

Abstract
An in situ method for sensitive detection of differences in the action of chlorhexidine against subpopulations of cells in Candida albicans biofilms is described. Detection relies on monitoring the kinetics of propidium iodide (PI) penetration into the cytoplasm of individual cells during dosing with chlorhexidine. Accurate estimation of the time for delivery of the dosing concentration to the substratum was facilitated by using a flow cell system for which transport to the interfacial region was previously characterized. A model was developed to quantify rates of PI penetration based on the shape of the kinetic data curves. Yeast were seeded onto the substratum, and biofilm formation was monitored microscopically for 3 h. During this period a portion of the yeast germinated, producing filamentous forms (both hyphae and pseudohyphae). When the population was subdivided on the basis of cell morphology, rates of PI penetration into filamentous forms appeared to be substantially higher than for yeast forms. Based on the model, rates of penetration were assigned to individual cells. These data indicated that the difference in rates between the two subpopulations was statistically significant (unpaired t test, P < 0.0001). A histogram of rates and analysis of variance indicated that rates were approximately equally distributed among different filamentous forms and between apical and subapical segments of filamentous forms.
Biofilms are communities of microbes colonizing a surface. Biofilm formation has been implicated as a factor that contributes significantly to the difficulty encountered in treating a number of infections. Classic examples include infections of the lungs of cystic fibrosis patients (26) and of the oral cavity (29) and those associated with medical implants (10). Biofilms have been shown to be resistant to antimicrobial agents in numerous in vitro studies, inspiring speculation that there are general mechanisms of biofilm resistance (40).
Candida albicans is an opportunistic pathogen that infects primarily immunocompromised hosts. It causes serious, life-threatening systemic as well as discomforting superficial infections. Superficial C. albicans infections such as thrush manifest as a complex biofilm consisting of yeast, hyphae, and desquamated epithelial cells (35). Biofilms on oral surfaces (dental plaque) are responsible for caries and periodontal disease. The role of surface-associated C. albicans in denture stomatitis is well documented, and it is possible that involvement of C. albicans in oral infections commonly attributed to bacterial biofilms, such as periodontitis, is underestimated (33).
Candida species are the principle etiological agent of nosocomial fungal infections, with C. albicans being the most common species (13). Acquisition can occur via contaminated infusates and biomedical devices (36). Use of indwelling catheters, total parenteral nutrition, and hemodialysis increases the risk of Candida infection (13). A relatively high level of risk is associated with the use of urinary and intravenous catheters (38, 44). C. albicans biofilms form readily on biomaterials in vitro (19) and were shown to have similar structures to those formed in vivo on catheters (8). Detachment of microbes from biofilms growing under flowing conditions occurs continuously. Thus, biofilms colonizing biomedical devices provide a reservoir of persistent contamination of catheter streams.
As both a former of biofilms and a commensal, C. albicans shares traits with Staphylococcus aureus and S. epidermidis, the bacteria found most frequently associated with medical implant infections. In vitro studies show that C. albicans biofilms are well adapted to survive in the low-iron environments found in the vicinity of implant devices (1). Fungal infections of prosthetic devices are less common than bacterial infections but are more difficult to treat (12).
C. albicans has both a budding and a filamentous life cycle. C. albicans biofilms incorporating both yeast and filamentous forms may be optimal for colonization of inert surfaces, with yeast forming an adhesive basal layer and the filaments providing the scaffolding for a multilayered architecture (2). C. albicans communities observed in clinical specimens consist typically of a pleomorphic mixture of budding and filamentous forms (35). Therefore, although there is an emphasis on the virulence of the filamentous form (30), successful pathogenesis may be enhanced by a mixed community structure similar to that typically observed in biofilms. C. albicans biofilms provide an ideal in vitro model system for characterizing physiological differences between morphological forms in these complex systems.
Chlorhexidine is a biocide used extensively as a topical antiseptic in hospitals (11) and for treatment of periodontal diseases (28). Chlorhexidine has a broad spectrum of activity against a variety of organisms, including C. albicans. Susceptibility of C. albicans biofilms to chlorhexidine was shown to be significantly reduced compared to its action against suspended organisms (9).
It has been pointed out that the resistance of biofilms originates typically from the recalcitrance of a small subpopulation (27). Analysis of the action of antimicrobial agents against biofilms by methods that preserve and discriminate the spatial arrangement of cells is needed to determine whether biofilms develop protective niches that allow some organisms to survive antimicrobial dosing. One method for in situ analysis is described here. The assay is based on the rate of propidium iodide (PI) penetration into the cytoplasm of cells as plasma membrane integrity is compromised by the action of chlorhexidine. PI was used previously as a C. albicans viability probe with respect to the action of amphotericin B, a polyene that disrupts the plasma membrane (17, 37).
MATERIALS AND METHODS
Growth media and chemicals.
The medium for batch cultures was as follows (per liter): 50 g of glucose, 10 g of Bacto Peptone (Difco), and 6 g of yeast extract (Difco). The medium for biofilm growth was as follows (per liter): 0.05 g of glucose, 0.1 g of Bacto Peptone, and 0.03 g of yeast extract (pH 6.5). Phosphate-buffered saline (PBS) was as follows (per liter): 0.34 g of monobasic sodium phosphate, 2.56 g of dibasic sodium phosphate, and 8.50 g of NaCl (pH 7.2). The solid medium was Sabouraud dextrose agar (Difco). PI and chlorhexidine digluconate (CHG) were from Sigma Chemical Co.
Organism.
C. albicans (CA-1) (18) is a clinical isolate obtained from the culture collection of Diane Brawner (Microbiology Department, Montana State University). Frozen stocks were maintained in 50% glycerol at −40°C. Slants were prepared from single 48-h colonies obtained from a streak of the frozen stock on Sabouraud dextrose agar.
Flow system.
The flow system enables analysis by conventional microscopy, Raman microscopy (Raman microprobe spectroscopy), and attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy (41, 42). The spectroscopic techniques were used previously to characterize transport into the interfacial region (ATR-FTIR) and into defined laterally discriminated regions of biofilms (Raman). Knowledge of the kinetics of transport into the interfacial region is necessary for interpretation of the data presented here. The flow channel is approximately rectangular (45 by 10 by 0.86 mm), with entrance and exit ports at each end of the long dimension. The top and bottom walls of the flow channel are comprised of a window for microscopic observation made from a coverslip and a single crystal, trapezoidal germanium (Ge) prism (Harrick Scientific Corp., Ossining, N.Y.) (50 by 20 by 2 mm3 and 45°) compatible with ATR-FTIR measurements, respectively. The thin walls of the flow cell cavity are composed of a sandwich of Teflon and viton spacers (McMaster-Carr Supply Co., Los Angeles, Calif.). The Teflon spacer contacts the prism. The Ge surface was cleaned by a protocol described previously (14). The flow system was modified to allow thermostatic regulation of the temperature (maintained at 37°C). This was accomplished by heating the aluminum base of the flow cell. A reservoir of water over the coverslip also equilibrated to this set temperature. The reservoir was insulated by a viton sheet overlayer (McMaster-Carr Supply Co., Los Angeles, Calif.) (except where the water immersion lens entered the water). Flow rate was regulated to 0.5 ml/min by making necessary adjustments in the pump rotation speed, with drop counts per time period in flow break tubes (calibrated by determination of the volume of liquid per drop) used as feedback.
Microscopy.
The microscope was an Olympus BX60. The stage was positioned so that the field of view was centered laterally with respect to the flow channel for all experiments. Differential interference contrast (DIC) optics was used to visualize cellular structures. A 40× water immersion (LUMPlanFl 40x/0.8W; Olympus) objective was used to monitor biofilm development (final magnification, ×400). It has a Teflon tip that minimizes heat conduction. Epi-illumination through a U-MWIG filter set was used to observe PI fluorescence, using a 20× objective (UPlanFl 20x/0.5W; Olympus) (final magnification, ×200). The lower magnification was used to increase the field of view (field size, 300 by 465 μm2). A black and white Photometrics charge-coupled device camera was used to digitally capture the images. Light from the Mercury lamp was reduced by a factor of 1/16 by a stack of four neutral density filters for excitation of PI fluorescence. In addition, a shutter was opened during a short period only (2 s) to allow image acquisition. These steps were taken to reduce exposure to the excitation light, thereby minimizing the effects of photobleaching. To provide a criterion for this minimization, heat-killed yeast cells were exposed to 20 μg of PI/ml in PBS. Under these conditions, all cells become maximally labeled in 1 to 2 min. The time course of decrease in PI fluorescence from individual cells was measured for different illumination conditions. Light exposure was reduced until this decrease was made negligible over a 10-min interval.
Data acquisition and image analysis.
Images were acquired and analyzed using Media Cybernetics Image-Pro Plus software (Media Cybernetics, Silver Spring, Md.). Gain (5) and exposure (325 ms) settings were adjusted so that pixel brightness was below saturation (pixel intensity value, 255) for all images. These settings were determined in preliminary experiments with maximally PI-labeled (heat-killed) yeast cells in a hemacytometer. Image acquisition for kinetic studies was automated using a macro routine. For measurement of changes in brightness of individual objects with respect to time, locations of objects were first acquired by viewing the brightest epi-fluorescence image (i.e., typically the last image in the time series). A square area (11 by 11 pixels) was demarcated that covered each object. The sum of the pixel intensity values (0 to 255) within the area was then calculated for each location in each image using a macro routine. For fields in which the objects were yeast cells seeded onto the surface, the locations were calculated automatically by identifying objects by using a brightness thresholding procedure and then computing the centroid. For seeded yeast, 200 cells were evaluated per field in all cases. For the 3-h biofilm, the locations were chosen manually by moving the square area to different locations of the image and recording these positions in a macro routine. Cell locations were stored and used in another macro routine that indicated each position, numerically ordered, in a DIC image acquired immediately before acquisition of the fluorescent images. The appearances of cellular structures in the epi-fluorescence and DIC images were compared on a one-to-one basis.
Seeding of yeast onto the substratum.
Batch cultures were grown in 100 ml of medium at 37°C with shaking at 160 rpm. An overnight batch culture, inoculated from a slant, served as a starter culture. Fresh, sterile medium was inoculated with 3 ml of the starter culture. After 14 h, 1 ml of the second batch culture was spun down and resuspended in 1 ml of PBS. Under these conditions no filamentous forms are present in batch cultures. A total of 100 μl of this cell suspension (containing approximately 2 × 108 cells/ml) was added to 35 ml of PBS in a flask connected to the autoclave-sterilized flow system described above. This dilute PBS cell suspension was pumped through the flow cell for 30 min at 0.5 ml/min. Flow was then diverted to introduce medium into the flow cell. This period of exposure to culture medium was extended for culturing of the biofilm.
Dosing with CHG.
Solutions were introduced into the flow cell by diverting the flow at a Teflon valve located proximal to the flow cell entrance port. Opening of this valve was clocked as time zero for timed experiments. Culture medium with PBS was introduced into the flow cell for 10 min, followed by 20 μg of PI/ml for 5 to 7 min in the PBS-buffered culture medium. Finally, the flow cell was dosed with CHG in this same PI-PBS culture medium solution. This final solution will be referred to as a CHG-PI solution.
Data analysis.
Kinetic data curves were fit with solutions to the partial differential equation (PDE) conventionally used to describe diffusion in one spatial dimension:
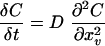 |
In this case, C is the PI fluorescence intensity as a function of time (t), and D can be considered to be the rate constant for the observed increase in PI fluorescence during CHG dosing. Initial conditions were C = 0 for 1> x >0, and boundary conditions were C = C0 at x = 1 and δC/δx = 0 at x = 0. Each of the curves in the family generated by the PDE has a sigmoidal shape that provides an empirical fit to the kinetic data curves. D is used to compare relative rates and is therefore not assigned a unit; i.e., it is considered to be a unitless constant. The estimate of D depends on the shape of the kinetic data curves only and not on the absolute intensities. Therefore, intensity values are normalized and are presented as relative pixel intensities throughout. The data were fit to a numerical solution of the PDE (equation 1) using a least-squares criterion to estimate D. The fits were calculated using a commercially available software package (AQUASIM 2.0; Peter Reichert, Computer Systems Sciences Department, Swiss Federal Institute for Environmental Science and Technology, Dübendorf, Switzerland). For some experiments, kinetic data curves from a number of cell locations were averaged to simplify the analysis and presentation. These are referred to as mean kinetic data curves.
RESULTS
Characterization of the kinetics of CHG delivery to the interface and CHG concentration dependence of PI penetration rates.
For these experiments, yeast were seeded onto the substratum and dosed with CHG as described in Materials and Methods. There was essentially no intervening growth period between seeding of the yeast onto the substratum and dosing with CHG.
PI in aqueous solution produced detectable background fluorescence. For the flow cell experiments, this resulted in a rapid (between 1 and 2 min), slight increase in fluorescence upon introduction of PI into the sampling volume of the microscope. The mean increase in PI fluorescence, as acquired from locations where cells were absent, is presented in Fig. 1a. The intensity values were normalized and are presented as relative pixel intensities. The estimated diffusion coefficient of PI (319 μm2/s) is close to the estimated diffusion coefficient of CHG (243 μm2/s) (45). Therefore, the kinetics of PI appearance in the sampling volume of the objective is expected to be similar to that of CHG. Thus, these data for PI suggest that CHG filled the sampling volume in about 2 min.
FIG. 1.

Yeast cells were seeded onto the substratum. PI was introduced into the flow cell before the CHG-PI solution. (a) Appearance of PI background fluorescence as PI fills the sampling volume (filled circles). The increase in fluorescence intensity of the cells that were labeled during the preexposure to PI (open squares) is shown. The broken line shows the decrease of PI background fluorescence as the CHG-PI solution fills the sampling volume, indicating that CHG and PI fill the sampling volume at approximately the same rate. (b) Changes in mean fluorescence intensity at positions of attached yeast cells during exposure to the CHG-PI solution (0.2 mg of CHG/ml). The arrow indicates the time at which CHG was expected to reach 90% of the dosing concentration, according to the findings of a previous study (41). (c) Increase in mean fluorescence intensity of yeast cells exposed to CHG-PI solutions with different CHG concentrations, with time zero set as the time when the interfacial region was saturated with CHG (90% dosing). Circles, 0.1 mg/ml; squares, 0.2 mg/ml; triangles, 0.4 mg/ml. (d) Model fits to data (broken lines).
During this preexposure to PI, a proportion of the cells (approximately 5% for seeded yeast) were labeled with PI immediately, as PI filled the sampling volume and saturated the interfacial region. These were cells with membranes that were compromised before exposure to CHG. The mean data curve for the time course of the change in intensity of these cells (13 yeast cells out a total of 285 cells in the field) is plotted in Fig. 1a. As described above, the intensity values were normalized and are presented as relative pixel intensities. The relatively small background PI fluorescence value was subtracted before the normalization. There was no trace of fluorescence from other cells in the field, even in contrast-enhanced images, until CHG entered the interfacial region. This preexposure to PI (with no CHG) was performed before each experiment as outlined in the Methods section. The results described above were typical for all experiments.
After exposure to PI, a CHG-PI solution with 0.2 mg of CHG/ml was introduced into the flow cell. Figure 1b shows the mean kinetic data curve for PI fluorescence values acquired from positions of attached yeast cells. Cells that were labeled during the preexposure to PI were omitted from the analysis. (This was the case for all experiments). The rise in the data curve originates from the increase in PI fluorescence as it penetrates into the cytoplasm of the cells and binds to nucleic acids. The arrow in Fig. 1b indicates the time at which, according to spectroscopic measurements, the interfacial CHG is expected to reach 90% of the dosing concentration under the same flow regime (41). There was a relatively small dip in the kinetic data curve just preceding this time point. This suggests that the quantum yield of the background PI fluorescence decreased due to the presence of 0.2 mg of CHG/ml in the solution. The portion of the curve containing the dip in pixel intensity exhibited in Fig. 1b was expanded and superimposed on data presented in Fig. 1a. The coincidence of the dip with the increase in PI background fluorescence is evident and is consistent with the interpretation that PI and CHG enter the sampling volume at approximately the same rate as expected. In summary, the time at which CHG attains 90% of the dosing concentration in the interfacial region can be estimated to be about 2 min (±30 s) based on (i) the kinetics of PI penetration into the sampling volume (Fig. 1a); (ii) the dip in PI background fluorescence upon introduction of the CHG-PI solution (Fig. 1a and b); and (iii) previous spectroscopic measurements, using an identical flow cell and flow regime, of CHG transport.
Fig. 1c shows mean kinetic data curves obtained from attached yeast cells (seeded yeast) exposed to CHG-PI solutions at three different CHG concentrations (0.1, 0.2, and 0.4 mg/ml). Each data set was acquired from a separate flow cell experiment. Time zero was adjusted to correspond to the time at which interfacial CHG reached 90% of the dosing concentration (2 min). The data curves have a similar sigmoidal shape. Figure 1d shows the model approximations to the mean data curves. Table 1 gives the D values for these data sets based on these theoretical curves. The relation between CHG concentration and rate of increase in PI fluorescence (D) as estimated by the model is nearly linear (r2 = 0.9834). Included in Table 1 are data for the fraction of cells in the field that were labeled during preexposure to PI (no CHG).
TABLE 1.
D estimates for yeast seeded into the substratum
| CHG (mg/ml) | D (102) | PIa |
|---|---|---|
| 0.1 | 2.02 (0.08)b | 0.050 |
| 0.2 | 7.15 (0.37) | 0.046 |
| 0.2 | 8.76 (0.74) | 0.055 |
| 0.4 | 25.12 (3.06) | 0.047 |
Fraction labeled during preexposure to PI.
Values in parentheses indicate standard deviations for the fit.
Development of the C. albicans biofilm on the Ge substratum and rate of CHG-induced PI penetration into biofilm cells.
Fig. 2 shows early stages of development of a C. albicans biofilm. All yeast forms present in the field of view and that were associated with filamentous forms appeared to be those of yeast that had spawned germ tubes rather than those of yeast that originated from budding of a filamentous form. (In more mature biofilms, cultured under identical conditions, filaments do acquire multiple yeast buds [41]). At 170 min, the biofilm was dosed with 0.2 mg of CHG/ml, as described in Materials and Methods. During preexposure to PI, two cells (yeast) in the entire field were labeled. Figure 3 shows the time course of increase in PI fluorescence in cellular structures as the biofilm is exposed to the CHG-PI solution. Images were acquired at ×200 to increase the field of view (and sample size). Time zero was adjusted to represent the estimated time of CHG saturation of the interfacial region.
FIG. 2.

Development of a C. albicans biofilm (×400 DIC images). Times (in minutes) after introduction of culture medium were as follows: (a) 42, (b) 63, (c) 82, (d) 124. Scale bar, 25 μm. The arrow in panel d is used for reference (see Fig. 4 legend).
FIG. 3.

Epi-fluorescent images (×200) of a C. albicans biofilm during exposure to a CHG-PI solution (0.2 mg of CHG/ml). Times (in minutes) after interfacial saturation with CHG (90% dosing) were as follows: (a) 0.5, (b) 1.0, (c) 1.5, (d) 2.0, (e) 4.0, (f) 8.0. Scale bar, 50 μm. Only a portion of the field of view is shown.
Figure 4a is an enlargement of the epi-fluorescence image presented in Fig. 3f, the last of the time course images presented in that figure. PI binds to both DNA and RNA (43). Since ribosomes are located in the cytoplasm, it labels the cytoplasm generally. Regions indicated by squares indicate locations where intensities were evaluated to construct kinetic data curves presented in Fig. 5. Figure 4b is a DIC image (×200) of the corresponding area of the biofilm, as acquired just previous to exposure to PI. Note the region of overlap between the images presented in Fig. 2d and 4b. Some growth occurred in the time interval (47 min) between the acquisitions of the first and the second DIC images. Figure 4b is less resolved than Fig. 2d due to the lower magnification (×200). Constrictions indicate that some filamentous forms (e.g., those numbered 2 and 3) were pseudohyphae. For some pseudohyphae, the PI fluorescence was distinctly dimmer in bands where constrictions appear in the DIC image (e.g., cell 3). For filamentous structures with no constrictions having lengths exceeding about 15 μm, PI fluorescence is sometimes more concentrated in certain regions of the cytoplasm (e.g., cell 5). Subapical hyphal compartments contain large vacuoles, while the occasional vacuoles that appear in apical compartments are located near the septum (16). PI is expected not to label vacuoles, which are devoid of cytoplasm. This may explain the nonuniform PI labeling. In that case, concentrated areas of PI fluorescence distal from the apex demarcate subapical compartments.
FIG. 4.

(a) Epi-fluorescence contrast-enhanced image of Fig. 3f, showing areas used to evaluate pixel intensities for kinetic data curves presented in Fig. 5. Scale bar, 25 μm. (b) DIC image of corresponding area of C. albicans biofilm, taken 12 min before exposure to CHG, with positions of cellular structures demarcated in the epi-fluorescence image indicated. Black arrows indicate yeast cells, and white arrows indicate filamentous forms. The cell labeled “r” corresponds to the cell indicated by the arrow in Fig. 2.
FIG. 5.

(a and b) Kinetics of PI labeling of cellular structures indicated in Fig. 4 during exposure to the CHG-PI solution for filamentous forms (a) and yeast forms (b). Legends indicate the correspondences between numbered arrows in Fig. 4b and the data symbols shown here. (c and d) Mean kinetic data curves for filamentous forms (c) and yeast forms (d) acquired from the entire field of view. Broken lines indicate model fit to the mean kinetic data curves. Inserts depict total data sets.
For the data presented in Fig. 5, hyphal and pseudohyphal forms were grouped into one category referred to as filamentous forms. The kinetics of the penetration of PI into the cellular structures shown in Fig. 4 as the structures are exposed to CHG is shown in Fig. 5a and b. Rates of penetration into filamentous forms appeared to be significantly greater than for yeast forms. Mean kinetic data curves acquired from cellular structures present in the entire field of view are shown in Fig. 5c (filamentous forms) and Fig. 5d (yeast forms). The inserts show the entire data set. The analysis was limited to cellular forms that were clearly distinguishable from surrounding structures. To simplify the statistical analysis, one compartment of each filament was evaluated. This resulted in analysis of the compartments of 33 filamentous forms. Only yeast forms with no visible germ tube were categorized as yeasts. Only one cell in each small cluster (doublets or triplets) was evaluated. This resulted in the location of 36 yeast cells. The model fit to the data curves is displayed with the mean data curves. The D values for fits to these mean data curves are 7.04 × 10−2 and 56.00 × 10−2 for yeast and filamentous forms, respectively. The same trend of relatively rapid kinetics of PI labeling for filamentous forms compared to that of yeast was maintained for this larger sample. To evaluate the statistical significance of this difference, kinetic data curves from each cell were fit with the model individually. The means of the D values for yeast and filamentous forms were 7.28 (4.90) × 10−2 and 60.20 (46.21) × 10−2, respectively, where standard deviations of the mean are given in parentheses. The means were significantly different, according to an unpaired t test (P < 0.0001).
For filamentous forms, D values associated with four categories of cells were grouped together. The categories and mean values of D for fits to kinetic data curves from individual cells in the biofilms for filamentous types were as follows: for the apical hyphal compartments (8 samples), 55.31 × 10−2 (standard deviation of the mean, 27.02 × 10−2); for the subapical hyphal compartments (6 samples), 75.69 × 10−2 (standard deviation of the mean, 22.57 × 10−2); for the apical pseudohyphal segments (12 samples), 56.49 × 10−2 (standard deviation of the mean, 49.12 × 10−2); and for the subapical pseudohyphal segments, where subapical hyphal compartments were defined and identified by nonuniform PI labeling as indicated above (7 samples), 47.52 × 10−2 (standard deviation of the mean, 17.98 × 10−2). Figure 6 presents a histogram of D values for the different filamentous forms. There is no obvious grouping of D values for any of the categories. One-way analysis of variance indicated that none of the pairs of means were significantly different (F3,29 = 0.54, P = 0.66). Grouping of the results into broader categories did not result in statistically significant differences in pairs of means: means of D values for all hyphal versus all pseudohyphal cells, or all apical versus all subapical cells, were not significantly different according to unpaired t tests (P > 0.40).
FIG. 6.

Histogram of D values for filamentous forms. Legend key: p-s, pseudohyphae, subapical; p-a, pseudohyphae, apical; h-s, hyphae, subapical; h-a, hyphae, apical. See text for criterion used to identify subapical hyphal compartments.
DISCUSSION
In previous key studies demonstrating resistance of C. albicans biofilms to antimicrobial agents, biofilms were treated as homogeneous entities (9, 20). It is important to obtain a description of interactions of antimicrobial agents with biofilms at a level that can allow researchers to distinguish differential responses that depend on the location within the biofilm, under conditions in which transport of substances within the biofilm can be characterized. This is a requirement for determining whether biofilm resistance originates from general biofilm characteristics, since a fundamental difference between biofilm communities and planktonic cultures is that biofilms are not well-mixed systems. That concentration gradients of both extrinsically and intrinsically derived substances can exist within biofilm populations is a major characteristic that distinguishes biofilms from planktonic cultures. Thus, biofilm organisms are likely to experience a variety of microniches depending on their positions within the biofilm, and this may, in turn, influence their susceptibility to an antimicrobial agent.
The methodology that has been presented here is part of a scheme to directly characterize the kinetics interaction of chlorhexidine with mature C. albicans biofilms, with both rate of transport to local regions and fungicidal action of chlorhexidine included in the description. The biofilm used as an example was in the initial stages of development. Since the biofilm coverage was relatively sparse, the rate of transport of chlorhexidine to all portions of the biofilm was identical to the rate of transport to the interfacial region in the absence of the biofilm. Thus, it provided a simple model system, which was used to demonstrate the viability of the method. For mature biofilms, transport to regions of the biofilm is expected to be hindered. A methodology for characterizing transport to local regions of a mature C. albicans biofilm, based on the experimental setup used here, was presented previously (41).
It has been fairly well established that the primary mode of chlorhexidine action in both bacteria and fungi is disruption of the membrane followed by rapid permeabilization (3, 5, 22-25, 31, 39, 46). The simplest explanation for the concentration dependence displayed in Fig. 1c and Table 1 is that the kinetics of appearance of PI fluorescence reflects the rate, or at least the extent, of membrane permeabilization that is a consequence of membrane disruption by chlorhexidine. The ability of the mathematical model to mimic the sigmoidal shape of the kinetic data curves suggests, but does not prove, that diffusion of PI through the permeabilized membrane, rather than the process of membrane disruption, is rate limiting.
In the 3-h-old biofilm studied here, yeast organisms were characteristically distinguishable from filamentous forms not only by the criterion of morphology but by the rate of development; filamentous forms were in the process of extension, while yeast did not appear to be actively replicating. It is reasonable to expect cells more actively undergoing processes necessary for growth and cell division to be more susceptible to antimicrobial agents that disrupt the membrane, since growth and division require synthesis and incorporation of new membrane and cell wall material. Maintenance of C. albicans yeast in stationary phase for prolonged periods (6 days) changes susceptibility to chlorhexidine in a complex manner (21). These changes in susceptibility were attributed to alteration in cell wall chemistry, based primarily on results from extensive investigation of membrane permeabilization by amphotericin B of C. albicans yeast. In the latter studies, it was found that the rate of potassium release induced by amphotericin B was decreased dramatically as cells moved from the late exponential stage into later stages of stationary phase (15). This decrease in susceptibility to permeabilization was attributed to changes in length and cross-linking of (1,3)-β-d-glucan chains in the cell wall. The fungicidal action of an imidazole, miconazole, is primarily due to disruption of the plasma membrane (4). For C. albicans yeast, susceptibility to miconazole decreases in the transition from exponential to early stationary phase due to changes in membrane lipid composition (4).
The differences in rates of permeabilization between yeast and filamentous forms in the biofilm may also originate from cell wall or membrane compositional differences inherent to the morphological forms, rather than from differences in growth phase. Investigation into differences in cell wall composition and molecular organization that regulate cell morphology is an active area of research (7). There are also differences in composition of the plasma membranes of yeast and filamentous forms of C. albicans (6, 32). Yeast organisms with germ tubes were more susceptible to the action of amphotericin B than nongerminated yeast organisms (34). However, it could be argued that the yeast organisms in the process of producing germ tubes were those more actively growing.
Figure 6 shows no obvious indication that a grouping of rates of PI penetration into filamentous forms can be associated with a particular morphology (pseudohyphal or hyphal) or spatial location (apical or subapical). The analysis of variance test indicated no significant difference in rates for the different categories. These results imply that cell attributes responsible for rate differences within the filamentous forms are not a primary determinant of cell morphology. With respect to growth, it is known that apical hyphal cells are more active than subapical cells, with extension occurring throughout the cell cycle (16). Since apical segments did not yield a higher mean rate, this suggests that differences in CHG-induced PI penetration rates within the filamentous population do not originate from cell wall or membrane characteristics that are highly correlated with growth rate. However, as hyphal compartments age, they may display decreased susceptibility similar to yeast.
In summary, the method described here enables identification of individual cells in a C. albicans biofilm exhibiting relative phenotypic resistance with respect to the action of chlorhexidine. Development of phenotypic resistance is considered to be a primary mechanism of biofilm resistance to both antibiotics (40) and antiseptics (31). The biofilm investigated here was in an early stage of development and provided a simple model system. Elucidating mechanisms of resistance of more complex, mature biofilms will likely involve determining spatial relationships between susceptible and resistant organisms.
Acknowledgments
This study was supported by grants from National Institute of Dental and Craniofacial Research (DE13231-02) (B. J. Tyler) and the National Science Foundation (EEC8907039) (Center for Biofilm Engineering).
REFERENCES
- 1.Baillie, G. S., and L. J. Douglas. 1998. Iron-limited biofilms of Candida albicans and their susceptibility to amphotericin B. Antimicrob. Agents Chemother. 42:2146-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baillie, G. S., and L. J. Douglas. 1999. Role of dimorphism in the development of Candida albicans biofilms. J. Med. Microbiol. 48:671-679. [DOI] [PubMed] [Google Scholar]
- 3.Barrett-Bee, K., L. Newboult, and S. Edwards. 1994. The membrane destabilising action of the antibacterial agent chlorhexidine. FEMS Microbiol. Lett. 119:249-253. [DOI] [PubMed] [Google Scholar]
- 4.Beggs, W. H. 1989. Development of phenotypic resistance to direct lethal miconazole action by Candida albicans entering stationary phase. Mycopathologia 108:201-206. [DOI] [PubMed] [Google Scholar]
- 5.Bobichon, H., and P. Bouchet. 1987. Action of chlorhexidine on budding Candida albicans: scanning and transmission electron microscopic study. Mycopathologia 100:27-35. [DOI] [PubMed] [Google Scholar]
- 6.Cannon, R. D., and D. Kerridge. 1988. Correlation between the sterol composition of membranes and morphology in Candida albicans. J. Med. Vet. Mycol. 26:57-65. [DOI] [PubMed] [Google Scholar]
- 7.Chaffin, W. L., J. L. Lopez-Ribot, M. Casanova, D. Gozalbo, and J. P. Martinez. 1998. Cell wall and secreted proteins of Candida albicans: identification, function, and expression. Microbiol. Mol. Biol. Rev. 62:130-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chandra, J., D. M. Kuhn, P. K. Mukherjee, L. L. Hoyer, T. McCormick, and M. A. Ghannoum. 2001. Biofilm formation by the fungal pathogen Candida albicans: development, architecture, and drug resistance. J. Bacteriol. 183:5385-5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chandra, J., P. K. Mukherjee, S. D. Leidich, F. F. Faddoul, L. L. Hoyer, L. J. Douglas, and M. A. Ghannoum. 2001. Antifungal resistance of candidal biofilms formed on denture acrylic in vitro. J. Dent. Res. 80:903-908. [DOI] [PubMed] [Google Scholar]
- 10.Dankert, J., A. H. Hogt, and J. Feijen. 1986. Biomedical polymers: bacterial adhesion, colonization, and infection. Crit. Rev. Biocompat. 2:219-301. [Google Scholar]
- 11.Doebbeling, B. N., G. L. Stanley, C. T. Sheetz, M. A. Pfaller, A. K. Houston, L. Annis, N. Li, and R. P. Wenzel. 1992. Comparative efficacy of alternative hand-washing agents in reducing nosocomial infections in intensive care units. N. Engl. J. Med. 327:88-93. [DOI] [PubMed] [Google Scholar]
- 12.Dougherty, S. H. 1989. Pathobiology of infection in prosthetic devices. Rev. Infect. Dis. 10:1102-1117. [DOI] [PubMed] [Google Scholar]
- 13.Fridkin, S. F., and W. R. Jarvis. 1996. Epidemiology of nosocomial fungal infections. Clin. Microbiol. Rev. 9:499-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frolund, B., P. A. Suci, S. Langille, R. M. Weiner, and G. G. Geesey. 1996. Influence of protein conditioning films on binding of a bacterial polysaccharide adhesin from Hyphomonas MHS-3. Biofouling 10:17-30. [DOI] [PubMed] [Google Scholar]
- 15.Gale, E. F. 1986. Nature and development of phenotypic resistance to amphotericin B in Candida albicans. Adv. Microb. Physiol. 27:277-320. [DOI] [PubMed] [Google Scholar]
- 16.Gow, N. A. 1997. Germ tube growth of Candida albicans. Curr. Top. Med. Mycol. 8:43-55. [PubMed] [Google Scholar]
- 17.Green, L., B. Petersen, L. Steimel, P. Haeber, and W. Current. 1994. Rapid determination of antifungal activity by flow cytometry. J. Clin. Microbiol. 32:1088-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han, Y., R. P. Morrison, and J. E. Cutler. 1998. A vaccine and monoclonal antibodies that enhance mouse resistance to Candida albicans vaginal infection. Infect. Immun. 66:5771-5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawser, S. P., and L. J. Douglas. 1994. Biofilm formation by Candida species on the surface of catheter materials in vitro. Infect. Immun. 62:915-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hawser, S. P., and L. J. Douglas. 1995. Resistance of Candida albicans biofilms to antifungal agents in vitro. Antimicrob. Agents Chemother. 39:2128-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hiom, S. J., J. R. Furr, A. D. Russell, and J. R. Dickinson. 1992. Effects of chlorhexidine diacetate on Candida albicans, C. glabrata and Saccharomyces cerevisiae. J. Appl. Bacteriol. 72:335-340. [DOI] [PubMed] [Google Scholar]
- 22.Hiom, S. J., J. R. Furr, A. D. Russell, and J. R. Dickinson. 1993. Effects of chlorhexidine diacetate and cetylpyridinium chloride on whole cells and protoplasts of Saccharomyces cerevisiae. Microbios 74:111-120. [PubMed] [Google Scholar]
- 23.Hugo, W. B., and A. R. Longworth. 1964. Some aspects of the mode of action of chlorhexidine. J. Pharm. Pharmacol. 16:655-662. [DOI] [PubMed] [Google Scholar]
- 24.Hugo, W. B., and A. R. Longworth. 1966. The effect of chlorhexidine on the electrophoretic mobility, cytoplasmic constituents, dehydrogenase activity and cell walls of Escherichia coli and Staphylococcus aureus. J. Pharm. Pharmacol. 18:569-578. [DOI] [PubMed] [Google Scholar]
- 25.Kuyyakanond, T., and L. B. Quesnel. 1992. The mechanism of action of chlorhexidine. FEMS Microbiol. Lett. 100:211-216. [DOI] [PubMed] [Google Scholar]
- 26.Lam, J., R. Chan, K. Lam, and J. W. Costerton. 1980. Production of mucoid microcolonies by Pseudomonas aeruginosa within infected lungs in cystic fibrosis. Infect. Immun. 28:546-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis, K. 2001. Riddle of biofilm resistance. Antimicrob. Agents Chemother. 45:999-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leyes Borrajo, J. L., V. L. Garcia, C. G. Lopez, I. Rodriguez-Nunez, F. M. Garcia, T. M. Gallas. 2002. Efficacy of chlorhexidine mouthrinses with and without alcohol: a clinical study. J. Periodontol. 73:317-321. [DOI] [PubMed] [Google Scholar]
- 29.Liljemarc, W., and F. Bloomquist. 1996. Human oral microbial ecology and dental caries and periodontal diseases. Crit. Rev. Oral. Biol. Med. 7:180-198. [DOI] [PubMed] [Google Scholar]
- 30.Lo, H. J., J. R. Kohler, B. DiDomenico, D. Loebenberg, A. Cacciapuoti, and G. R. Fink. 1997. Nonfilamentous C. albicans mutants are avirulent. Cell 90:939-949. [DOI] [PubMed] [Google Scholar]
- 31.McDonnell, G., and A. D. Russell. 1999. Antiseptics and disinfectants: activity, action, and resistance. Clin. Microbiol. Rev. 12:147-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mishra, P., J. Bolard, and R. Prasad. 1992. Emerging role of lipids of Candida albicans, a pathogenic dimorphic yeast. Biochim. Biophys. Acta 1127:1-14. [DOI] [PubMed] [Google Scholar]
- 33.Nikawa, H., T. Hamada, and T. Yamamoto. 1998. Denture plaque—past and recent concerns. J. Dent. 26:299-304. [DOI] [PubMed] [Google Scholar]
- 34.Nugent, K. M., K. R. Couchot, and L. D. Gray. 1987. Effect of Candida morphology on amphotericin B susceptibility. Antimicrob. Agents Chemother. 31:335-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Odds, F. C. 1988. Candida and candidosis, p. 1-468. Bailliere Tindall, London, England.
- 36.Pfaller, M. A. 1996. Nosocomial candidiasis: emerging species, reservoirs, and modes of transmission. Clin. Infect. Dis. 22(Suppl. 2):S89-S94. [DOI] [PubMed] [Google Scholar]
- 37.Ramani, R., A. Ramani, and S. J. Wong. 1997. Rapid flow cytometric susceptibility testing of Candida albicans. J. Clin. Microbiol. 35:2320-2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richards, M. J., J. R. Edwards, D. H. Culver, and R. P. Gaynes. 1999. Nosocomial infections in medical intensive care units in the United States. National Nosocomial Infections Surveillance System. Crit. Care Med. 27:887-892. [DOI] [PubMed] [Google Scholar]
- 39.Sheppard, F. C., D. J. Mason, S. F. Bloomfield, and V. A. Gant. 1997. Flow cytometric analysis of chlorhexidine action. FEMS Microbiol. Lett. 154:283-288. [DOI] [PubMed] [Google Scholar]
- 40.Stewart, P. S., and J. W. Costerton. 2001. Antibiotic resistance of bacteria in biofilms. Lancet 358:135-138. [DOI] [PubMed] [Google Scholar]
- 41.Suci, P. A., G. G. Geesey, and B. J. Tyler. 2001. Integration of Raman microscopy, differential interference contrast microscopy, and attenuated total reflection Fourier transform infrared spectroscopy to investigate chlorhexidine spatial and temporal distribution in Candida albicans biofilms. J. Microbiol. Methods 46:193-208. [DOI] [PubMed] [Google Scholar]
- 42.Suci, P. A., K. J. Siedleki, R. J. Palmer, Jr., D. C. White, and G. G. Geesey. 1997. Combined light microscopy and attenuated total reflection Fourier transform spectroscopy for integration of biofilm structure, distribution, and chemistry at solid-liquid interfaces. Appl. Environ. Microbiol. 63:4600-4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tas, J., and G. Westerneng. 1981. Fundamental aspects of the interaction of propidium diiodide with nuclei acids studied in a model system of polyacrylamide films. J. Histochem. Cytochem. 29:929-936. [DOI] [PubMed] [Google Scholar]
- 44.Tiraboschi, I. N., J. E. Bennett, C. A. Kauffman, J. H. Rex, C. Girmenia, J. D. Sobel, and F. Menichetti. 2000. Deep Candida infections in the neutropenic and non-neutropenic host: an ISHAM symposium. Med. Mycol. 38(Suppl. 1):199-204. [PubMed] [Google Scholar]
- 45.Treybal, R. E. 1980. Mass transfer operations, p. 35. McGraw-Hill Book Co., London, England.
- 46.Walters, T. H., J. R. Furr, and A. D. Russel. 1983. Antifungal action of chlorhexidine. Microbios 38:195-204. [Google Scholar]