

Abstract
To determine if saquinavir mesylate (saquinavir) is a substrate of human multidrug resistance-associated protein 1 (hMRP1 [ABCC1]) or hMRP2 (cMOAT, or ABCC2), MDCKII cells that overexpress either hMRP1 (MDCKII-MRP1) or hMRP2 (MDCKII-MRP2) were used to investigate saquinavir's cytotoxicity and transport in comparison with those of control MDCKII wild-type (MDCKII/wt) cells. Cytotoxicity was assessed with the mitochondrial marker MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium], and saquinavir transport was measured directly through the cell monolayers. GF120918 (an inhibitor of P glycoprotein, but not of the MRP family) and MK-571 (an MRP family inhibitor) were used to delineate the specific contributions of these transporters to saquinavir cytotoxicity and transport. In the presence of GF120918 and increasing saquinavir concentrations, the MDCKII-MRP1 (50% lethal dose [LD50] = 10.5 μM) and MDCKII-MRP2 (LD50 = 27.1 μM) cell lines exhibited statistically greater viability than the MDCKII/wt cells (LD50 = 7.8 μM). Saquinavir efflux was directional, not saturable, and was inhibited by MK-571 (35 and 75 μM) in all cell lines. The ratios of saquinavir (3 μM) basolateral to apical permeability (i.e., efflux ratios) for the MDCKII/wt, MDCKII-MRP1, and MDCKII-MRP2 cell monolayers were 2.6, 1.8, and 6.8, respectively. The MDCKII-MRP1 cells have a significantly reduced saquinavir efflux ratio relative to MDCKII/wt cells, due to basolaterally directed transport by hMRP1 competing with endogenous, apically directed canine MRP2. The MDCKII-MRP2 cells have a significantly increased saquinavir efflux ratio relative to MDCKII/wt cells, due to the additive effects of the apically directed transport by hMRP2 and endogenous MRP2. Collectively, the cytotoxicity and transport results provide direct evidence that saquinavir is transported by MRP1 and MRP2.
The oral bioavailabilities of the human immunodeficiency virus (HIV) protease inhibitors (saquinavir, ritonavir, indinavir, nelfinavir, and amprenavir) are low and/or variable, with limited penetration into the central nervous system (CNS) (18). Saquinavir mesylate was the first drug approved in this class. The two marketed saquinavir capsule formulations have mean oral bioavailabilities that range from 4 to 16% and are highly variable, as indicated by area under the concentration-time curve (AUC) coefficients of variation that are ≥30% (11). Saquinavir's low and variable bioavailability is primarily attributed to metabolism by cytochrome P-450 3A4 (27). However, there is increasing understanding that membrane transporters contribute significantly to the biopharmaceutic characteristics of saquinavir and this entire class of drugs.
To relate bioavailability to molecular transport characteristics, we, like many others, speculated that an efflux (countertransport) mechanism might contribute to the low and variable bioavailability of saquinavir and other HIV protease inhibitors. Saquinavir efflux (basolateral to apical [BL→AP] permeability > AP→BL permeability) and the inhibition of saquinavir efflux with verapamil hydrochloride, a substrate for multiple transporters and a nonspecific efflux inhibitor, were demonstrated with a Caco-2 cell model (3). In preliminary work, we demonstrated that saquinavir efflux from rat intestinal tissue is an active process (27). However, these studies did not specifically identify the transporter or transporters involved within these complex tissues. Since Caco-2 cells and rat intestinal tissue are known to express multiple transporters, the observed saquinavir transport behavior may be related to multiple transporters with multiple affinities.
Recent studies have shown that the HIV protease inhibitors are substrates for the P glycoprotein (Pgp [ABCB1]) efflux pump and have demonstrated reduced saquinavir cytotoxicity due to saquinavir transport by Pgp (17). Saquinavir transport by Pgp has also been correlated with reduced bioavailability and CNS penetration (18). However, saquinavir transport by Pgp does not rule out saquinavir being a substrate for other putative membrane transporters. To this end, other investigators have demonstrated that saquinavir inhibits multidrug resistance-associated protein (MRP) family (MRP1 and MRP2)-mediated transport (15, 20, 24) and that a fluorescent saquinavir derivative is transported by MRP2 (14). Additionally, intracellular saquinavir concentrations were shown to be reduced in MRP1-expressing human lymphocytes relative to those in control cells (15, 16).
To determine if unmodified saquinavir is a substrate of human MRP1 (hMRP1 [ABCC1]) or the human canalicular multispecific organic anion transporter hMRP2 (cMOAT, or ABCC2), MDCKII cells that overexpress either hMRP1 (MDCKII-MRP1) or hMRP2 (MDCKII-MRP2) were used to investigate saquinavir's cytotoxicity and transport in comparison with those of control MDCKII wild-type (MDCKII/wt) cells (15, 16). The results of this study demonstrate saquinavir transport by hMRP1 and hMRP2, implicating these transporters in the reduced oral bioavailability and distribution of saquinavir.
MATERIALS AND METHODS
Chemicals.
Saquinavir mesylate (hereafter, “saquinavir”) and [14C]saquinavir were provided by Roche Laboratories (Nutley, N.J.) (28). GF120918 and MK-571 were provided by GlaxoSmithKline, Inc. (Research Triangle Park, N.C.), and Merck Laboratories, Inc. (Whitehouse, N.J.), respectively. [3H]mannitol and [3H]propranolol were obtained from Sigma Chemicals (St. Louis, Mo.). [14C]polyethylene glycol 400 (PEG 400) was obtained from American Radiolabeled Chemicals, Inc. (St. Louis, Mo.). [3H]vincristine and [3H]vinblastine were obtained from Amersham Pharmacia Biotech, Inc. (Piscataway, N.J.). Medium, fetal bovine serum (FBS), nonessential amino acids, trypsin, and transport buffer components were purchased from Fisher Scientific (Pittsburgh, Pa.). Transwell plates were purchased from Corning Costar Corp. (Cambridge, Mass.). All other chemicals were commercial products of reagent or enzymatic grade and were obtained from Fisher Scientific or Sigma Chemicals.
Cell culture.
These studies were performed with wild-type Madin-Darby canine kidney cells type II (MDCKII/wt), a well-characterized, polarized epithelial cell line) and stable MDCKII transfectants overexpressing hMRP1 (MDCKII-MRP1 cells), hMRP2 (MDCKII-MRP2 cells), and hPgp (MDCKII-PGP cells) (generously provided by R. Evers and P. Borst, The Netherlands Cancer Institute, Amsterdam) (4, 8, 9, 19). These cell lines were selected for use because they were the first polarized epithelial cells stably expressing these human transporters, enabling direct comparison of their functional behaviors without the confounding effects of multiple transporters expressed in cell lines such as Caco-2.
Studies were performed with the following cell passage numbers from our initial stock: MDCKII/wt (passages 6 to 21), MDCKII-MRP1 (passages 4 to 23), MDCKII-MRP2 (passages 8 to 18), and MDCKII-PGP (passages 12 to 19). The MDCKII cell lines were cultured in T-75 flasks with Dulbecco's modified Eagle's medium (with high glucose and glutamine concentrations) supplemented with 10% FBS, 1% nonessential amino acids, 100 μg of penicillin per ml, and 100 μg of streptomycin per ml. They were incubated at 37°C in 5% CO2 at 95% humidity. The medium was changed three times per week at regular intervals; cells were harvested via trypsinization at 80 to 90% confluence (about 4 days of growth) and passaged or seeded. Polycarbonate membranes (Transwell, 12-mm) were coated with type 1 rat tail collagen (100 μg/cm2), equilibrated with medium, and seeded at a density of 5 × 104 cells per cm2. Following seeding, medium was changed daily, and transport studies were performed 4 or 5 days after seeding.
Transporter expression and localization.
With these cell lines, the BL expression of hMRP1 and AP expression of hMRP2 and hPgp were previously demonstrated by Western blot analysis (4, 8, 9, 19). The expression of hMRP1, hMRP2, hPgp, and endogenous canine Pgp (cPgp) in these four different cell lines (MDCKII-MRP1, MDCKII-MRP2, MDCKII-PGP, and MDCKII/wt) was confirmed in our laboratory by Western blot and reverse transcription-PCR analyses (13). The expression of cMRP2 was previously identified in MDCKII/wt cells obtained from the same source by Western blot analysis (10).
In this study, the expression of total MRP2 (canine plus human) in the MDCKII/wt, MDCKII-MRP1, and MDCKII-MRP2 cell lines was confirmed by slot blotting. Actin levels were also assessed as an internal control for protein level. The MDCKII/wt, MDCKII-MRP1, and MDCKII-MRP2 cell lines were cultured as described above and seeded onto polycarbonate membranes (Transwell; 44-cm2 growth area) that had previously been coated with collagen. Concurrently, a blank control was prepared with a collagen-coated polycarbonate membrane (Transwell, 44-cm2 growth area) that was only treated with medium. The medium was changed daily, and protein was collected 4 days after seeding. Briefly, protein collection was performed with 1 ml of radioimmunoprecipitation assay buffer (RIPA; Santa Cruz Biotechnology, Santa Cruz, Calif.) per plate and scraping the membrane growth surfaces with cell scrapers. Protein levels were assayed, and 40 μg of protein was applied per lane per treatment group. After being blocked overnight with 5% nonfat powdered milk in Tris-buffered saline (TBS), antibody solutions were prepared and applied to the blots. Antibody solutions, prepared in 1% nonfat powdered milk in TBS, contained the following: a 1:500 dilution of MRP2-specific primary antibody H-17 (a goat polyclonal antibody raised against a peptide mapping near the hMRP2 amino terminus, which reacts with multiple MRP2 mammalian species; Santa Cruz Biotechnology), a 1:100 dilution of actin primary antibody I-19 (a goat polyclonal antibody raised against a peptide mapping near the human actin carboxy terminus, which reacts with multiple actin mammalian species; Santa Cruz Biotechnology), and a 1:1,000 dilution of the horseradish peroxidase-conjugated antigoat secondary antibody sc-2020 (Santa Cruz Biotechnology). Detection was performed by enhanced chemiluminescence (SuperSignal West Femto Maximum Sensitivity Substrate; Pierce Chemical Co., Rockford, Ill.) with a charge-coupled device (CCD) camera (NucleoTech 920 chemiluminescence imaging system equipped with an 8-bit, Peltier cooled digital camera).
Cytotoxicity testing.
Cytotoxicity studies were performed to screen the cell lines for differences in saquinavir cytotoxicity as a biological indicator of saquinavir transport by hMRP1 and hMRP2. Cytotoxicity studies of the MDCKII/wt, MDCKII-MRP1, MDCKII-MRP2, and MDCKII-PGP cell lines were performed with the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium] test (17). The MTT test for cytotoxicity is a colorimetric test that measures cell survival as a percentage of survival of untreated controls. Cells were seeded into 96-well plates at a density of 2 × 103 cells per cm2 (600 cells per 200 μl per well) and exposed to saquinavir (0, 0.4, 2.0, 10.0, 20.0, and 50.0 μM, the upper solubility limit in medium) for 4 days, with daily changes of medium. These studies were performed with and without 0.1 μM GF120918. GF120918 is an inhibitor of Pgp and breast cancer resistance protein (BCRP), but not of MRP family transporters. GF120918 has been shown to sensitize cells expressing Pgp and BCRP (7, 26). After 4 days of exposure, the cells were treated with MTT. MTT is a soluble tetrazolium salt that is cleaved in viable cells by active mitochondrial dehydrogenases forming blue or purple insoluble formazan crystals. The crystals are dissolved in 200 μl of dimethyl sulfoxide (DMSO) per well and measured with a 96-well plate reader at 570 nm. The blue color development associated with the 0 μM saquinavir treatment group (minus the blank) was designated 100% viable, and all comparisons were made relative to that reference. Vincristine (a positive control for MRP1 and also an MRP2 substrate) and vinblastine (an MRP2 substrate) (5) were evaluated as positive controls and confirmed the functionality of hMRP1, hMRP2, and hPgp in these cell lines under these test conditions (data not shown). All studies were performed at least three times, with six replicates (n = 6) per treatment group. Representative results are presented for each study.
Transport studies.
To investigate the differences in saquinavir cytotoxicity between these cell lines and relate them to efflux, saquinavir transport was evaluated with monolayers of each of these cell lines. All transport studies were performed with phosphate-buffered saline containing glucose (1 g/liter) at pH 7.4 (290 mOsmol). Saquinavir and inhibitor solutions were prepared immediately prior to performance of transport studies. Saquinavir and each inhibitor were separately dissolved in DMSO to make concentrated stock solutions (saquinavir, 20 mM; GF120918, 1 mM; MK-571, 50 mM) and then diluted in transport buffer to the specified working concentrations. The final DMSO concentration in all test solutions was constant at 0.50% (vol/vol). Saquinavir solutions, spiked with [14C]saquinavir, ranged from 1 to 47 μM (upper limit of solubility in transport buffer). To monitor monolayer integrity and serve as a permeability internal standard, [3H]mannitol (0.01 μM) was added to every test solution for every experiment. The AP and BL test solution volumes were 0.5 and 1.5 ml, respectively.
AP→BL transport and BL→AP transport were determined for each cell line. System equilibration and transport studies were performed at 37°C with rotation on an orbital shaker at 40 rpm. Prior to the addition of test solutions, cultured monolayers were rinsed and equilibrated for 30 min with transport buffer. Following the addition of test solutions, samples were withdrawn at 30, 45, 60, 75, 90, and 105 min with replacement buffer added to maintain constant volumes during each experiment. The AP and BL sample volumes were 50 and 150 μl, respectively, and all sample concentrations were corrected for dilution with replacement buffer during sampling. Samples were transferred to scintillation vials containing 5 ml of scintillation fluid, and all quantitations were performed by liquid scintillation counting. All transport studies were performed at least twice with four replicates (n = 4) per treatment group. Representative results are presented for each study.
Monolayer integrity.
To assess monolayer integrity, the permeabilities of [3H]mannitol, [3H]propranolol, and [14C]PEG 400 were tested for each cell line. PEG 400 was chosen as an additional paracellular marker because its molecular weight is closer to saquinavir's (weight, 767.0) than mannitol (weight, 182.2).
Transport inhibition.
To eliminate the potential contributions of endogenous Pgp and BCRP (MXR, or ABCP) to saquinavir transport, all transport studies were performed in the presence of GF120918. To chemically inhibit Pgp and BCRP, the cell monolayers were equilibrated with GF120918 (1 μM in transport buffer) on the AP and BL surfaces for 30 min prior to beginning the transport studies. All saquinavir solutions also contained 1 μM GF120918. This concentration of GF120918 was selected on the basis of a previous demonstration that equilibration of Caco-2 cells with 500 nM GF120918 chemically and effectively inhibits the contribution of Pgp (21). In addition, 1 μM GF120918 was previously shown to sensitize Pgp- and BCRP-expressing cell lines (7).
MRP family transporters were selectively inhibited with MK-571, a specific leukotriene D4 (LTD4) receptor antagonist. MK-571 specifically inhibits at least MRP1 and MRP2, but not Pgp. MK-571 has been shown to sensitize MRP1- and MRP2-expressing cell lines (6, 12, 25). To specifically inhibit the MRP family transporters, the cell monolayers were equilibrated with MK-571 (35 or 75 μM in transport buffer) for 30 min prior to beginning transport studies. The saquinavir (3 μM) solutions used in these studies also contained 35 or 75 μM MK-571 with 1 μM GF120918.
Functional transporter expression in cell monolayers.
To confirm the functional expression of hMRP1 and hMRP2 in these cell monolayers, the permeabilities of vincristine (positive control for MRP1 and also an MRP2 substrate) and vinblastine (an MRP2 substrate) were evaluated with the MDCKII-MRP1 and MDCKII-MRP2 cell lines (5). These confirmatory studies were performed in the presence of GF120918 (1 μM), to rule out the potential contributions of endogenous Pgp and BCRP. Directional transport of vincristine (200 nM, spiked with [3H]vincristine) and vinblastine (200 nM, spiked with [3H]vinblastine) was assessed for the MDCKII-MRP1 and MDCKII-MRP2 cell lines, respectively.
Numerical and statistical analyses.
The effective permeabilities (Pe, in centimeters per second) were calculated as follows:
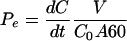 |
where dC/dt = the slope of the linear region of each concentration versus time plot, V = receiver chamber volume, C0 = initial drug concentration in the donor chamber, and A = filter (monolayer) surface area. To facilitate comparisons between the MDCKII/wt and MDCKII-MRP1 cell lines and to correct for small differences in paracellular transport, saquinavir permeabilities were normalized to representative mannitol permeabilities. For all studies, the MDCKII/wt and MDCKII-MRP1 cell line saquinavir permeabilities were normalized to a mannitol permeability of 3.0 × 10−6 cm/s. All MDCKII-MRP2 cell line saquinavir permeabilities were normalized to a mannitol permeability of 1.2 × 10−5 cm/s. Efflux ratios (the BL→AP/AP→BL permeability ratios) were calculated.
For illustrative purposes only, the concentration dependence data were fit by using the Michaelis-Menten equation:
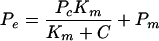 |
where Pc (carrier permeability) = (Jmax/Km), Pm = passive permeability, Jmax = maximal flux, and Km = the Michaelis constant. Trend lines were defined with weighted (1/standard deviation) nonlinear regression (SigmaPlot, version 7.0; SPSS, Inc., Chicago, Ill.). For concentration-dependent, but nonsaturable data, transport parameters cannot be delineated and are not reported.
Statistical assessments were performed with t tests for comparisons of two sample means and analysis of variance (Tukey's tests) for multiple comparisons (SigmaStat, version 2.03; SPSS, Inc.). The significance level was defined as a minimum P value of 0.05.
RESULTS
MRP2 expression.
Slot blot analysis (Fig. 1) confirmed that cMRP2 was expressed in the MDCKII/wt and MDCKII-MRP1 cell lines. The figure shows the relative expression of total MRP2 (canine plus human) and actin in the MDCKII/wt, MDCKII-MRP1, and MDCKII-MRP2 cell lines, as indicated by specific polyclonal antibodies. The similar actin levels obtained for these cell lines confirmed the use of comparable total protein loads.
FIG. 1.

Relative expression of total MRP2 (canine plus human) and actin in the MDCKII/wt, MDCKII-MRP1, and MDCKII-MRP2 cell lines. This blot confirmed that endogenous cMRP2 is expressed in the MDCKII/wt and MDCKII-MRP1 cell lines. The similar actin levels obtained for these cell lines confirmed the use of comparable total protein loads.
Monolayer integrity.
The average mannitol permeabilities for the studies in this report for each cell line are as follows: MDCKII/wt, 3.1e− 6 cm/s; MDCKII-MRP1, 2.7e− 6 cm/s; and MDCKII-MRP2, 1.1e− 5 cm/s. The mannitol permeabilities of the MDCKII/wt and MDCKII-MRP1 cell lines are not different from each other and are consistent with typical literature values for epithelial cell transport (2). However, the MDCKII-MRP2 mannitol permeabilities are significantly higher than those of the other two cell lines. The trend in PEG 400 (paracellular) transport was consistent with the mannitol data, demonstrating significantly higher MDCKII-MRP2 paracellular permeability (2.1e− 5 cm/s) than those for the MDCKII/wt (2.9e− 6 cm/s) and MDCKII-MRP1 (2.3e− 6 cm/s) cell lines. Within each cell line, the AP→BL and BL→AP mannitol and PEG (paracellular) permeabilities were not different (data not shown). The AP→BL propranolol (transcellular) permeabilities of these three cell lines were similar (ranging from 2.7 to 2.8e− 5 cm/s) and correspond with typical literature values for epithelial cell transport (1). Consistent interday mannitol and propranolol permeability results were obtained with all cell lines for a period of 4 to 6 days following seeding (data not shown).
Functional transporter expression in cell monolayers.
Directional transport of vincristine was observed with an efflux ratio of 2.2, which is consistent with active transport. While directional transport was expected, an efflux ratio of <1 was expected due to transport by BL-directed hMRP1. By using the MDCKII-MRP2 cell line, directional transport of vinblastine was observed, with an efflux ratio of 2.3, which is consistent with the AP expression of hMRP2.
Effect of saquinavir concentration on comparative cytotoxicity of MDCKII/wt, MDCKII-MRP1, MDCKII-MRP2, and MDCKII-PGP cells.
Figure 2 illustrates that the hMRP1-, hMRP2-, and hPgp-overexpressing cell lines exhibited statistically greater viability with increasing saquinavir concentration than that of the wild-type cells. GF120918 significantly reduced the viability of the MDCKII-PGP cell line (Fig. 2), but had no effect on the viabilities of MDCKII-MRP1, MDCKII-MRP2, and wild-type cell lines (Fig. 3). In the presence and absence of GF120918, the 50% lethal dose (LD50) values were 7.8 ± 0.6 μM for the wild type, 10.5 ± 1.3 μM for MDCKII-MRP1, and 27.1 ± 1.2 μM for MDCKII-MRP2. The LD50 values for the MDCKII-PGP cell line were 33.7 ± 1.4 μM in the absence of GF120918 and 7.2 ± 0.8 μM in the presence of GF120918. These results were consistent with saquinavir's known transport by Pgp and suggested saquinavir transport by hMRP1 and hMRP2.
FIG. 2.

Comparative viabilities of MDCKII cell lines in the presence of saquinavir. An asterisk indicates that the MDCKII/wt, MDCKII-PGP, MDCKII-MRP1, and MDCKII-MRP2 treatment groups were tested without GF120918. Each data point represents a mean value (n = 6). The following significant differences are indicated: a, at 10 and 20 μM saquinavir, the MDCKII-PGP*, MDCKII-MRP2* and MDCKII-MRP1* cells have greater viability than the MDCKII/wt* cells (P < 0.05); b, at 10 and 20 μM saquinavir the MDCKII-PGP* cells (without GF120918) have greater viability than the MDCKII-PGP cells with 0.1 μM GF120918 (P < 0.05).
FIG. 3.

Comparative viabilities of MDCKII/wt, MDCKII-MRP1, and MDCKII-MRP2 cell lines in the presence of saquinavir, with (+) or without (−) 0.1 μM GF120918. GF120918 had no effect on the viability of these cell lines, confirming the lack of contributions by Pgp and BCRP to the viability of the MDCKII-MRP1, MDCKII-MRP2, and MDCKII/wt cell lines under these conditions. Each data point represents a mean value (n = 6).
Concentration dependence and directionality of saquinavir transport.
To investigate these differences in saquinavir cytotoxicity, the AP→BL and BL→AP permeabilities for saquinavir were determined with MDCKII/wt, MDCKII-MRP1, and MDCKII-MRP2 cell monolayers. The permeabilities and efflux ratios for each cell line as a function of saquinavir concentration are illustrated in Fig. 4. In all cell lines, saquinavir transport was directional, with BL→AP permeabilities exceeding AP→BL permeabilities. The saquinavir efflux ratios ranged from 3.1 to 1.4 (MDCKII/wt) and 2.2 to 1.1 (MDCKII-MRP1) with increasing concentration. The MDCKII-MRP2 efflux ratios for saquinavir exhibited no trend with respect to increasing concentration and had a mean efflux ratio of 6.0 ± 0.9 (range, 4.9 to 7.4).
FIG. 4.

Saquinavir AP→BL and BL→AP permeability and efflux ratio concentration dependence following equilibration with GF120918 (1 μM) on both surfaces of MDCKII/wt (A), MDCKII-MRP1 (B), and MDCKII-MRP2 (C) cell monolayers. The trend lines are weighted (1/standard deviation) Michaelis-Menten fits. Each data point represents the mean ± standard deviation (n = 4).
For the MDCKII/wt and MRP1 cell lines, saquinavir transport appeared to be concentration dependent. Both cell lines showed trends of decreasing BL→AP permeabilities with corresponding trends of increasing AP→BL permeabilities as a function of increasing saquinavir concentration. However, transport was not saturable in this concentration range (47 μM is the upper solubility limit of saquinavir in this buffer system). This apparent concentration-dependent behavior suggested the involvement of a carrier-mediated transport process or processes. Saquinavir transport was not concentration dependent for the MDCKII-MRP2 cell monolayers, but had a constant efflux ratio of 6.0 over this concentration range, which also suggested a carrier-mediated transport process. In a separate study with the MDCKII-PGP cell line, but without GF120918, saquinavir (3 μM) had an efflux ratio of 71. These results were consistent with the cytotoxicity data in suggesting saquinavir transport by hMRP1 and hMRP2 and were additionally consistent with saquinavir's known transport by Pgp.
Inhibition of saquinavir efflux by MK-571.
To confirm that MRP family transporters mediated the transport of saquinavir in the MDCKII/wt, MDCKII-MRP1, and MDCKII-MRP2 cell lines, a series of inhibition studies were performed with MK-571 (0, 35, and 75 μM) in the presence GF120918 (1 μM).
The efflux ratios of the MDCKII/wt cells were 2.5 (no MK-571), 1.3 (35 μM MK-571), and 0.9 (75 μM MK-571). The MDCKII-MRP1 cells had efflux ratios of 1.8 (no MK-571), 1.1 (35 μM MK-571), and 1.0 (75 μM MK-571). The MDCKII-MRP2 cells had efflux ratios of 7.2 (no MK-571), 3.9 (35 μM MK-571), and 1.2 (75 μM MK-571). Complete and concentration-dependent inhibition of saquinavir transport by MK-571 in the presence of GF120918 indicated that an MRP family member was responsible for saquinavir transport (Fig. 5).
FIG. 5.

Inhibition of saquinavir (3 μM) efflux following equilibration with MK-571 and GF120918 (1 μM) on both surfaces of MDCKII/wt (A), MDCKII-MRP1 (B), and MDCKII-MRP2 (C) cell monolayers. AP→BL and BL→AP permeabilities are designated. Each data point represents the mean ± standard deviation (n = 4). The following significant differences are indicated: a, the efflux ratio for 0 μM MK-571 > 35 and 75 μM MK-571 (P < 0.05); b, the efflux ratio for 0 μM MK-571 > 35 and 75 μM MK-571 (P < 0.05); c, the efflux ratio for 0 μM MK-571 > 35 and 75 μM MK-571 (P < 0.05); d, the efflux ratio for 35 μM MK-571 > 75 μM MK-571 (P < 0.05).
DISCUSSION
The primary outcome of this work is the demonstration that saquinavir is transported by hMRP1 and hMRP2. These results are based on two experimental responses: a biological indicator, cytotoxicity, and the direct measurement of saquinavir transport. Saquinavir transport was measured with MDCKII-MRP1 and MDCKII-MRP2 cells with specific Pgp and MRP family inhibitors that rule out the potential contributions of other known transporters. Since saquinavir is known to be a substrate for Pgp, these studies were performed in the presence of GF120918 to specifically inhibit the contributions of Pgp and BCRP. Therefore, saquinavir transport in these studies cannot be attributed to Pgp or BCRP.
Earlier investigators have used the term “substrate” to describe molecules that bind to and are translocated by membrane transporters. However, others have used this term to describe molecules that bind to and inhibit transport, but are not themselves translocated. A recent example of this distinction is the finding that saquinavir inhibited tetraethylammonium transport by a human organic cation transporter (hOCT1), but is not transported by hOCT1. (29) While it was formerly shown that saquinavir inhibits MRP family (MRP1 and MRP2)-mediated transport (15, 20, 24), this does not provide evidence that saquinavir is translocated by MRP1.
In addition, a fluorescent saquinavir derivative was previously shown to be transported by MRP2 (14). Since it is known that changes in chemical structure, including derivatization, may alter transport characteristics (23), this does not provide evidence that saquinavir is translocated by MRP2. Although suggestive of saquinavir transport by MRP2, the derivatization of saquinavir may have potentially confounded the study result by creating a new molecule with transport properties different from those of the parent saquinavir molecule.
In this report, the saquinavir cytotoxicity results for the hMRP1 and hMRP2 cell lines suggested that saquinavir efflux might be the mechanism of increased cell viability. In the absence of inhibitors, the magnitudes of the LD50 values for these cell lines have the same rank order as the magnitudes of the efflux ratios for these cell lines (MDCKII-PGP > MDCKII-MRP2 > MDCKII-MRP1), establishing a clear relationship between saquinavir transport and cytotoxicity. The reduced viability of the MDCKII-PGP cells in the presence of GF120918 confirms the utility of GF120918 as a Pgp inhibitor in these studies. In addition, the lack of effect of GF120918 on the viability of MDCKII-MRP1 and MDCK-MRP2 cells is consistent with saquinavir transport by hMRP1 and hMRP2.
The directionality and apparent concentration dependence of saquinavir transport additionally support the involvement of MRP family carrier-mediated transport. Concentration-dependent efflux inhibition by MK-571 further suggests that the transporters in each cell line belong to the MRP family. Since MRP2 is the only known MRP family member that is apically directed, these results confirmed hMPR2 transport in the MDCKII-MRP2 cell line and suggested that saquinavir transport was comediated by cMRP2 in the MDCKII/wt and MDCKII-MRP2 cell lines. The observed saquinavir efflux in the wild-type cell line is consistent with the apical expression of endogenous cMRP2 that we have observed and that was previously observed in this cell line (10).
The concentration-dependent inhibition of saquinavir efflux by MK-571 in these cell lines suggests that the transporters mediating saquinavir efflux are members of the MRP family. These results are consistent with the expression of endogenous cMRP2 in the wild-type cells and confirm that saquinavir is transported by hMRP2. When these results are considered together with the increased viability of the hMRP1 cell line relative to the wild type, they also suggest that saquinavir is transported by hMRP1.
Figure 6 summarizes the mean efflux ratios for saquinavir (3 μM) and these cell lines, while also highlighting the apparent roles of the multiple transporters expressed in these cell lines. For example, the MDCKII/wt cells had a mean saquinavir efflux ratio of 2.6, presumably due to translocation by AP-expressed endogenous cMRP2. However, the MDCKII-MRP2 cells had a significantly increased efflux ratio, 6.8, relative to that of the wild type due to the additive effects of AP-directed transport by hMRP2 and cMRP2. Additionally, the MDCKII-MRP1 cells had a significantly reduced efflux ratio, 1.8, relative to that of the wild type due to BL-directed transport by hMRP1 in competition with AP-directed cMRP2. Under these conditions, the MDCKII-MRP1 cell monolayers demonstrate reduced efflux in the AP direction, but augmented total cellular efflux relative to the wild-type cells. The MDCKII-MRP1 total cellular efflux is bidirectional, representing the sum of AP→BL plus BL→AP transport. The increased viability of the MDCKII-MRP1 cells relative to the wild-type cells supports this model of augmented total cellular efflux and is consistent with the reduced intracellular saquinavir concentrations that were previously observed with MRP1-expressing human lymphocyte cells relative to those of control cells (15, 16).
FIG. 6.

Comparative saquinavir (3 μM) efflux ratios for MDCKII cell monolayers following GF120918 (1 μM) equilibration on both surfaces. This model illustrates the competing and additive directional transport of saquinavir by hMRP1, cMRP2, and hMRP2 in these cell lines. Each bar represents the mean ± standard deviation (n = 12 to 16). The following significant differences are indicated: a, the efflux ratio for MDCKII-MRP1 < MDCKII/wt (P < 0.05); b, the efflux ratio for MDCKII-MRP2 > MDCKII/wt (P < 0.05).
Furthermore, this model explains the efflux ratio obtained for vincristine. Vincristine is a substrate for MRP1 and MRP2 (5). When applied to MDCKII-MRP1 cell monolayers, vincristine, like saquinavir, is subject to competing AP and BL transporters. These results demonstrate the roles of multiple transporters—transfected and endogenous and known and unknown—and their effects on the interpretation of whole-cell transport data. While these data consistently support the contributions of hMRP1, hMRP2, and cMRP2 to saquinavir transport, they do not rule out the potential contributions of additional transport processes. To examine the roles of domain-specific transport events and eliminate potentially confounding factors, it is likely that membrane vesicle studies would have to be performed.
The MDCKII-MRP2 transport and cytotoxicity data suggest that saquinavir is readily transported by hMRP2. However, the increased saquinavir efflux ratio for the MDCKII-MRP2 cells relative to that of the wild type seems small compared to the large relative increase observed in cell viability. It is hypothesized that the increased paracellular permeability of the MDCKII-MRP2 monolayers (average mannitol permeability, 1.1e− 5cm/s) relative to the MDCKII/wt and MDCKII-MRP1 cell lines (average mannitol permeability, 2.9e to 6cm/s) obscures the measurement of saquinavir efflux, resulting in the observation of a reduced efflux ratio for the MDCKII-MRP2 monolayers. In one follow-up study with MDCKII-MRP2 cell monolayers, reduced mannitol permeability did appear to correlate with increased saquinavir efflux ratios (data not shown). Collectively, these facts strongly suggest that saquinavir has a significant paracellular transport component. However, the enhanced viability of the MDCKII-MRP1 and MDCKII-MRP2 cell lines along with MK-571's complete and concentration-dependent inhibitions of saquinavir efflux in these cell lines specifically indicates hMRP1 and hMRP2 transport, which differentiates these MRP family-mediated transport events from differences in paracellular transport. It is also clear from the significantly different paracellular permeabilities of these cell lines that the MDCKII-MRP2 cell tight junctional proteins are expressed with compositions and/or functionalities different from those of the other MDCKII cell lines, which suggests that there may be additional differences in expression and properties.
In conclusion, our results provide direct evidence that saquinavir is transported by hMRP1 and hMRP2. Saquinavir transport by hMRP1 and hMRP2, in addition to that by hPgp, has an apparent rank order of hPgp > hMRP2 ≫ hMRP1 and suggests that the roles of multiple transporters may help to explain the low and/or variable oral absorption of saquinavir and the other HIV protease inhibitors. The BL localization of intestinal MRP1 may facilitate saquinavir absorption, reducing the net secretory effect of Pgp and MRP2. Additionally, MRP1, MRP2, and Pgp may contribute to CNS barrier functions. (22, 24) A mechanism-based approach to improving the oral absorption and distribution of the HIV protease inhibitors requires additional characterization of their transport processes, the contributions of putative transporters, and their physiological relevance.
Acknowledgments
Financial support was provided by NIH grant AI 42007. This work was also supported by Roche Laboratories, GlaxoSmithKline, and Merck Laboratories.
We thank Raymond Evers and Piet Borst for providing us with the MDCKII cell lines. We additionally thank Kurt Amsler, Joseph Polli, and Hugh Wiltshire for assistance.
REFERENCES
- 1.Adson, A., P. Burton, T. Raub, C. Barsuhn, K. Audus, and N. Ho. 1995. Passive diffusion of weak organic electrolytes across Caco-2 cell monolayers: uncoupling the contributions of hydrodynamic, transcellular, and paracellular barriers. J. Pharm. Sci. 84:1197-1204. [DOI] [PubMed] [Google Scholar]
- 2.Adson, A., T. Raub, P. Burton, C. Barsuhn, A. Hilgers, K. Audus, and N. Ho. 1994. Quantitative approaches to delineate paracellular diffusion in cultured epithelial cell monolayers. J. Pharm. Sci. 83:1529-1536. [DOI] [PubMed] [Google Scholar]
- 3.Alsenz, J., H. Steffen, and R. Alex. 1998. Active apical secretory efflux of the HIV protease inhibitors saquinavir and ritonavir in Caco-2 cell monolayers. Pharm. Res. 15:423-428. (Erratum, 15:958.) [DOI] [PubMed] [Google Scholar]
- 4.Bakos, E., R. Evers, G. Szakacs, G. Tusnady, E. Welker, K. Szabo, M. de Haas, L. van Deemter, P. Borst, A. Varadi, and B. Sarkadi. 1998. Functional multidrug resistance protein (MRP1) lacking the N-terminal transmembrane domain. J. Biol. Chem. 273:32167-32175. [DOI] [PubMed] [Google Scholar]
- 5.Borst, P., R. Evers, M. Kool, and J. Wijnholds. 1999. The multidrug resistance protein family. Biochim. Biophys. Acta 1461:347-357. [DOI] [PubMed] [Google Scholar]
- 6.Chen, Z.-S., T. Kawabe, M. Ono, S. Aoki, T. Sumizawa, T. Furukawa, T. Uchiumi, M. Wada, M. Kuwano, and S.-I. Akiyama. 1999. Effect of multidrug resistance-reversing agents on transporting activity of human canalicular multispecific organic anion transporter. Mol. Pharmacol. 56:1219-1228. [DOI] [PubMed] [Google Scholar]
- 7.de Bruin, M., K. Miyake, T. Litman, R. Robey, and S. Bates. 1999. Reversal of resistance by GF120918 in cell lines expressing the ABC half-transporter, MXR. Cancer Lett. 146:117-126. [DOI] [PubMed] [Google Scholar]
- 8.Evers, R., N. Cnubben, J. Wijnholds, L. van Deemter, P. van Bladeren, and P. Borst. 1997. Transport of glutathione prostaglandin A conjugates by the multidrug resistance protein 1. FEBS Lett. 419:112-116. [DOI] [PubMed] [Google Scholar]
- 9.Evers, R., M. Kool, L.van Deemter, H. Janssen, J. Calafat, L. Oomen, C. Paulusma, R. Elferink, F. Baas, A. Schinkel, and P. Borst. 1998. Drug export activity of the human canalicular multispecific organic anion transporter in polarized kidney MDCK cells expressing cMOAT (MRP2) cDNA. J. Clin. Investig. 101:1310-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flanagan, S., C. Cummins, M. Susanto, X. Liu, L. Takahashi, and L. Benet. 2002. Comparison of furosemide and vinblastine secretion from cell lines overexpressing multidrug resistance protein (P-glycoprotein) and multidrug resistance-associated proteins (MRP1 and MRP2). Pharmacology 64:126-134. [DOI] [PubMed] [Google Scholar]
- 11.Flexner, C. 1998. HIV-protease inhibitors. N. Engl. J. Med. 338:1281-1292. [DOI] [PubMed] [Google Scholar]
- 12.Gekeler, V., W. Ise, K. Sanders, W. Ulrich, and J. Beck. 1995. The leukotriene LTD4 receptor antagonist MK571 specifically modulates MRP associated multidrug resistance. Biochem. Biophys. Res. Commun. 208:345-352. [DOI] [PubMed] [Google Scholar]
- 13.Guo, A., W. Marinaro, P. Hu, and P. Sinko. 2002. Delineating the contribution of secretory transporters in the efflux of etoposide using Madin-Darby canine kidney (MDCK) cells overexpressing P-glycoprotein, multidrug resistance-associated protein (MRP1), and canalicular multispecific organic anion transporter (cMOAT). Drug Metab. Dispos. 30:457-463. [DOI] [PubMed] [Google Scholar]
- 14.Gutmann, H., G. Fricker, J. Drewe, M. Toeroek, and D. Miller. 1999. Interactions of HIV protease inhibitors with ATP-dependent drug export proteins. Mol. Pharmacol. 56:383-389. [DOI] [PubMed] [Google Scholar]
- 15.Jones, K., P. Bray, S. Khoo, R. Davey, E. Rhiannon Meaden, S. Ward, and D. Back. 2002. P-glycoprotein and transporter MRP1 reduce HIV protease inhibitor uptake in CD4 cells: potential for accelerated viral drug resistance? AIDS 15:1353-1358. [DOI] [PubMed] [Google Scholar]
- 16.Jones, K., P. Hoggard, S. Sales, S. Khoo, R. Davey, and D. Back. 2001. Differences in the intracellular accumulation of HIV protease inhibitors in vitro and the effect of active transport. AIDS 15:675-681. [DOI] [PubMed] [Google Scholar]
- 17.Kim, A. E., J. M. Dintaman, D. S. Waddell, and J. A. Silverman. 1998. Saquinavir, an HIV protease inhibitor, is transported by P-glycoprotein. J. Pharmacol. Exp. Ther. 286:1439-1445. [PubMed] [Google Scholar]
- 18.Kim, R. B., M. F. Fromm, C. Wandel, B. Leake, A. J. Wood, D. M. Roden, and G. R. Wilkinson. 1998. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J. Clin. Investig. 101:289-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Louvard, D. 1980. Apical membrane aminopeptidase appears at site of cell-cell contact in cultured kidney epithelial cells. Proc. Natl. Acad. Sci. USA 77:4132-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller, D., S. Nobmann, H. Gutmann, M. Toeroek, J. Drewe, and G. Fricker. 2000. Xenobiotic transport across isolated brain microvessels studied by confocal microscopy. Mol. Pharmacol. 58:1357-1367. [DOI] [PubMed] [Google Scholar]
- 21.Polli, J., J. Jarrett, S. Studenberg, J. Humphreys, S. Dennis, K. Brouwer, and J. Woolley. 1999. Role of P-glycoprotein on the CNS disposition of amprenavir (141W94), an HIV protease inhibitor. Pharm. Res. 16:1206-1212. [DOI] [PubMed] [Google Scholar]
- 22.Rao, V., J. Dahlheimer, M. Bardgett, A. Snyder, R. Finch, A. Sartorelli, and D. Piwnica-Worms. 1999. Choroid plexus epithelial expression of MDR1 P glycoprotein and multidrug resistance-associated protein contribute to the blood-cerebrospinal-fluid drug-permeability barrier. Proc. Natl. Acad. Sci. USA 96:3900-3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sinko, P., and P. Balimane. 1998. Carrier-mediated intestinal absorption of valacyclovir, the l-valyl ester prodrug of acyclovir. 1. Interactions with peptides, organic anions and organic cations in rats. Biopharm. Drug Dispos. 19:209-217. [DOI] [PubMed] [Google Scholar]
- 24.Srinivas, R., D. Middlemas, P. Flynn, and A. Fridland. 1998. Human immunodeficiency virus protease inhibitors serve as substrates for multidrug transporter proteins MDR1 and MRP1 but retain antiviral efficacy in cell lines expressing these transporters. Antimicrob. Agents Chemother. 42:3157-3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Aubel, R., M. van Kuijck, J. Koenderink, P. Deen, C. van Os, and F. Russel. 1998. Adenosine triphosphate-dependent transport of anionic conjugates by the rabbit multidrug resistance-associated protein MRP2 expressed in insect cells. Mol. Pharmacol. 53:1062-1067. [PubMed] [Google Scholar]
- 26.Wallstab, A., M. Koester, M. Bohme, and D. Keppler. 1999. Selective inhibition of MDR1 P-glycoprotein-mediated transport by the acridone carboxamide derivative GG918. Br. J. Cancer 79:1053-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams, G. C., and P. J. Sinko. 1999. Oral absorption of the HIV protease inhibitors: a current update. Adv. Drug Del. Rev. 39:211-238. [DOI] [PubMed] [Google Scholar]
- 28.Wiltshire, H. R., K. J. Prior, J. Dhesi, F. Trach, M. Schlageter, and H. Schonenberger. 1998. The synthesis of labelled forms of saquinavir. J. Label. Compd. Radiopharm. 41:1103-1126. [Google Scholar]
- 29.Zhang, L., W. Gorset, C. Washington, T. Blaschke, D. Kroetz, and K. Giacomi. 2000. Interactions of HIV protease inhibitors with a human organic cation transporter in a mammalian expression system. Drug Metab. Dispos. 28:329-334. [PubMed] [Google Scholar]