single-pair FRET efficiency and construction of mutant subunit F are described in Materials and methods.
It has been shown that the subcomplex A3B3DF rotates faster than the subcomplex A3B3D; thus, subunit F is clearly related to the rotation efficiency of the prokaryotic V-ATPase complex (Imamura et al, 2004). In the F-ATPase from Bacillus stearothermophilus PS3, the C-terminal helix of the ɛ subunit shows an ‘up' and ‘down' conformational change that is regulated by nucleotide binding (Suzuki et al, 2003). Although the structure and function of subunit F are quite different from the ɛ subunit, subunit F is located at a similar position as the ɛ subunit. It is possible that subunit F regulates the activity of A3B3D minimum rotor unit due to the nucleotide-induced conformational change like ɛ subunit. The physiological importance of this conformational change in subunit F in the presence of ATP is not clear. However, the structural similarity of subunit F to CheY leads us to speculate that this subunit may have a regulatory role in the rotation of the V-ATPase. In eukaryotic V-ATPase, Armbrüster et al (2005) reported that subunit C possesses a low-affinity ATP-binding site, suggesting that the activity of the eukaryotic V-ATPase might be controlled by nucleotide binding to the subunit. Further studies are necessary to understand the role of nucleotide binding to these regulatory subunits and the conformational changes in V-ATPases.
Covalent crosslinking of subunit F using 4-(N-maleimido) benzophenone (MBP)
To identify the location of subunit F relative to the other subunits in the holoenzyme, we produced seven site-directed cysteine mutants of subunit F and one of subunit C and performed crosslinking experiments using the photoactivatable crosslinker MBP. In the presence of MBP, these introduced cysteine residues are crosslinked to an adjacent residue by UV irradiation. A unique cysteine residue was introduced at Glu-13, Ala-21, Ala-48, Ala-55, Pro-58, Ala-80 of subunit F, and Thr-105 of subunit C. For three of the seven mutants, we obtained crosslinked products, which were immunostained using anti-subunit C or F antibodies (Figure 5). A faint band stained by anti-F was seen for F/A48C, but this crosslinked product was not reproducible. Crosslinked products for Thr-105 (subunit C) and Glu-13 (subunit F) had a molecular mass of 50 kDa, and showed crossreactivity with both anti-subunit F and C antibodies, indicating that these residues are at the interface of subunits C and F. These results clearly indicate that the ‘active site' of the subunit F is facing subunit C, which is a part of the V0 domain. Single-molecular analysis and reconstitution experiments clearly indicated that subunit F bound peripherally to subunit D (Xu et al, 1999; Imamura et al, 2003, 2004). The crosslinked product for Ala-80 of subunit F has a molecular mass of 38 kDa (Figure 5). Taken together, it is reasonable to conclude that the crosslinked product of 38 kDa is composed of subunits F (12 kDa) and D (25 kDa), and thus strand β4 in subunit F might be facing subunit D.
Figure 5.
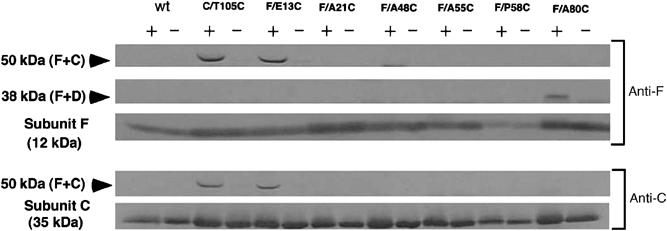
Crosslinking of subunit F and other subunits of the V-ATPase using the photoactivatable sulfhydryl reagent MBP. Western blot using an anti-F antibody (upper) or anti-C antibody (lower). All mutants (mutations are indicated in the figure) are based on the cysteine-less mutant of the subunit (DC). Each of the isolated mutant V-ATPases (50 mg of protein) was incubated in the presence (+) or absence (−) of 1 mM MBP and subjected to 15% SDS–PAGE. Indicated to the left of the panel are the molecular masses estimated from molecular marker proteins.
Location of subunit F in the holoenzyme
Subunit F is part of the central stalk of the V-ATPase. As the crosslinking study shows, this subunit is associated with subunit C, which has a socket-like structure in the V0 domain. The position of subunit C was determined in previous studies and is based on the results of the EM single-particle analysis and the crosslinking experiments (Iwata et al, 2004). These results indicate that subunit C caps the pore that runs through the center of the rotor (subunit L ring) of the V0 domain on the V1-ATPase side (Figure 6A). The location of subunit C is also supported by the structure of the V-ATPase from Enterococcus hirae (Murata et al, 2005). In the electron density maps of the prokaryotic V-ATPase from T. thermophilus (Bernal and Stock, 2004), there is a bulge attached to the density of subunit C on the V1 side (Figure 6A and B). The results of the crosslinking experiments strongly suggest that subunit F should be located within this density.
Figure 6.
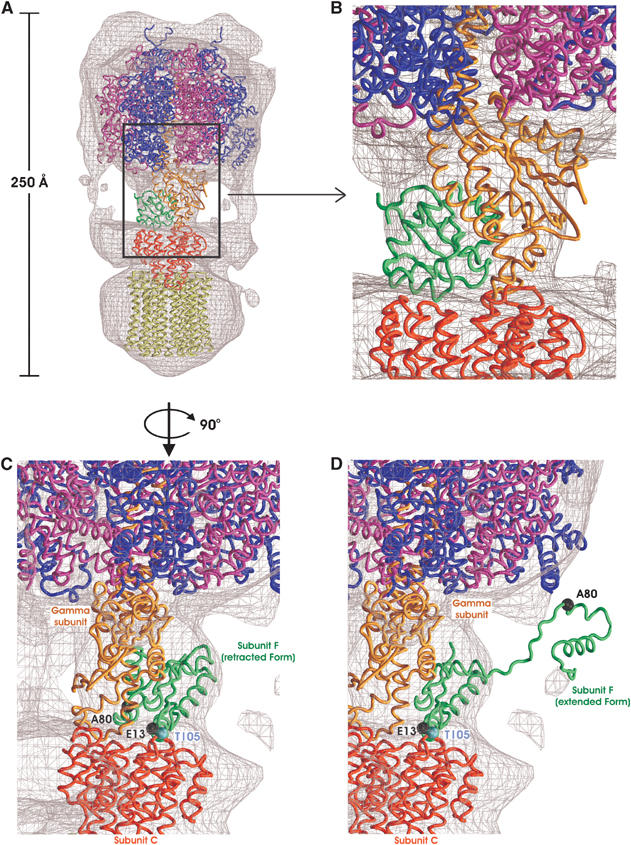
A model of V-ATPase. (A) Fitting of the known X-ray coordinates was carried out using the electron density map of the V-ATPase that was determined by single-particle analysis (mention two different contour levels). Subunit F (green) is shown with the subunit L ring model (Iwata et al, 2004). Bovine mitochondrial F1-ATPase α3β3 (blue and magenta) γ (orange) complex (PDB ID 1E79; Gibbons et al, 2000) and the subunit C (red) (PDB ID 1R5Z; Iwata et al, 2004) of prokaryotic V-ATPase are also fitted to the V0 domain as a reference. (B) The close-up view of the interface between V1 domain and V0 domain. The subunits at the interface between V1 domain and V0 domain are colored as in A). (C) The retracted form of subunit F in a model of V-ATPase. Subunit F (retracted form) in a model of V-ATPase is represented with crosslinked residues (residues E13 and A80 of subunit F, T105 of subunit C) in Figure 5. The crosslinked residues are represented with ball-and-stick representation. Each subunit is colored as in A). (D) The extended form of subunit F in a model of V-ATPase. Subunit F (extended form) in a model of V-ATPase is represented with the same crosslinked residues in Figure 5. The crosslinked residues are represented with a ball-and-stick representation. Each subunit is colored as in A).
We have modeled the T. thermophilus subunit F structure into the EM density together with subunit C, the α3β3γ subcomplex of bovine F1-ATPase (PDB entry 1E79) (Figure 6A). The α3β3γ F1-ATPase subcomplex is equivalent to the A3B3D subcomplex of the V-ATPase. Although there is no clear sequence homology between the subunit D of the V-ATPase and subunit γ of the F-ATPase, subunit D is predicted to have two long α helices, and it is likely that it shares a similar overall structure as that of the γ subunit of the F1-ATPase. In the fitted model, subunit F is located in a similar position as the ɛ subunit of the F1-ATPase. This model corresponds to the results of the crosslinking experiments as follows: Glu-13, which is in the ‘active site' of subunit F, and Thr-105 of subunit C are placed at the interface between subunits C and F and residue Ala-80 of subunit F is placed at the interface between subunits D and F (Figure 6C). In this orientation, the subunit D docking site in subunit F is presumably at the equivalent position to the FliM docking site of CheY (Figure 3A). Therefore, the potential subunit docking sites of subunit F elucidated from the similarity to CheY seem to be used for the subunit assembly of the V-ATPase. In this model, the ‘active site' of subunit F is facing the direction of subunit C. It is possible that the ‘active site' of subunit F, which is a part of the V0–V1 domain interface, might play an important role in the regulatory mechanism that results in the reversible dissociation of the V1 domain from the V0 domain in vivo. We also tried to fit the extended structure of subunit F to the density (Figure 6D). Although the structure does not fit the density, the modeling results show that the C-terminal domain can extend far enough to interact with the subunits A and D, which might be important for the regulatory function of this subunit.
Materials and methods
Protein preparation and crystallization
Subunit F was expressed and purified as described previously (Imamura et al, 2004; Iwata et al, 2004). The subunit F used for crystallization was concentrated to 20 mg/ml in a solution containing 20 mM MOPS, pH 8.0, and 100 mM KCl and stored at −80°C until use. Crystallization trials were performed using the hanging drop vapor diffusion technique. A volume of 1 μl of protein solution was mixed with 1 μl of reservoir solution (0.1 M MES, pH 5.6, 0.2 M calcium acetate, 30% (v/v) PEG 400) and left to equilibrate at 20°C. Crystals reached maximum dimensions of 0.1 × 0.2 × 0.03 mm3 within 2 weeks. Prior to freezing, the crystals were transferred into a cryo-solution containing 34% (w/v) PEG 400, 20 mM MOPS, pH 8.0, 100 mM KCl, 0.1 M MES, pH 5.6, and 0.2 M calcium acetate. The crystals diffracted X-rays beyond 2.1 Å. The crystals belong to the orthorhombic space group P21212 with cell dimensions of a=81.7 Å, b=138.3 Å, and c=66.1 Å. The crystallographic asymmetric unit contains six subunit F molecules.
Data collection and structure determination
A MAD data set was collected using an ADSC Quantum 210 CCD detector at the European Synchrotron Radiation Facility (ESRF) beamline ID14-4. A high-resolution data set was collected using a Mar165 CCD detector at the Swiss Light Source (SLS) beamline X06SA. All data sets were collected from frozen crystals at 100 K. Wavelengths for optimal data collection were determined by an absorption edge scan using a single crystal. Image data were processed using the HKL program package (Otwinowski and Minor, 1997). The structure of subunit F was determined by the MAD technique using 12 Se sites in the asymmetric unit. Se sites were determined using the program SnB (Weeks and Miller, 1999). The CCP4 program suite (CCP4, 1994) was used for the phasing and density modification. Initial phases were calculated to 3.0 Å using the program MLPHARE (Otwinowski, 1991) with an overall figure of merit of 0.47, and were extended to 2.2 Å with the program DM using solvent flattening, histogram matching and averaging options (Cowtan, 1994). The map after phase extension was readily interpretable. Automated model building in combination with phase extension to 2.2 Å was performed using the ARP/wARP program suite (Perrakis et al, 1997). The program suite was able to place 87 of 654 residues with Rcryst of 29% and Rfree of 59%. The rest of the protein residues and the cofactors were modeled manually using the program O (Jones et al, 1991). Further refinement, including water molecule placement, was performed using the programs ARP (Perrakis et al, 1997) and Refmac 5 with TLS refinement (Murshudov et al, 1999). Subunit F is composed of two domains and the relative orientations of these two domains are slightly different in all six subunit F molecules in the asymmetric unit. Therefore, a modest NCS restraint between the six molecules was applied separately for the N- and C-terminal domains. The r.m.s. deviation of the Cα positions for the six superimposed N-terminal domains is ∼0.95 and ∼0.76 Å for the C-terminal domains. The current R-factor and Rfree is 21.4 and 26.2%, respectively. The model contains 654 residues, 306 water molecules, and four Ca ions (four subunit F molecules are associated with a Ca ion and two are Ca free). The refinement statistics and the model stereochemistry are summarized in Table II.
Table 2.
Refinement statistics
| Number of nonhydrogen atoms | 5290 |
| Number of water molecules | 306 |
| Average temperature factor (Å2) | 59.5 |
| Resolution rangea (Å) | 25–2.2 |
| R-factora,b (%) | 21.4 (26.1) |
| Number of reflectionsa | 37 320 |
| Rfreea,b (%) | 26.2 (35.0) |
| Number of reflectionsa | 1206 |
| Estimated overall coordinate error | |
| Based on Rfree, Å | 0.219 |
| R.m.s. deviations from ideal values | |
| Bond length (Å) | 0.033 |
| Bond angles (deg) | 2.492 |
| Ramachandran plot (non-Gly, non-Pro residues) (%) | |
| Residues in most favored regions | 86.3 |
| Residues in additionally allowed regions | 13.0 |
| Others | 0.7 |
| aValues for the highest resolution shell are given in parentheses. | |
| bR factor=∑∣∣Fobs∣–∣Fcalc∣∣/∑∣Fobs∣, where Fobs and Fcalc are the observed and calculated structure factors, respectively. | |
| cRfree was calculated for 3% of reflections randomly excluded from the refinement. | |
The slightly high free-R factor is due to the disorder of the C-terminal domain, which is shown to be mobile in the FRET experiments. Owing to this disorder, the effective resolution of the region is lower than the nominal resolution and the model is therefore less accurate in this region (Supplementary Figure 1).
Mutagenesis and crosslinking experiment
For crosslinking experiments, mutations (A-His8-tags/ΔCys or /C-T105C or /F-E13C or /F-A21C or /F-A48C or /F-A55C or /F-P58C or /F-A80C) were introduced into the atp operon of the T. thermophilus genomic DNA using the integration vector system (Tamakoshi et al, 1997, 1998, 1999). The mutated V-ATPases were solubilized from the membranes of recombinant T. thermophilus at 0.7 mg/ml in a buffer containing 20 mM Tris–Cl, pH 8.0, 0.1 mM EDTA, and 0.05% Triton X-100. Crosslinking experiments using 4-(N-maleimido)benzophenone (MBP) were carried out as described previously (Iwata et al, 2004). The crosslink formation was analyzed by 15% (w/v) SDS–PAGE and by Western blot analysis using anti-subunit C or F antibody.
Single-pair FRET
To probe the conformations of the subunit F in V1 by single-pair FRET, two mutant F subunits (V2C/A55C or A55C/H89C) and A3B3D (A-His8-tags/ΔCys/A-S232A/A-T235S) were prepared. The expression and purification of subunit F and the A3B3D complex were carried out as described previously (Imamura et al, 2004). The mutant F subunit was incubated in buffer A (20 mM MOPS, pH 7.0, 100 mM NaCl) with 10 μM each of Cy3- and Cy5-maleimide (Amersham Biosciences). In this condition, the introduced cysteine residues are randomly labeled by Cy3- or Cy5-maleimide. After a 60-min incubation at 25°C, excess reagent was removed on a PD-10 gel filtration column that had been equilibrated with buffer A. An excess of the double-labeled subunit F was incubated with A3B3D for 60 min at 25°C, and then passed through a Superdex 200 HR column (Amersham Biosciences) equilibrated with buffer A to remove free subunit F. The labeled subunit F was reconstituted into a V1 complex with the A3B3D complex as well as wild-type subunit F, and the reconstituted complex generally exhibited the same range of ATPase activity (30 s−1) as wild-type V1 complex (35 s−1).
Single-pair FRET was measured under an objective-type total internal reflection fluorescence microscope (for the detailed instrumental setup, see Supplementary Figure 2). The donor dye (Cy3) was excited at 532 nm and fluorescence from the donor and acceptor dyes was simultaneously imaged using dual-view optics at a video rate of 30 frames/s (Kinosita et al, 1991). The effect of various nucleotides was examined as follows. The labeled V1 (≈10 pM) in buffer B (50 mM Tris–Cl, pH 8.0, 100 mM NaCl, 2 mM MgCl2) was infused into a flow chamber constructed of two coverslips separated by ≈50 μm spacers. After a 1-min infusion, unbound V1 was washed out with 20 volumes of buffer B containing the indicated nucleotides and oxygen-scavenging system (6 mg/ml glucose, 0.036 mg/ml catalase, 0.22 mg/ml glucose oxidase, and 0.5% 2-mercaptoethanol). Under ATP hydrolysis conditions, a regenerating system (5 mM phosphoenolpyruvate and 0.2 mg/ml pyruvate kinase) was supplemented. All observations were carried out at room temperature (24–26°C). FRET efficiency was calculated with the equation, IA(t)/[ID(t)+IA(t)], where ID(t) and IA(t) are the fluorescence intensities of the single donor and acceptor pair at time t, after background intensities near the pair were subtracted, respectively. To generate the histograms shown in Figure 5, the median value of the FRET efficiency for each pair, calculated at each video frame, was used.
Estimation of the Förester distance and the distance between FRET pairs
The Förester distance was calculated using the quantum yield of Cy3 (0.18, 0.22, and 0.24 for F/V2C, F/A55C, and F/H89C, respectively), the overlap integral between Cy3 and Cy5 (7.2 × 10−13 M−1 cm3), the refractive index of water (1.33), and assuming the value of the orientation factor as 2/3 (expected value when the relative orientation of the donor and acceptor transition dipoles is dynamically randomized during the excited state lifetime of the donor). As a result, the Förester distance was 55±1 Å. The steady-state fluorescence anisotropies of Cy3 were 0.34, 0.34, and 0.28 for F/V2C, F/A55C, and F/H89C, respectively, and those of Cy5 were 0.32, 0.31, and 0.34, respectively. These anisotropies yield a maximum range of 0.1–3.2 for orientation factor for the combination of F/55C-Cy3 and F/89C-Cy5 (Dale et al, 1979). This range corresponds to a −27% (40 Å) to +30% (72 Å) maximum uncertainty in the Förester distance. Furthermore, note that Cy3 and Cy5 conjugated with cysteine residues have a linker of ∼10 Å from the fluorophore to the cysteine sulfur.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
We thank Drs Clemens Schulze-Briese and Takashi Tomizaki (PX06SA SLS, Paul Scherrer Institute, Villigen, Switzerland) and Raimond Ravelli and Andrew McCarthy (ID14-4 European Synchrotron Radiation Facility) for technical support with data collections and Dr Bernadette Byrne for critical reading of the manuscript. Part of this work was supported by the Biotechnology and Biological Sciences Research Council (to SI). The atomic coordinates and the structure factors have been deposited in the Protein Data Bank for structure (PDB ID 2D00).
References
- Armbrüster A, Hohn C, Hermesdorf A, Schumacher K, Borsch M, Grüber G (2005) Evidence for major structural changes in subunit C of the vacuolar ATPase due to nucleotide binding. FEBS Lett 579: 1961–1967 [DOI] [PubMed] [Google Scholar]
- Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, Eddy SR (2004) The Pfam protein families database. Nucleic Acids Res 32: D138–D141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal RA, Stock D (2004) Three-dimensional structure of the intact Thermus thermophilus H+-ATPase/synthase by electron microscopy. Structure (Cambridge) 12: 1789–1798 [DOI] [PubMed] [Google Scholar]
- Bond CS (2003) TopDraw: a sketchpad for protein structure topology cartoons. Bioinformatics 19: 311–312 [DOI] [PubMed] [Google Scholar]
- Boyer PD (1993) The binding change mechanism for ATP synthase—some probabilities and possibilities. Biochim Biophys Acta 1140: 215–250 [DOI] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project, Number 4) (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Cowtan K (1994) Joint CCP4 and ESF-EACBM newsletter on protein. Crystallography 31: 34–38 [Google Scholar]
- Dale RE, Eisinger J, Blumberg WE (1979) The orientational freedom of molecular probes. The orientation factor in intramolecular energy transfer. Biophys J 26: 161–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz AA, Laurence TA, Dahan M, Chemla DS, Schultz PG, Weiss S (2001) Ratiometric single-molecule studies of freely diffusing biomolecules. Annu Rev Phys Chem 52: 233–253 [DOI] [PubMed] [Google Scholar]
- Diez M, Zimmermann B, Borsch M, Konig M, Schweinberger E, Steigmiller S, Reuter R, Felekyan S, Kudryavtsev V, Seidel CA, Graber P (2004) Proton-powered subunit rotation in single membrane-bound F0F1-ATP synthase. Nat Struct Mol Biol 11: 135–141 [DOI] [PubMed] [Google Scholar]
- Esnouf RM (1997) An extensively modified version of MolScript which includes greatly enhanced colouring capabilities. J Mol Graph 15: 132–134 [DOI] [PubMed] [Google Scholar]
- Falke JJ, Bass RB, Butler SL, Chervitz SA, Danielson MA (1997) The two-component signaling pathway of bacterial chemotaxis: a molecular view of signal transduction by receptors, kinases, and adaptation enzymes. Annu Rev Cell Dev Biol 13: 457–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förester T (1948) Zwischenmolekulare Energiewanderung und Fluoreszenz. Ann Phys 2: 55–70 [Google Scholar]
- Forgac M (2000) Structure, mechanism and regulation of the clathrin-coated vesicle and yeast vacuolar H+-ATPases. J Exp Biol 203: 71–80 [DOI] [PubMed] [Google Scholar]
- Gibbons C, Montgomery MG, Leslie AG, Walker JE (2000) The structure of the central stalk in bovine F1-ATPase at 2.4 Å resolution. Nat Struct Biol 7: 1055–1061 [DOI] [PubMed] [Google Scholar]
- Graham LA, Hill KJ, Stevens TH (1994) VMA7 encodes a novel 14-kDa subunit of the Saccharomyces cerevisiae vacuolar H+-ATPase complex. J Biol Chem 269: 25974–25977 [PubMed] [Google Scholar]
- Ha T (2001) Single-molecule fluorescence resonance energy transfer. Methods 25: 78–86 [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C (1996) Mapping the protein universe. Science 273: 595–603 [DOI] [PubMed] [Google Scholar]
- Imamura H, Ikeda C, Yoshida M, Yokoyama K (2004) The F subunit of Thermus thermophilus V1-ATPase promotes ATPase activity but is not necessary for rotation. J Biol Chem 279: 18085–18090 [DOI] [PubMed] [Google Scholar]
- Imamura H, Nakano M, Noji H, Muneyuki E, Ohkuma S, Yoshida M, Yokoyama K (2003) Evidence for rotation of V1-ATPase. Proc Natl Acad Sci USA 100: 2312–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata M, Imamura H, Stambouli E, Ikeda C, Tamakoshi M, Nagata K, Makyio H, Hankamer B, Barber J, Yoshida M, Yokoyama K, Iwata S (2004) Crystal structure of a central stalk subunit C and reversible association/dissociation of vacuole-type ATPase. Proc Natl Acad Sci USA 101: 59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kane PM, Parra KJ (2000) Assembly and regulation of the yeast vacuolar H+-ATPase. J Exp Biol 203: 81–87 [DOI] [PubMed] [Google Scholar]
- Kinosita K Jr, Itoh H, Ishiwata S, Hirano K, Nishizaka T, Hayakawa T (1991) Dual-view microscopy with a single camera: real-time imaging of molecular orientations and calcium. J Cell Biol 115: 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Lakowicz JR (1999) Principles of Fluorescence Spectroscopy, 2nd edn. New York: Kluwer Academic/Plenum Publishers [Google Scholar]
- Lee SY, Cho HS, Pelton JG, Yan D, Berry EA, Wemmer DE (2001a) Crystal structure of activated CheY. Comparison with other activated receiver domains. J Biol Chem 276: 16424–16431 [DOI] [PubMed] [Google Scholar]
- Lee SY, Cho HS, Pelton JG, Yan D, Henderson RK, King DS, Huang L, Kustu S, Berry EA, Wemmer DE (2001b) Crystal structure of an activated response regulator bound to its target. Nat Struct Biol 8: 52–56 [DOI] [PubMed] [Google Scholar]
- Lewis RJ, Muchova K, Brannigan JA, Barak I, Leonard G, Wilkinson AJ (2000) Domain swapping in the sporulation response regulator Spo0A. J Mol Biol 297: 757–770 [DOI] [PubMed] [Google Scholar]
- Mekler V, Kortkhonjia E, Mukhopadhyay J, Knight J, Revyakin A, Kapanidis AN, Niu W, Ebright YW, Levy R, Ebright RH (2002) Structural organization of bacterial RNA polymerase holoenzyme and the RNA polymerase-promoter open complex. Cell 108: 599–614 [DOI] [PubMed] [Google Scholar]
- Merritt EA, Murphy ME (1994) Raster3D version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr D 50: 869–873 [DOI] [PubMed] [Google Scholar]
- Müller V, Grüber G (2003) ATP synthases: structure, function and evolution of unique energy converters. Cell Mol Life Sci 60: 474–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T, Takase K, Yamato I, Igarashi K, Kakinuma Y (1997) Purification and reconstitution of Na+-translocating vacuolar ATPase from Enterococcus hirae. J Biol Chem 272: 24885–24890 [DOI] [PubMed] [Google Scholar]
- Murata T, Takase K, Yamato I, Igarashi K, Kakinuma Y (1999) Properties of the V0V1 Na+-ATPase from Enterococcus hirae and its V0 moiety. J Biochem (Tokyo) 125: 414–421 [DOI] [PubMed] [Google Scholar]
- Murata T, Yamato I, Kakinuma Y, Leslie AG, Walker JE (2005) Structure of the rotor of the V-type Na+-ATPase from Enterococcus hirae. Science 308: 654–659 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Lebedev A, Wilson KS, Dodson EJ (1999) Efficient anisotropic refinement of macromolecular structures using FF. Acta Crystallogr D 55: 247–255 [DOI] [PubMed] [Google Scholar]
- Nishi T, Forgac M (2002) The vacuolar (H+)-ATPases—nature's most versatile proton pumps. Nat Rev Mol Cell Biol 3: 94–103 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z (1991) Maximum likelihood refinement of heavy atom parameters. In Isomorphous Replacement and Anomalous Scattering, Proceedings of the CCP4 Study Weekend, Wolf W, Evans PR, Leslie AGW (eds), pp 80–86. Warrington, UK: SERC Daresbury Laboratory [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Parra KJ, Kane PM (1998) Reversible association between the V1 and V0 domains of yeast vacuolar H+-ATPase is an unconventional glucose-induced effect. Mol Cell Biol 18: 7064–7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A, Sixma TK, Wilson KS, Lamzin VS (1997) wARP: improvement and extension of crystallographic phases by weighted averaging of multiple refined dummy atomic models. Acta Crystallogr D 53: 448–455 [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16: 276–277 [DOI] [PubMed] [Google Scholar]
- Rice S, Lin AW, Safer D, Hart CL, Naber N, Carragher BO, Cain SM, Pechatnikova E, Wilson-Kubalek EM, Whittaker M, Pate E, Cooke R, Taylor EW, Milligan RA, Vale RD (1999) A structural change in the kinesin motor protein that drives motility. Nature 402: 778–784 [DOI] [PubMed] [Google Scholar]
- Shih WM, Gryczynski Z, Lakowicz JR, Spudich JA (2000) A FRET-based sensor reveals large ATP hydrolysis-induced conformational changes and three distinct states of the molecular motor myosin. Cell 102: 683–694 [DOI] [PubMed] [Google Scholar]
- Silversmith RE, Guanga GP, Betts L, Chu C, Zhao R, Bourret RB (2003) CheZ-mediated dephosphorylation of the Escherichia coli chemotaxis response regulator CheY: role for CheY glutamate 89. J Bacteriol 185: 1495–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock AM, Mottonen JM, Stock JB, Schutt CE (1989) Three-dimensional structure of CheY, the response regulator of bacterial chemotaxis. Nature 337: 745–749 [DOI] [PubMed] [Google Scholar]
- Stryer L (1978) Fluorescence energy transfer as a spectroscopic ruler. Annu Rev Biochem 47: 819–846 [DOI] [PubMed] [Google Scholar]
- Suzuki T, Murakami T, Iino R, Suzuki J, Ono S, Shirakihara Y, Yoshida M (2003) F0F1-ATPase/synthase is geared to the synthesis mode by conformational rearrangement of epsilon subunit in response to proton motive force and ADP/ATP balance. J Biol Chem 278: 46840–46846 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Yasunaga T, Ohkura R, Wakabayashi T, Sutoh K (1998) Swing of the lever arm of a myosin motor at the isomerization and phosphate-release steps. Nature 396: 380–383 [DOI] [PubMed] [Google Scholar]
- Tamakoshi M, Uchida M, Tanabe K, Fukuyama S, Yamagishi A, Oshima T (1997) A new Thermus–Escherichia coli shuttle integration vector system. J Bacteriol 179: 4811–4814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamakoshi M, Yamagishi A, Oshima T (1998) The organization of the leuC, leuD and leuB genes of the extreme thermophile Thermus thermophilus. Gene 222: 125–132 [DOI] [PubMed] [Google Scholar]
- Tamakoshi M, Yaoi T, Oshima T, Yamagishi A (1999) An efficient gene replacement and deletion system for an extreme thermophile, Thermus thermophilus. FEMS Microbiol Lett 173: 431–437 [DOI] [PubMed] [Google Scholar]
- Weeks CM, Miller R (1999) The design and implementation of SnB version 2.0. J Appl Crystallogr 32: 120–124 [Google Scholar]
- Weninger K, Bowen ME, Chu S, Brunger AT (2003) Single-molecule studies of SNARE complex assembly reveal parallel and antiparallel configurations. Proc Natl Acad Sci USA 100: 14800–14805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Vasilyeva E, Forgac M (1999) Subunit interactions in the clathrin-coated vesicle vacuolar (H(+))-ATPase complex. J Biol Chem 274: 28909–28915 [DOI] [PubMed] [Google Scholar]
- Yasuda R, Masaike T, Adachi K, Noji H, Itoh H, Kinosita K Jr (2003) The ATP-waiting conformation of rotating F1-ATPase revealed by single-pair fluorescence resonance energy transfer. Proc Natl Acad Sci USA 100: 9314–9318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama K, Akabane Y, Ishii N, Yoshida M (1994) Isolation of prokaryotic V0V1-ATPase from a thermophilic eubacterium Thermus thermophilus. J Biol Chem 269: 12248–12253 [PubMed] [Google Scholar]
- Yokoyama K, Muneyuki E, Amano T, Mizutani S, Yoshida M, Ishida M, Ohkuma S (1998) V-ATPase of Thermus thermophilus is inactivated during ATP hydrolysis but can synthesize ATP. J Biol Chem 273: 20504–20510 [DOI] [PubMed] [Google Scholar]
- Yokoyama K, Nagata K, Imamura H, Ohkuma S, Yoshida M, Tamakoshi M (2003a) Subunit arrangement in V-ATPase from Thermus thermophilus. J Biol Chem 278: 42686–42691 [DOI] [PubMed] [Google Scholar]
- Yokoyama K, Nakano M, Imamura H, Yoshida M, Tamakoshi M (2003b) Rotation of the proteolipid ring in the V-ATPase. J Biol Chem 278: 24255–24258 [DOI] [PubMed] [Google Scholar]
- Yokoyama K, Ohkuma S, Taguchi H, Yasunaga T, Wakabayashi T, Yoshida M (2000) V-type H+-ATPase/synthase from a thermophilic eubacterium, Thermus thermophilus. Subunit structure and operon. J Biol Chem 275: 13955–13961 [DOI] [PubMed] [Google Scholar]
- Yokoyama K, Oshima T, Yoshida M (1990) Thermus thermophilus membrane-associated ATPase. Indication of a eubacterial V-type ATPase. J Biol Chem 265: 21946–21950 [PubMed] [Google Scholar]
- Yoshida M, Muneyuki E, Hisabori T (2001) ATP synthase—a marvellous rotary engine of the cell. Nat Rev Mol Cell Biol 2: 669–677 [DOI] [PubMed] [Google Scholar]
- Zhao R, Collins EJ, Bourret RB, Silversmith RE (2002) Structure and catalytic mechanism of the E. coli chemotaxis phosphatase CheZ. Nat Struct Biol 9: 570–575 [DOI] [PubMed] [Google Scholar]
- Zhuang X, Bartley LE, Babcock HP, Russell R, Ha T, Herschlag D, Chu S (2000) A single-molecule study of RNA catalysis and folding. Science 288: 2048–2051 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3