

Abstract
Several pathological and disease conditions can alter the mechanical properties of the extracellular matrix (ECM). Conversely, some diseases may arise from changes in the density or rigidity of the ECM. This necessitates the use and development of in vitro models to understand how both biophysical and biochemical signals regulate complex cellular behaviors. T47D breast epithelial cells will differentiate into duct-like tubules when cultured in a floating three-dimensional (3D) collagen gel, but not a 3D collagen gel that is left attached to the culture dish. This paper details several protocols we have developed for analyzing breast cell biology in 3D matrices, including culturing cells in 3D collagen gels, immunostaining cellular structures, and performing biochemical procedures directly from cells embedded in collagen gels.
Keywords: Cell Culture Techniques; Signal Transduction; Mechanotransduction, Cellular; Epithelial Cells
Introduction
Breast epithelial cells in vivo respond to cues from the extracellular matrix (ECM) to differentiate into polarized, growth-arrested, and highly organized multicellular structures that form a functional tissue (Fig. 1A). During cancer progression, however, cells lose their normal interactions with the ECM and breast structure is compromised as cells de-differentiate, proliferate, and migrate (Fig. 1B and C). A leading risk factor for breast carcinoma is increased breast density, which accounts for approximately 30% of breast cancers (1). Dense breast tissue is characterized by an increased deposition of ECM proteins and fibroblasts in the stroma surrounding the epithelial cells. An increase in breast density leads to a four to six-fold increased risk of developing breast cancer (1). However, the mechanisms by which stromal density could promote breast carcinoma are unknown.
Fig. 1. Normal and cancerous breast morphology.
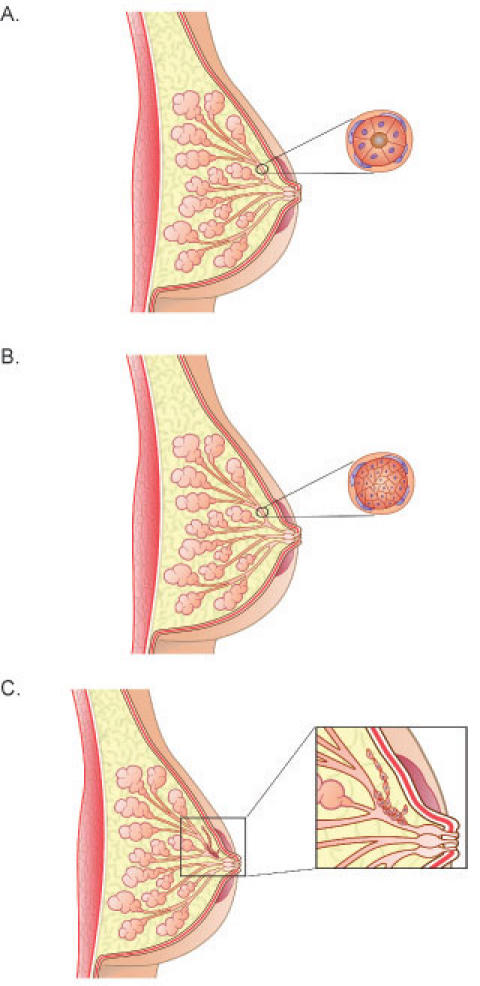
A) Diagram showing the normal organization of ducts and acini in the human breast. The cross-section demonstrates luminal epithelial cells aligned in a polar manner so their apical side faces and surrounds the lumen. The luminal epithelial cells are surrounded by a non-continuous layer of myoepithelial cells. Surrounding these cells is the basement membrane. Fibroblasts align the basement membrane and this entire structure is surrounded by the stroma, which is predominantly, but not exclusively, composed of type I collagen. B) During ductal carcinoma in situ (DCIS), the normal polar organization of the luminal epithelial cells is lost, as these cells de-differentiate and proliferate. The cross-section shows the epithelial cells completely filling the lumen. In less severe cases of DCIS, the luminal epithelial cells do not completely block the lumen. In DCIS, the transformed epithelial cells do not cross the basement membrane, but remain within the duct. C) DCIS sometimes leads to invasive, or infiltrating, carcinoma, in which the epithelial cells migrate and invade through the basement membrane and into the surrounding stroma.
In order to study the molecular mechanisms by which breast cells become transformed, tissue culture cell lines are often used because they are homogenous, can be genetically altered, and allow for large harvests of cells for biochemical procedures. However, when breast cell lines are cultured on normal tissue culture two-dimensional (2D) surfaces, they do not recapitulate the differentiated structures seen in vivo. It was therefore recognized that in vitro three-dimensional (3D) model systems were needed in order to study breast epithelial biology in a more relevant context (2, 3). Since then, several excellent in vitro systems have been designed for studying breast cell behavior and tumorigenesis in a three-dimensional context. These include primary mouse epithelial cells in/on collagen gels (2-5) or on reconstituted basement membrane (6), normal murine mammary gland (NMuMG) cells in collagen gels (7), nonmalignant HMT-3522 S-1 breast cells and their tumorigenic progeny HMT-3522 T4-2 cells in reconstituted basement membrane (8-10), MCF-10A breast epithelial cells in Matrigel (11), and many others.
Each of these systems promotes breast cell differentiation in vitro. The selection of a 3D system should be based on which one will best address the questions to be studied. In our case, there are two main features we desired in a 3D model system. First, given that invasive ductal carcinoma is the most common type of breast cancer, we want to study the formation and disruption of ductal morphogenesis. Thus, we need a cell line that recapitulates ductal structures and we need to culture the cells in an appropriate ECM protein to support the formation of ducts. Although the T47D breast cell line originates from an invasive ductal carcinoma, it is classified as a well-differentiated cell line (12). In addition, T47D cells retain the ability to differentiate into duct-like tubules when cultured in floating 3D collagen gels (13).
Type I collagen, type IV collagen, and laminin are abundant components of the ECM and basement membrane in the breast, with type I collagen being the predominant structural component of connective tissue. The use of type I collagen gels instead of Matrigel was best for our model for several reasons. First, type I collagen is localized around the mouse (14) and human (15) mammary ducts, suggesting a physiological relevance for collagen in the development and maintenance of these structures. Laminin and type IV collagen (components of Matrigel), however, are more abundant around alveoli and the growing terminal endbuds of the mouse mammary gland (14) and around the acini in human breast (16). Based on this data, it is not surprising that the culture of breast epithelial cells in Matrigel normally promotes the formation of cyst-like, acinar structures (11, 17). For these reasons, culturing cells in type I collagen better supports the in vitro ductal environment.
The second feature we desired in a 3D system was the manipulation of the physical properties of the matrix in order to study mechanotransduction. Mechanosensing is defined as the ability of a cell or tissue to detect, and respond to, the imposition of force (18). Mechanotransduction refers to the ability of a cell to transform mechanical signals into biochemical signals (18). For these purposes, collagen was again advantageous because one can physically detach a collagen gel to impose a different mechanical environment on the cells, compared to a gel that is left attached to the culture dish (19, 20). In addition, one can easily alter the density of 3D collagen gels by changing the concentration of collagen (21, 22). Conversely, Matrigel is too pliable and yielding for these experiments and does not lend itself to simple manipulation of density.
In addition, type I collagen is the key determinant of tensile properties in connective tissue (21), indicating that type I collagen regulates the mechanical properties of connective tissue. The observation that type I collagen is abundant around ductal structures (14, 15) further supports the notion that type I collagen regulates the in vivo biophysical properties of the ECM surrounding the ducts. Thus, the choice to culture cells in 3D gels composed of type I collagen satisfied both the biological and mechanical parameters of our model. The purpose of this paper is to explain the methods we have developed to study T47D breast epithelial cell morphogenesis and signal transduction in 3D collagen gels. We will also discuss the difficulties and intricacies of working with this 3D model system.
Materials and Methods
Collagen gel culture
T47D breast epithelial cells were maintained and cultured in type I collagen gels at a final concentration of 1.3mg/ml, with 1 x 105 cells/ml cultured per gel, as previously described (13). One day after the gels were poured, one set of gels was left attached to the dish and one set of gels was detached from the sides and bottom of the dish (these gels are referred to as “floating”). Media was added to the gels at this time. The gels were fed approximately every four days. Cells were grown for seven to ten days, and phase contrast pictures taken to assess morphology using a Nikon 35mm camera attached to a TE300 Nikon inverted microscope. Detailed methods for T47D cell general maintenance and collagen gel culture are listed in Protocols 1 and 2, respectively.
Rho activity assays and immunoprecipitations
Subconfluent T47D cells were harvested in 0.5mM EDTA in PBS, resuspended in 5mg/ml fatty acid free BSA (IMP Biomedicals) in RPMI, and approximately 15-20 x 106 cells used for each condition to be analyzed. Conditions we typically use are: no collagen, culture on a collagen-coated petri plate (60-100mm) ("2D"), and culture in attached or floating 3D collagen gels. For culture in 3D gels, the gels were made with BSA/RPMI instead of full T47D media in order to determine the effect of collagen alone on the cells, in the absence of serum. (However, we have also performed assays in the presence of serum). Gels were allowed to polymerize for one hour at 37°C before 1ml BSA/RPMI was added to the gels and the gels rendered floating in BSA/RPMI or left attached to the culture dish. The gels were then incubated for another hour. After this incubation, the activity assay or immunoprecipitation was performed. For the Rho activity assay, cells in gels were lysed in TBS buffer containing 0.1% SDS, 1% Triton X-100, protease inhibitor cocktail (Sigma), and sodium pervanadate. RBD-GST pull down assays were performed as described (22) and detailed protocols are included as Protocols 6-8. Samples were run on a SDS-PAGE gel, transferred to a PVDF membrane, and the membrane was probed with anti-Rho (Santa Cruz, 1:250) followed by anti-mouse HRP (Jackson Immunolabs, 1:5000). Rho was detected using ECL substrate (Amersham Biosciences).
For immunoprecipitations, 4 μg antibody and 25 μl GammaBind Beads (Amersham Biosciences) were added to the cleared cell lysate and rotated, at 4°C, for two hours to overnight, depending on which antibody used. Samples were run on a SDS-PAGE gel and transferred to a PVDF membrane. Immunoblotting from lysates and immunoprecipitaton methods are detailed in Protocols 4 and 5, respectively.
Immunofluorescence
Cells were grown in collagen gels for seven to ten days. The gels were fixed in 4% paraformaldehyde in PBS for 15 minutes at room temperature, washed for 10 minutes with PBS while shaking, and then incubated in 0.15M glycine in PBS for 10 minutes. After washing again with PBS for 10 minutes, 0.02% Triton X-100 in PBS was added for 10 minutes. The gels were washed with PBS for 10 minutes and then blocked overnight at 4°C with 1% fatty acid free BSA, 1% donkey serum in PBS. The gels were then soaked in 100 ml primary antibody diluted in PBS with 1% donkey serum for 30 minutes at room temperature or overnight at 4°C. After extensive washing in PBS, 100 ml secondary antibody and 1% donkey serum in PBS was added for 30 minutes at room temperature. The gels were washed twice in PBS for 10 minutes, in water for 10 minutes, and mounted on a slide with Prolong Antifade mounting medium (Molecular Probes). Immunostaining was analyzed by epifluorescence using a TE300 Nikon inverted microscope equipped with a Photometrics CoolSnap fx CCD camera. Images were collected and 3D deconvolution performed using Inovision software. This method is detailed in Protocol 3.
Results and Discussion
Morphogenesis and characterization of T47D cells
The methods described here use T47D breast epithelial cells. However, these protocols can be used for other cell types cultured in collagen gels and, we anticipate, for cells cultured in other 3D matrices as well.
The T47D cell line was originally isolated from a 54 year old female patient with an infiltrating ductal carcinoma. Although the cells will form colonies in soft agar (23, 24), they are weakly tumorigenic in mice ((25) and our unpublished observations). These cells are classified as a well-differentiated cell line since they are estrogen receptor-positive (26) and retain the ability to form organized structures in 3D ECM (13).
When T47D cells are grown on 2D tissue culture plastic, they proliferate, migrate, and do not differentiate (Fig. 2). Grown at low confluency, these cells will spread to develop focal adhesions and various actin structures, such as lamellipodia and stress fibers. When the cells are grown to greater confluency, they show a more epithelial-like cobblestone morphology (Fig. 2), although not to the extent of other “normal” breast cell lines.
Fig. 2. T47D breast epithelial cell morphology.

T47D cells differentiate into tubules when cultured in a floating 3D collagen gel for seven days, but not when cultured in an attached collagen gel, or when cultured on a 2D surface (at low or high confluency). Scale bar, 100 μm.
T47D cells cultured in a 3D type I collagen gel become mechanically loaded and develop isometric tension because the matrix is restrained and can resist the force exerted on it by the cells (19, 20, 27, 28). Cells grown in the 3D gels attached to the culture dish will not form differentiated structures (Fig. 2), and will continue to proliferate (22). However, if this gel is released, the cells become mechanically unloaded because the matrix can no longer resist the force exerted on it by the cells (19, 20). The collagen gel is contracted by the cells, and the cells decrease proliferation (22) and undergo tubulogenesis (Fig. 2). We describe this as tubulogenesis, and not simply a clustering of cells, because T47D cells that form these structures display lumens that stain with an antibody against the human milk fat globule, as described previously (13).
T47D tubule formation in floating collagen gels occurs as single cells proliferate to form a branched structure. Single cells do not migrate towards each other to form tubules, as suggested by the observation that adding more cells to a floating collagen gel does not result in faster tubule formation (13). In addition, we have tracked single cells in a floating collagen gel over seven days to confirm that cells grow into, rather than coalesce into, tubule structures (Fig. 3). This observation is significant because both human and murine mammary gland development proceeds in this manner, by the outward growth of terminal end buds into the mammary fat pad (reviewed in (29, 30)).
Fig. 3. Development of T47D tubules from single cells.

T47D cells were cultured in a floating 1.3 mg/ml 3D collagen gel. Every day for seven days, the same gel location was identified and photographed. T47D cells grow into tubules—they do not cluster together to form tubule structures. Scale bar, 100 μm.
Differentiation primarily in floating collagen gels (vs. attached) is not a T47D cell-line specific phenomenon. MCF10-A and NMuMG breast epithelial cell lines and primary mouse and rat breast epithelial cells also form differentiated structures in floating, but not attached, 3D collagen gels. Moreover, all of these breast epithelial cells contract a floating collagen gel (2-4, 22), further supporting the theory that differentiation requires cellular contractility.
It was also observed that human breast luminal epithelial cells taken from primary cultures form inside-out acinar-like structures in attached collagen gels (31), again suggesting that culture in an attached collagen gel does not support proper breast differentiation. Interestingly, addition of laminin or breast myoepithelial cells to the luminal cells in an attached collagen gel promotes proper acinar differentiation (31). Because primary breast epithelial cells can polarize and differentiate when cultured in a floating 3D collagen gel, and indeed can deposit a basement membrane (2, 3, 5), these observations suggest that changes in the physical properties of a collagen gel may also regulate signal transduction pathways controlling ECM deposition. The deposited ECM could then direct consequent cell polarization and differentiation in a floating collagen gel.
As mentioned above, an increase in breast stromal density is a risk factor for breast carcinoma, suggesting that breast cells respond to mechanical cues in their ECM in vivo. This system proved useful to determine if changes in collagen density alone, without stromal fibroblasts and growth factors, could alter breast cell behavior. This question was addressed by culturing cells in 3D gels of varying collagen concentrations, which has been shown to vary collagen density (21). Cells cultured in floating high-density collagen gels showed disrupted tubulogenesis (22) and increased proliferation (unpublished data) in a manner similar to cells cultured in attached, lower density (1.3mg/ml) 3D collagen gels.
Using this 3D system, we have also found that the ECM does not simply “set up” breast cells for differentiation or proliferation. Rather, breast cells are continually sensing and responding to the mechanical properties of the ECM. For example, cells that have been cultured in an attached collagen gel for days do not form tubules. However, if the same gel is detached from the dish, the cells will begin to form tubules within 24 hours (unpublished data), suggesting breast cells are constantly relaying information to and from the ECM. In addition, inhibitors of contractility, such as inhibitors of Rho, ROCK, or myosin, added to already-formed tubules in floating 3D collagen gels will disrupt tubule structure ((22) and unpublished data). We have proposed that T47D breast cells continually sense changes in their environment through the ability to contract the surrounding ECM, which alters cell signaling events regulating proliferation and differentiation.
Immunofluorescent staining of breast cells in 3D collagen gels
In order to visualize the localization of specific proteins and structures, cells 3D matrices are often snap frozen and cryosectioned for immunostaining. We originally tried this approach, but found that the ductual structures were damaged from the sectioning. The tubules formed by T47D cells are not that large (about 50 μm cross-section of a duct); however, the ducts themselves are long (from 300-1000 μm in length) and we did not want to compromise this structure. Therefore, the immunofluorescent staining protocol we have designed for cells in 3D collagen gels does not require cryosectioning and is a modification to protocols used for cells cultured on 2D surfaces.
There are several factors to take into consideration when immunostaining breast cells in a 3D collagen gel, the most important being that fixation solutions and antibodies must reach cellular structures embedded in the collagen gel. An advantage of working with collagen gels is that solutions and molecules can diffuse through collagen rather easily. One step we have taken to ensure specific staining with little background is to considerably increase washing times. In between each fixing and permeabilization step, the gels are washed with PBS for 10 minutes, with agitation, and after antibody treatment, the gels are washed extensively (30 minutes up to overnight). Another modification we have made is to block the cells overnight at 4°C. This greatly helps to decrease non-specific staining. For some antibodies that still show high background, we found that background fluorescence could be reduced by staining overnight at 4°C, instead of 30 minutes at room temperature, and increasing washing time after antibody treatment.
Using this protocol, we have found that focal adhesion formation is down-regulated in cells in floating 3D collagen gels (Fig. 4) (22). This procedure can be used not only to determine the localization of molecules, but also to assess other cellular behaviors, such as proliferation. For example, we have used this procedure to immunostain for Ki67, a protein required for cell cycle progression (32), in order to determine the relative proliferation of cells in floating vs. attached collagen gels and have found that cells in floating gels have reduced proliferation (22).
Fig. 4. Immunostaining T47D breast cells cultured in 3D collagen gels.
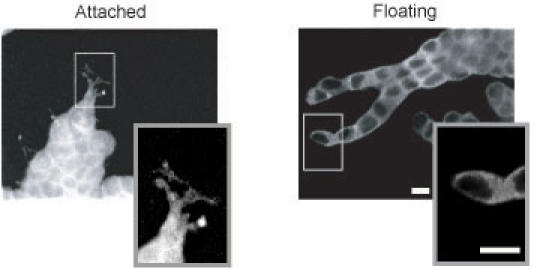
Cells were cultured in attached or floating 3D collagen gels and seven days later, the gels were immunostained with anti-paxillin as detailed in the protocols. Using this procedure, we have observed that cells in floating collagen gels down-regulate focal adhesion formation (see insets for more detail). Note that although it appears that cells are in a single plane, this is an optical section of the 3D structures formed by T47D cells. Scale bar, 25 μm.
Biochemical procedures in breast cells cultured in 3D collagen gels
In order to determine the signaling events that are regulated in cells in floating vs. attached 3D collagen gels, it is necessary to perform biochemistry in cells cultured in 3D matrices. There are three major considerations to take into account when performing biochemical procedures in cells cultured in 3D collagen gels. The first is that the cells are embedded, and not on top of, a 3D matrix. Collagenase can be used to digest the collagen and collect the cells. However, collagenase digestion takes approximately 30 minutes and sensitive signaling events, such as phosphorylation, would likely not be retained within this time period. Therefore, cells need to by lysed rapidly while embedded in a collagen gel.
We developed a protocol to allow the collagen gel to polymerize into a gel around the cells for an hour, then render the gels floating or attached for one hour. A one hour time point was chosen in order to look at early signaling events regulated in response to physical changes in the ECM. At this time, the entire collagen gel can be lysed with 2X lysis buffer and pipetted to break apart the gel. Much collagen protein is retained in the lysate, which would interfere with protein quantitation. However, because equal numbers of cells are cultured in the gels at the beginning of the experiment and the cells are only in the collagen gel for two hours, additional protein quantitation and standardization is not needed. After the cells are lysed in the gel and the lysate is cleared by centrifugation, standard biochemical procedures such as GST-pulldown assays and immunoprecipitations can be performed (Fig. 5).
Fig. 5. Performing biochemical procedures directly from lysed T47D cells cultured in 3D collagen gels.

Cells were cultured in attached or floating 3D collagen gels for one hour and a FAK/PI3-K co-immunoprecipitation performed as detailed in the protocols. Association between FAK and PI3-K is decreased in cells cultured in floating 3D collagen gels.
A second consideration when performing biochemical procedures in 3D collagen gels is volume. Pulldown assays and immunoprecipitations are normally performed in a total volume of under 1 ml lysate. However, the volume of the collagen gel is 1 ml, and addition of an equal volume of 2X lysis buffer makes the total volume 2 ml. We originally tried pouring smaller collagen gels (i.e., 0.5 ml), but they fell apart easily and the floating gels had a tendency to reattach to the dish. Therefore, 6-well plates were used and the gel volume was kept at 1ml.
Finally, because the total volume of the lysate is twice the volume normally used, the amount of protein, and thus the number of cells, must be increased. For an immunoprecipitation, we typically use 7-10 million cells per condition, whereas a pulldown assay requires 15-20 million cells. These numbers can vary and the minimum number of cells that can be used should be determined for each pulldown/immunoprecipitation. Because so many cells are necessary, cells can only be cultured for a maximum of 24 hours. (Such a large number of cells would outgrow the gel in a few days). Therefore, because a large number of cells are used for these assays, we currently cannot analyze later signaling events (at a few days, for example).
Biochemical analyses require many more cells than are typically used for morphogenesis studies. Therefore, an important consideration is that there may be differences in cell:cell junctions compared to collagen gels with fewer cells. However, we believe this is a valid way to analyze the difference in biochemical interactions because signaling changes are observed between cells in floating and attached 3D gels (Fig. 4 and (%R[22}%)) that are consistent with morphogenesis and immunostaining results. We are currently investigating the use of ELISA-based assays in order to analyze biochemical events. The advantage of the ELISA is that less protein (and thus less cells) can be used, which will allow one to directly compare tubulogenesis with biochemical events. In addition, using ELISA-based assays, we anticipate the ability to analyze signaling events that occur later in the differentiation process.
In conclusion, this paper has described methods used to analyze signal transduction in T47D breast epithelial cells cultured in 3D collagen gels. As it is becoming more apparent that signal transduction differs for cells cultured in 3D matrices as compared to 2D conditions, these approaches will likely become more regularly used and adjusted as technology advances.
Acknowledgments
The authors are grateful to Dr. Keith Burridge and members of his laboratory, who provided a RBD:GST purification and Rho activity assay protocol, which we modified for our use. We also thank Emily Vaughan and Dr. Scott Gehler for critically reading the manuscript. This work was supported by the American Cancer Society (RPG-00-339 to P.J.K).
Appendix
Protocols
Protocol 1: General maintenance of T47D breast epithelial cells
Reagents
T47D Media
RPMI (Cellgro)
2mM L-glutamine
8μg/ml insulin
10% heat-inactivated FBS
0.05% trypsin, 0.53mM EDTA (Cellgro)
15ml conical tubes
75cm2 tissue culture flask
Protocol
Aspirate the media. (Optional: rinse cells with PBS.)
To detach cells, add 3ml trypsin to each flask. Make sure the trypsin covers all the cells in the flask. The cells should detach and start to float in about five minutes. Gently tap the flask to ensure all the cells are detached.
Add 3ml media to the flask and gently pipette to collect all the cells and break up cell clumps. Add the 6ml cell/trypsin mixture to a 15ml conical tube.
Centrifuge the cells for three minutes at 1,100 rpm at room temperature.
While cells are centrifuging, prepare 75cm2 flasks by adding 10ml media to each flask.
Add media to the cells and divide them into new flasks. Grow the cells at 37°C, 5% CO2. A confluent flask split 1:3 will be confluent in about two days, and a confluent flask split 1:4 will be ready in about three days.
Guidelines
Keep track of passage number. In our hands, T47D cells begin to look and behave atypically around passage 45-50. At this time, new cells should be thawed and cultured.
Do not let T47D cells become too confluent (for example, when they become very small in size). Because they tend to grow in clusters, cells may need to be passed before the flask is completely confluent with cells.
Feed the cells about every two days, or when the media just begins to turn orange. If cells are confluent and are not fed or passed, the cells will begin to cluster together closely and pull off the plate. If this happens, do not continue to use these cells; instead, thaw new cells to be cultured.
Protocol 2: Culture of T47D breast epithelial cells in 3D collagen gels
Reagents
T47D media
RPMI (Cellgro)
2mM L-glutamine
8 μg/ml insulin
10% heat-inactivated FBS
0.05% trypsin, 0.53mM EDTA (Cellgro)
Collagen, Type I (BD Biosciences)
Neutralizing solution
100mM Hepes
pH 7.3 in 2X PBS
**Keep collagen and neutralizing solution at 4°C until needed.
15ml conical tubes
6-well plate
Protocol
Detach the cells to be cultured using trypsin. Spin the cells out of trypsin, resuspend in regular growth medium, and count cells. Plan to use 1-1.5 x 105 cells/gel.
Transfer the appropriate volume of cells to a tube.
Determine the volume of collagen and neutralizing solution needed as follows: Calculate the volume of collagen needed to make a collagen gel of 1.3mg/ml final concentration. The total volume is the number of gels, since each gel has a volume of 1ml. We typically make extra gel solution (~0.5ml extra) to account for the viscosity of collagen, and to ensure we have enough for each gel. The amount of neutralizing solution you will add is equal to the volume of collagen. The remaining volume is made up with regular growth medium. Below is an example of the correct volumes to combine for five gels, assuming 5 x 105 cells in 0.3ml medium and type I collagen with a stock concentration of 4mg/ml:
0.3ml cells
1.64ml media
1.78ml neutralizing solution
1.78ml collagen
Total=5.5ml for 5 gels
To create the collagen gels, add the appropriate volume of media to the tube containing cells. Next, add the neutralizing solution, and finally add the collagen. The neutralizing solution will control the pH of the final collagen gel. Make sure to keep the neutralizing solution and collagen cold until you are ready to use them, as higher temperatures increase the rate of collagen polymerization. (You may want to work on ice at first.)
Quickly, mix all solutions together. The final solution will become viscous and take on a yellow/pinkish hue. Pipette 1ml per well in a 6-well plate. Swirl the plate so the collagen solution covers the entire well and immediately place the plate in the incubator, at 37°C.
The collagen will polymerize around the cells in approximately 30 minutes-one hour. Therefore, at this time or the following day, you can release the gels and feed the cells. (Regardless of when the gel is floated, we see no differences in subsequent tubulogenesis.) Because we normally compare attached to floating gels, we will only float about half of the gels we pour. To float a collagen gel, use a small pipette tip (a 10 or 20μl tip) and insert it on the side of the dish, next to the gel. Carefully, rim the entire edge of the gel. Next, add 2ml media to the gel and carefully shake and swirl the plate to fully release the gel. For attached gels, simply add 2ml media carefully on top of the gel. Inhibitors can also be added at this time in the media; remember that the gel itself has a volume of 1ml, so add enough inhibitor for a 3ml volume in the 2mls media.
The cells in collagen gels will need to be fed about every four days, or whenever the media turns orange in color. Make sure to look at the cells every day to observe their morphology and to make sure the floating gels remain floating. (This is normally not a problem, but if the gels are not manipulated for a few days, they sometimes will reattach to the bottom of the well.)
Guidelines
Smaller gel volumes may be made in 12-well plates (in order to conserve collagen); however, we have found that these smaller gels, when floated in media, tend to reattach to the bottom of the dish more often compared to the larger gels made in a 6-well plate.
In floating gels, you can expect to see the cells rounding and the beginning of tubules at about 3-4 days. A more developed, extensive tubule network is usually seen around 7-10 days. In attached gels, the cells will not differentiate into tubules, and will proliferate more (vs. cells in floating gels), and develop cell extensions containing 3D matrix adhesions.
It is important to test the serum and collagen lots, as certain lots of serum may contain more or fewer growth factors. Lots of collagen may vary slightly also. We normally test lots of both and when a good quality lot is found, we order as much as we can of that lot, so there is no inconsistency in collagen gel culture.
To determine the extent to which breast cells can contract their surrounding gel, the diameter of the gels (in mm) can be measured and recorded every day for 10-12 days and then statistical analysis performed.
In order to examine the effect of collagen gel concentration on cell morphology, we culture cells in collagen gels the same way, except a higher concentration of stock collagen must be used. For these experiments, we special order high concentration collagen type I (9-12mg/ml) from BD Biosciences.
Protocol 3: Immunofluorescent staining of breast cells cultured in 3D collagen gels
Reagents
PBS
4% paraformaldehyde in PBS
0.15M glycine in PBS
0.02% Triton X-100 in PBS
Blocking solution
1% fatty acid-free BSA
1% donkey serum in PBS
Mounting medium, such as ProLong Antifade Solution (Molecular Probes)
Primary and seconday antibodies—the table below details antibodies we have successfully used:
Table 1.
Successfully used antibodies
| Antibody / Stain | Species | Supplier | Time | Dilution |
| α-actinin | Mouse | Sigma | 30 min | 1:100 |
| Bisbenzimide | n/a | Sigma | (with 2°) | 1:1000 |
| FAK | Mouse, clone 4.47 | Upstate | 30 min | 1:200 |
| Ki67 | Rabbit | Zymed | 30 min | 1:80 |
| Paxillin | Mouse | Transduction Labs | o/n | 1:100 |
| Phalloidin | n/a | Jackson Immunolabs | (with 2°) | 1:1000 |
| Phospho-FAK Y397 | Rabbit | Biosource | 30 min | 1:100 |
| Phospho-Src Y416 | Rabbit | Biosource | o/n | 1:100 |
| Talin | Mouse | Sigma | 30 min | 1:500 |
| Tensin | Mouse | Transduction Labs | o/n | 1:100 |
| Vinculin | Mouse | Sigma | 30 min | 1:400 |
Razor blade
Tweezers
12-well plate
Slides and coverslips
Protocol
Culture breast cells in attached and/or floating 3D collagen gels for the desired amount of time.
Using a sterile razor blade, cut the gel into 4 sections of equal size and move the individual pieces into a 12-well plate. (You will only use ¼ of the gel for staining. Therefore, you can stain for four different proteins using one gel.)
Wash the gels with PBS and then fix the gels with ~2ml 4% paraformaldehyde in PBS for 15 minutes at room temperature.
Wash the gels again in PBS for 10 minutes while shaking.
Add 0.15M glycine in PBS to the gels for 10 minutes to quench the formaldehyde.
Wash the gels in PBS for 10 minutes, and then add 0.02% Triton X-100 in PBS to the gels for 10 minutes to permeabilize the cells.
Wash with PBS for 10 minutes and block the cells with blocking solution overnight at 4°.
The following day, prepare 100μl diluted primary antibody per gel. Dilute the antibody in PBS with 1% donkey serum. Lay down a piece of parafilm, add the 100μl antibody solution, and then, using tweezers, add the piece of gel to the solution. This can be incubated overnight at 4°C, or for 30 minutes at room temperature, depending on the antibody used.
Wash the gels in PBS for 10 minutes 3-4 times.
Dilute the secondary antibody, anti-mouse or anti-rabbit TRITC or FITC (Jackson Immunolabs) 1:100 in PBS with 1% donkey serum. TRITC or FITC-conjugated phalloidin (Jackson Immunolabs) or bisbenzimide (Sigma) can also be added to this solution, both at a 1:1000 dilution. Incubate the diluted secondary antibody with the gels for 30 minutes at room temperature.
Wash the gels in PBS for 10 minutes, 2-4 times.
Wash the gels in water for 5 minutes, 2 times.
To mount the gels, pick up the piece of gel with tweezers and carefully blot it with a delicate task wiper. (Do not soak up too much liquid or the gel will shrivel; if this happens, simply rehydrate in water.) Lay the gel down on a glass slide and, using pipet tips, carefully pull out the gel so it lies flat on the slide—the gel will be about 0.5-1mm thick. Add 30μl ProLong Antifade Solution (Molecular Probes) on top of the gel. Lay a coverslip on top and carefully push down. Do not push down too hard or you will stretch out the gel and distort the tubule structures.
Let the Antifade Solution dry overnight, in the dark.
Guidelines
Cutting the gel into four pieces is optional. We see no distortion in tubule structure after cutting the gel prior to fixation and staining.
To capture images of immunostained cells in 3D collagen gels, use a deconvolution or confocal microscopy system. Z stack series can be collected in 50μm steps. Use two iterations for deconvolution.
Protocol 4: Immunoblotting for phospho-proteins from breast cells cultured in 3D collagen gels
Reagents
0.5mM EDTA in calcium and magnesium-free PBS
2X Lysis Buffer (a basic lysis buffer is provided below and may need to be modified depending on the protein being immunoblotted)
3X Laemmli Sample Buffer
Protocol
Detach cells using 0.5mM EDTA.
Culture breast cells in attached and/or floating 3D collagen gels in a total volume of 1ml/gel for the desired amount of time.
Carefully pipette off the media so only the gel is in the well.
To make a cell lysate, add 1ml 2X Lysis Buffer to the gel and pipette up and down. The collagen gel should start to break up and the solution will be slightly cloudy.
Let the cells lyse on ice for 10 minutes.
Centrifuge the lysate at 4°C for 12 minutes, at full speed, to spin out the cellular debris, which will be in the pellet. The supernatant is the cell lysate.
Add 30μl 3X Laemmli sample buffer to 60μl supernatant.
Heat the sample for 3 minutes at 100°C.
The sample can now be loaded into an SDS-PAGE gel for protein analysis.
Guidelines
0.5mM EDTA is used instead of trypsin in biochemical experiments because it does not proteolyse the integrins or other cell-surface receptors.
If a comparison between 2D and 3D is needed, petri plates can be coated with 30μg/ml collagen overnight and blocked with 5mg/ml fatty acid-free BSA in RPMI for 30 minutes. The plates are treated with BSA in RPMI in order to block any areas not fully coated with collagen. Fatty acid-free BSA is used because the fatty acids in normal BSA can bind to the phospholipids in the cell membrane and this may disrupt cell morphology and behavior. Equal numbers of cells can be cultured 2D vs. 3D, both in a total volume of 1ml.
The bands from at least three individual experiments can be quantitated by densitometry and then statistically analyzed.
Protocol 5: Immunoprecipitation and co-immunoprecipitation from cells cultured in 3D collagen gels
Reagents
0.5mM EDTA in calcium and magnesium-free PBS
BSA-RPMI: 5mg/ml fatty acid free BSA (MP Biomedicals) in RPMI
2X Lysis/RIPA buffer**
50mM HEPES, pH 7.4
150mM NaCl
2mM EDTA
2mM NaF
2% NP-40
0.5% DOC
Protease inhibitor cocktail (Sigma) (add before use)
2X NaVO4
**This is a basic lysis buffer that may have to be modified, depending on the protein being immunoprecipitated.
100X NaVO4
18mg NaVO4 in 1ml 10mM Tris, pH 10
Boil for 10 minutes
100X: 866μl water, 34μl 30% H2O2, 100μl boiled NaVO4
3X Laemmli Sample Buffer
Antibody (4μg per experimental condition)
GammaBind beads (Amersham) or other immunoprecipitation beads
Protocol
Detach cells in 0.5mM EDTA.
Culture breast cells in attached and/or floating 3D collagen gels in a total volume of 1ml/gel. Because more cells are typically needed for immunoprecipitations and pulldowns (approximately 10-20 million), the cells can be resuspended in a total volume of 600μl. To make a 1ml collagen gel, 400μl of a 2.5mg/ml collagen solution can be added to create a final collagen concentration of 1mg/ml. (For example, using a 10.9mg/ml stock of collagen, 2ml of 2.5mg/ml collagen can be made by adding 1.08ml BSA-RPMI, 460μl Neutralizing Solution, and 460μl collagen.) Allow the gels to polymerize for one hour at 37°C.
Carefully release one gel and add 1ml BSA in RPMI to each well. Inhibitors may also be added at this time. If the gel is stuck to the dish, gently release it using a cell scraper.
Incubate the gel for one hour at 37°C.
Carefully pipette off the media so only the gel is in the well.
Add 1ml 2X Lysis Buffer to the gel and pipette up and down. The collagen gel should start to break up and the solution will be slightly cloudy.
Let the cells lyse on ice for 10 minutes.
Centrifuge the lysate at 4°C for 12 minutes, at full speed, to spin out the cellular debris.
Make a control lysate sample by adding 30μl 3X Laemmli sample buffer to 60μl supernatant. Heat the sample for 3 minutes at 100°C.
To perform an immunoprecipitation, add 4μg antibody and 25μl GammaBind beads (Amersham) to the lysate (supernatant).
Incubate the lysate for 2h to overnight at 4°C, with rotation. The length of incubation depends on the antibody used and what is being immunoprecipitated.
Centrifuge the sample for 20sec at full speed, at 4°C. The supernantant can be discarded or frozen at -80°C.
Wash the beads three times with 1X lysis buffer.
Add 30μl 1X Laemmli sample buffer to the beads. Heat the sample for 3 minutes at 100°C.
The samples are now ready for SDS-PAGE analysis.
Guidelines
If a comparison between 2D and 3D is needed, petri plates can be coated with 30μg/ml collagen overnight and blocked with 5mg/ml fatty acid-free BSA in RPMI for 30 minutes. Equal numbers of cells can be cultured 2D vs. 3D, both in a total volume of 1ml.
The bands from at least three individual experiments can be quantitated by densitometry and then statistically analyzed.
Protocol 6: PBD:GST protein purification (for use in Rac activity assays)
Reagents
Glycerol stock of bacteria expressing PBD:GST fusion protein
IPTG
10X Tris Saline: 20mM Tris, pH 7.4
150mM NaCl
***Dilute to 1X before use. Store 1X and 10X at 4°.
250mM PMSF in absolute ethanol
10% Triton X-100
20% SDS
Glutathione sepharose beads (Amersham), 50% slurry
Protocol
Grow a 5ml overnight culture (+antibiotic).
The next morning, add the entire 5 ml culture to each flask (+antibiotic). Grow for about 3 or 4 hours at 37°C, or until the OD600 is about 0.6.
Induce each flask with 0.4mM final concentration of IPTG. (Make IPTG in fresh sterile water.) Induce for 4 hours at 37°C.
Harvest bacteria by centrifuging at 4,000rpm for 10 minutes at 4°. NOTE: At this point, the bacterial pellets can be frozen at -80°C until you are ready to do the purification.
Resuspend the bacteria in 1X Tris Saline, in a 50ml conical tube. Aim for a final volume of about 32ml.
Add PMSF at a 1:250 concentration. Close cap quickly.
Sonicate on ice two times, 60 pulses each time, at 50% duty cycle. Make sure to keep the bacterial solution cold.
Immediately after sonication, add Triton X-100 to a final concentration of 1%.
Incubate for 15 minutes at 4°C, with rotation.
Remove insoluble debris by centrifuging at 13,000 rpm for 12 minutes, at 4°C.
Incubate the supernatant with 800μl (50% slurry) glutathione sepharose beads for 30 minutes, with rotation, at 4°C. To prepare the beads, they are shipped in 30% ethanol so wash with 1X Tris Saline extensively before use. Add an equal volume 1X Tris Saline to the beads and this is your 50% slurry. Mix well before you add beads to the supernatant.
Wash the beads 4 times, each time with about 40ml Tris Saline.
Transfer to an eppendorf and adjust to a 50% slurry with 1X Tris Saline and store on ice at 4°.
Quantify protein concentration by running the protein on a SDS-PAGE gel with protein standards of known concentration. The PBD:GST fusion protein will be about 33kDa.
Guidelines
Both PBD and RBD:GST beads are only useful for one week unless 10% glycerol is added, in which case they can be frozen at -80°C.
For both proteins, the amount of bacteria you must grow to obtain enough protein depends on several parameters, such as the bacterial clone and the quality of protein purification.
Protocol 7: RBD:GST protein purification (for use in Rho activity assays)
Reagents
Glycerol stock of bacteria expressing RBD:GST fusion protein
Lysis buffer
50mM Tris, pH 7.4
50mM NaCl
5mM MgCl2
1% Triton X-100
1mM DTT
1mM PMSF
Protease inhibitor cocktail (add before use)
Glutathione sepharose beads
Protocol
Grow a 5ml overnight culture (+antibiotic).
The next morning, add the entire 5 ml culture to each flask (+antibiotic). Grow for about 3 or 4 hours at 37°C, or until the OD600 is about 0.6.
Induce each flask with 0.4mM final concentration of IPTG. (Make IPTG in fresh sterile water.) Induce for 4 hours at 37°C.
Harvest bacteria by centrifuging at 4,000rpm for 10 minutes at 4°C. NOTE: At this point, the bacterial pellets can be frozen at -80°C until you are ready to do the purification.
Resuspend the bacteria in Lysis Buffer, in a 50ml conical tube. Aim for a final volume of about 32ml.
Sonicate on ice four times, 30 pulses each time, at 50% duty cycle.
Remove insoluble debris by centrifuging at 13,000 rpm for 15 minutes, at 4°C.
Incubate the supernatant with 800μl (50% beads in lysis buffer without detergent or protease inhibitors) glutathione sepharose beads for one hour, with rotation, at 4°C.
Wash the beads three times in Lysis Buffer
Wash the beads three times in Lysis Buffer with no detergent.
Transfer to an eppendorf and adjust to a 50% slurry with Lysis Buffer (with no detergent) and store on ice at 4°C.
Quantify protein concentration by running the protein on a SDS-PAGE gel with protein standards of known concentration. The RBD:GST fusion protein will be about 33kDa.
Protocol 8: GTPase activity assays/GST pulldowns from cells cultured in 3D collagen gels
Reagents
0.5mM EDTA in calcium and magnesium-free PBS
BSA-RPMI: 5mg/ml fatty acid-free BSA (MP Biomedicals) in RPMI
2X Lysis buffer for Rho activity assays
50mM Tris, pH 7.6
400mM NaCl
0.5mM MgCl2
1% Triton X-100
0.1% SDS
0.5% DOC
Protease inhibitor cocktail (add before use)
2X NaVO4 (add before use)
2X Lysis buffer for Rac activity assays
100mM HEPES, pH 7.4
150mM NaCl
2mM EDTA
2μM NaF
0.2% SDS
0.5% DOC
2% NP-40
Protease inhibitor cocktail (add before use)
2X NaVO4 (add before use)
Wash buffer for Rho activity assays
50mM Tris, pH 7.6
150mM NaCl
0.5mM MgCl2
1% Triton X-100
Wash buffer for Rac activity assays
1X Lysis Buffer
3X Laemmli Sample Buffer
Purified GST fusion protein (GST:RBD or GST:PBD) on glutathione sepharose beads
Protocol
Detach cells using 0.5mM EDTA.
Culture breast cells in attached and/or floating 3D collagen gels in a total volume of 1ml/gel as described above. Allow the gels to polymerize for one hour at 37°C.
Carefully release one gel and add 1ml BSA in RPMI to each well. Inhibitors may also be added at this time. If the gel is stuck to the dish, gently release it using a cell scraper.
Incubate the gel for one hour at 37°C.
Carefully pipette off the media so only the gel is in the well.
Add 1ml 2X Lysis Buffer to the gel and pipette up and down. The collagen gel should start to break up and the solution will be slightly cloudy.
Let the cells lyse on ice for 10 minutes.
Centrifuge the lysate at 4°C for 12 minutes, at full speed, to spin out the cellular debris.
Make a control lysate sample by adding 30μl 3X Laemmli sample buffer to 60μl supernatant. Heat the sample for 3 minutes at 100°C.
To perform the pulldown, add 30μg GST:RBD or GST:PBD to the cleared lysate.
Incubate the lysate for 30 minutes at 4°C, with rotation.
Spin the lysate for ~ 20 seconds, at 4°C, to bring down the beads.
Wash the beads with 1ml Wash Buffer three times.
Add 30μl 1X Laemmli Sample Buffer to the beads. Heat the sample for 3 minutes at 100°C.
The samples are now ready for SDS-PAGE analysis—run the samples on a 15% gel, transfer to a PVDF membrane for 100 minutes at ~180mamps. Block overnight with 5% Blotto. For Rho activity assays, use anti-RhoA antibody (Transduction Laboratories) at a 1:250 dilution for 30 minutes, at 37°C. For Rac activity assays, use anti-Rac (Transduction Laboratories) at a 1:1000 dilution for 2 hours at room temperature.
Guidelines
If a comparison between 2D and 3D is needed, petri plates can be coated with 30μg/ml collagen overnight and blocked with 5mg/ml fatty acid-free BSA in RPMI for 30 minutes. Equal numbers of cells can be cultured 2D vs. 3D, both in a total volume of 1ml.
The bands from at least three individual experiments can be quantitated by densitometry and then statistically analyzed.
References
- Boyd NF, Lockwood GA, Byng JW, Tritchler DL, Yaffe MJ. Mammographic densities and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 1998;7(12):1133–1144. [PubMed] [Google Scholar]
- Emerman JT, Pitelka DR. Maintenance and induction of morphological differentiation in dissociated mammary epithelium on floating collagen membranes. In Vitro. 1977;13(5):316–328. doi: 10.1007/BF02616178. [DOI] [PubMed] [Google Scholar]
- Lee EY, Parry G, Bissell MJ. Modulation of secreted proteins of mouse mammary epithelial cells by the collagenous substrata. J Cell Biol. 1984;98(1):146–155. doi: 10.1083/jcb.98.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry G, Lee EY, Farson D, Koval M, Bissell MJ. Collagenous substrata regulate the nature and distribution of glycosaminoglycans produced by differentiated cultures of mouse mammary epithelial cells. Exp Cell Res. 1985;156(2):487–499. doi: 10.1016/0014-4827(85)90556-7. [DOI] [PubMed] [Google Scholar]
- Streuli CH, Bissell MJ. Expression of extracellular matrix components is regulated by substratum. J Cell Biol. 1990;110(4):1405–1415. doi: 10.1083/jcb.110.4.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcellos-Hoff MH, Aggeler J, Ram TG, Bissell MJ. Functional differentiation and alveolar morphogenesis of primary mammary cultures on reconstituted basement membrane. Development. 1989;105(2):223–225. doi: 10.1242/dev.105.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall HG, Farson DA, Bissell MJ. Lumen formation by epithelial cell lines in response to collagen overlay: a morphogenetic model in culture. Proc Natl Acad Sci USA. 1982;79(15):4672–4676. doi: 10.1073/pnas.79.15.4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand P, Petersen OW, Van Deurs B. A new diploid nontumorigenic human breast epithelial cell line isolated and propagated in chemically defined medium. In Vitro Cell Dev Biol. 1987;23(3):181–188. doi: 10.1007/BF02623578. [DOI] [PubMed] [Google Scholar]
- Weaver VM, Howlett AR, Langton-Webster B, Petersen OW, Bissell MJ. The development of a functionally relevant cell culture model of progressive human breast cancer. Semin Cancer Biol. 1995;6(3):175–184. doi: 10.1006/scbi.1995.0021. [DOI] [PubMed] [Google Scholar]
- Weaver VM, Petersen OW, Wang F et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137(1):231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3(9):785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freake HC, Marcocci C, Iwasaki J, MacIntyre I. 1,25-dihydroxyvitamin D3 specifically binds to a human breast cancer cell line (T47D) and stimulates growth. Biochem Biophys Res Commun. 1981;101(4):1131–1138. doi: 10.1016/0006-291x(81)91565-5. [DOI] [PubMed] [Google Scholar]
- Keely P, Fong A, Zutter M, Santoro S. Alteration of collagen-dependent adhesion, motility, and morphogenesis by the expression of antisense a2 integrin mRNA in mammary cells. J Cell Science. 1995;108:595–607. doi: 10.1242/jcs.108.2.595. [DOI] [PubMed] [Google Scholar]
- Keely P, Wu JE, Santoro S. The spatial and temporal expression of the alpha 2 beta 1 integrin and its ligands, collagen I, collagen IV, and laminin, suggest important roles in mouse mammary morphogenesis. Differentiation. 1995;59(1):1–13. doi: 10.1046/j.1432-0436.1995.5910001.x. [DOI] [PubMed] [Google Scholar]
- al Adnani MS, Taylor S, al-Bader AA, al-Zuhair AG, McGee JO. Immunohistochemical localization of collagens and fibronectin in human breast neoplasms. Histol Histopathol. 1987;2(3):227–238. [PubMed] [Google Scholar]
- Hewitt RE, Powe DG, Morrell K et al. Laminin and collagen IV subunit distribution in normal and neoplastic tissues of colorectum and breast. Br J Cancer. 1997;75(2):221–229. doi: 10.1038/bjc.1997.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30(3):256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Janmey PA, Weitz DA. Dealing with mechanics: mechanisms of force transduction in cells. Trends Biochem Sci. 2004;29(7):364–370. doi: 10.1016/j.tibs.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Grinnell F. Fibroblast biology in three-dimensional collagen matrices. Trends Cell Biol. 2003;13(5):264–269. doi: 10.1016/S0962-8924(03)00057-6. [DOI] [PubMed] [Google Scholar]
- Grinnell F. Fibroblast-collagen-matrix contraction: growth-factor signalling and mechanical loading. Trends Cell Biol. 2000;10(9):362–365. doi: 10.1016/S0962-8924(00)01802-X. [DOI] [PubMed] [Google Scholar]
- Roeder BA, Kokini K, Sturgis JE, Robinson JP, Voytik-Harbin SL. Tensile mechanical properties of three-dimensional type I collagen extracellular matrices with varied microstructure. J Biomech Eng. 2002;124(2):214–222. doi: 10.1115/1.1449904. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. 2003;163(3):583–595. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng MT, Safa AR. Light, fluorescent, and electron microscopic analysis of cultured breast tumor cells (T-47D) treated with 9,10-anthracenedicarboxaldehyde bis[(4,5-dihydro-1H-imidazol-2-yl)hydrazone] dihydrochloride. Cancer Res. 1983;43(12 Pt 1):5910–5914. [PubMed] [Google Scholar]
- Korah RM, Sysounthone V, Scheff E, Wieder R. Intracellular FGF-2 promotes differentiation in T-47D breast cancer cells. Biochem Biophys Res Commun. 2000;277(1):255–260. doi: 10.1006/bbrc.2000.3655. [DOI] [PubMed] [Google Scholar]
- Schafer JM, Lee ES, O'Regan RM, Yao K, Jordan VC. Rapid development of tamoxifen-stimulated mutant p53 breast tumors (T47D) in athymic mice. Clin Cancer Res. 2000;6(11):4373–4380. [PubMed] [Google Scholar]
- Keydar I, Chen L, Karby S et al. Establishment and characterization of a cell line of human breast carcinoma origin. Eur J Cancer. 1979;15(5):659–670. doi: 10.1016/0014-2964(79)90139-7. [DOI] [PubMed] [Google Scholar]
- Kolodney MS, Wysolmerski RB. Isometric contraction by fibroblasts and endothelial cells in tissue culture: a quantitative study. J Cell Biol. 1992;117(1):73–82. doi: 10.1083/jcb.117.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvoye P, Wiliquet P, Leveque JL, Nusgens BV, Lapiere CM. Measurement of mechanical forces generated by skin fibroblasts embedded in a three-dimensional collagen gel. J Invest Dermatol. 1991;97(5):898–902. doi: 10.1111/1523-1747.ep12491651. [DOI] [PubMed] [Google Scholar]
- Hovey RC, Trott JF. Morphogenesis of mammary gland development. Adv Exp Med Biol. 2004;554:219–228. doi: 10.1007/978-1-4757-4242-8_19. [DOI] [PubMed] [Google Scholar]
- Howard BA, Gusterson BA. Human breast development. J Mammary Gland Biol Neoplasia. 2000;5(2):119–137. doi: 10.1023/a:1026487120779. [DOI] [PubMed] [Google Scholar]
- Gudjonsson T, Ronnov-Jessen L, Villadsen R, Rank F, Bissell MJ, Petersen OW. Normal and tumor-derived myoepithelial cells differ in their ability to interact with luminal breast epithelial cells for polarity and basement membrane deposition. J Cell Sci. 2002;115(Pt 1):39–50. doi: 10.1242/jcs.115.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D, Gatter K. KI67 protein: the immaculate deception? Histopathology. 2002;40(1):2–11. doi: 10.1046/j.1365-2559.2002.01343.x. [DOI] [PubMed] [Google Scholar]