

Abstract
Mutations in the X-linked gene doublecortin lead to “double cortex” syndrome (DC) in females and to X-linked lissencephaly (XLIS) in males. Because most patients with DC and XLIS are sporadic, representing de novo doublecortin mutations, we considered that some of these patients could be somatic or germline mosaics. Among a population of 20 patients and their families, we found evidence for mosaic doublecortin mutations in 6 individuals. Germline mosaicism was identified in two unaffected women, each with two affected children. Additionally, one affected male with DC was found to be a somatic mosaic, which presumably spared him from the more severe phenotype of lissencephaly. The high rate of mosaicism indicates that there may be a significant recurrence risk for DC/XLIS in families at risk, even when the mother is unaffected.
Introduction
Double cortex (DC [MIM 600348 and MIM 300067]) and X-linked lissencephaly syndrome (XLIS [MIM 300067]) is an X-linked dominant disorder in which mothers with DC transmit DC to their daughters and XLIS to their sons (Pinard et al. 1994). Mutations in doublecortin (DCX) account for the majority of cases (des Portes et al. 1998a, 1998b; Gleeson et al. 1998; 1999b). In XLIS, cortical neuronal migration is severely disrupted, leading to a rudimentary four-layered cortex (Berg et al. 1998). In DC, one population of neurons forms a relatively normal cortex whereas a second population apparently arrests during migration, leading to a collection of neurons beneath the cortex. Because of lyonization, affected females with a DCX mutation are mosaics for two populations of neurons, which presumably segregate according to which of the copies of DCX is active. Because DC is seen with heterozygous DCX mutations and is a less severe phenotype than lissencephaly, DC is considered, on the basis of lyonization, to be a mosaic phenotype.
In families with a single affected child with DC/XLIS that are considering having other children, the risk of recurrence in future children is unclear. It previously had been suggested that, in the setting of a single affected girl with DC or a single boy with XLIS, the mother should have magnetic-resonance imaging (MRI) of the brain, to determine whether she is subtly affected and therefore likely to carry the DCX mutation (Clark and Noebels 1999). If evidence of DC is detected by the brain MRI, then the mother likely carries the DCX mutation, and her future children would have a 50% chance of inheriting it. Because a male with a DCX mutation should be clinically affected, brain MRI typically is not performed on the father. Of course, these recommendations do not consider either the possibility that either parent carries the mutation but is clinically unaffected or the possibility that either parent is an unaffected somatic or germline mosaic for the mutation, each of which is associated with up to a 50% incidence of recurrence of DC or XLIS in future children.
Two families that have unaffected parents and a sib pair in which one child has DC and the other has XLIS have been identified, indicating that the mother in each of these families transmitted the mutation to two offspring. The most likely explanations for the finding that these mothers were unaffected are that they either (1) had skewed X inactivation or (2) were germline or somatic mosaics for the DCX mutation. We investigated both of these possibilities in each of these individuals, and we present data suggesting that the mother in each family carries a germline DCX mosaic mutation, which presumably spares them from displaying clinical features of DC.
On the basis of these results, we searched for the possibility of mutation mosaicism in settings in which it was likely to occur. We searched for evidence of (a) somatic mosaicism among patients with DC, (b) somatic or germline mosaicism among unaffected parents of patients with DC, and (c) somatic mosaicism among the few males identified as being affected with DC. We report that mutation mosaicism was identified in each of these settings, suggesting that germline- and somatic-mutation mosaicism is not uncommon among patients with DCX mutations and that the recurrence risk in families with a single affected child with DC/XLIS may be significant.
Patients and Methods
Patients
Two families displaying non-Mendelian inheritance (i.e., two affected children with DC/XLIS who were from unaffected parents) were included in this study. For one of these families, the mutation has been reported elsewhere (Gleeson et al. 1999b). Six females with sporadic DC from whom parental DNA was available (mutations for which had been reported elsewhere [Gleeson et al. 1999b]) were included in this study, to allow a search for mutation mosaicism among unaffected parents of patients with DC. Ten additional females with sporadic DC from whom parental DNA was not available also were included, to allow a search for mutation mosaicism among patients affected with DC. Additionally, two males with sporadic DC who had not previously been evaluated for a DCX mutation were included in this study. Family or patient identification numbers from previous publications have been retained (Gleeson et al. 1998, 1999b). A brain MRI was performed on all clinically affected individuals, as well as on mothers with either somatic or germline mosaicism, to determine affection status.
Mutation Analysis
This research was approved by the Human Subjects Committees at Children's Hospital (Boston), Beth Israel Deaconess Medical Center, the University of Minnesota, and the University of California, San Diego. Genomic DNA was extracted from lymphocytes, as described elsewhere (Gleeson et al. 1998). Each exon was amplified by PCR and was analyzed for single-stranded conformational polymorphism (SSCP), on a nondenaturing polyacrylamide gel. To increase the sensitivity of mutation detection, each exon was also analyzed on an MDE (mutation-detection enhancement) gel, according to the recommendations of the manufacturer (FMC BioProducts). Bands were visualized by silver staining according to the manufacturer’s directions (Promega). Polymorphic bands were excised from the gel, reamplified by PCR, and sequenced in both directions, to identify mutations, described elsewhere (Gleeson et al. 1998). The DNA from the unaffected parents of each patient with sporadic DC also was analyzed by SSCP and was run in a lane next to that containing the patient’s DNA, to test whether the mutation derived from somatic mosaicism in one of the parents. Somatic mosaicism was identified by the presence, on an SSCP gel, of mutant bands at an intensity lower than that of the wild-type bands.
Band intensity was quantitated by three methods. First, each genomic PCR reaction was repeated in the presence of 1 μCi of α[P32]-dCTP per reaction, over three different cycle numbers (25, 30, and 35 cycles), to ensure that the amplification was in the linear range, followed by SSCP analysis of PCR products and exposure to a Phosphoimager cassette (Molecular Dynamics) for 2–3 d. Band intensity was quantitated by GENEQUANT software (Molecular Dynamics). Second, each genomic PCR reaction was repeated, without radioactivity, and was analyzed by SSCP, visualized by silver staining, photographed at several varying exposure times, to ensure that the bands were not saturated, and digitized by computer scanning, and then band intensity was quantitated by GENEQUANT software. Using this method, we could determine mosaicism at a level as low as 5%, as determined by mixture of known amounts of wild-type and mutant alleles. Third, each genomic PCR reaction was repeated and directly sequenced by Big Dye (ABI) dye terminators and an ABI 377 or 310 sequencer. After automated sequence analysis, the area under the curve at the site of the mutation mosaicism was estimated, to ensure that it was consistent with results from the two previously described methods of quantification. Because the peaks from fluorescent sequencing are not quantitative, the area under the curve was not measured.
To be certain that preferential amplification of the wild-type allele versus the mutant allele did not account for the allele intensity differences, the two alleles were excised from the SSCP gel, diluted to equimolar concentrations, and reamplified over 20, 25, and 30 cycles, and the intensity was reanalyzed by SSCP and band quantification. Additionally, PCR products derived from amplification of patients' DNA were cloned to separate the two alleles, by polishing the ends with T4 DNA polymerase, followed by cloning with ZeroBlunt cloning vector (Invitrogen). A clone representing the wild-type and the mutant alleles was identified for each mutation, by direct sequencing of several clones from each patient. PCR with 0.1 ng of miniprep DNA and the same pair of primers, over 20 or 25 cycles, was performed in the presence of 1 μCi of α[P32]-dCTP, and the band intensity was quantitated by Phosphoimager analysis.
Results
Mosaicism Detection
All patients and their parents were analyzed for mosaicism, by PCR of the DCX coding region. In patients with a previously identified mutation, only the mutant exon was reanalyzed. The percentage of mosaicism was determined by three independent methods, the results of which which, for any given patient, were quantitatively within 5% of one another. The results reported in table 1 are from the first method. Germline mosaicism was inferred on the basis of the absence of a DCX mutation in peripheral blood lymphocytes in an obligate carrier (Zlotogora 1998). Family I displayed an A400T point mutation, leading to a premature stop codon at amino acid position 134. Male 1 with sporadic DC displayed a G628T point mutation, leading to a V210F amino acid substitution. All mutations and a summary of mutation-mosaicism findings are presented in table 1.
Table 1.
Families and Patients with Sporadic DC and either DCX Somatic- or Germline-Mutation Mosaicism[Note]
| Individual Displaying Mosaicisma | Comments on Pedigree | Frequency of Mosaicism in Blood Lymphocytes(%) | doublecortin Mutation | Resultant Doublecortin Abnormality | Phenotype of Mosaic Individual | Probable Type of Mosaicism |
| Mother in family H (I-1) | Unaffected parents, affected daughter and son | <5 | C907T | 303stop | Unaffected | Maternal germline |
| Mother in family I (I-2) | Unaffected parents, affected daughter and son | <5 | A400T | 134stop | Unaffected | Maternal germline |
| Mother of female 7 with sporadic DC (I-1) | Unaffected parents, affected daughter | 22 | del684CT | Frameshift at 229, protein stop at 240 | Unaffected | Somatic |
| Female 17 with sporadic DC (II-1) | Unaffected parents, affected daughter | 43 | del803A | Frameshift at 268, protein stop at 274 | DC | Somatic |
| Female 64 with sporadic DC (II-1) | Unaffected parents, affected daughter | 31 | del691CT | Frameshift at 231, protein stop at 240 | DC | Somatic |
| Male 1 with sporadic DC (II-1) | Unaffected parents, affected son | 31 | G628T | V210F | DC | Somatic |
Note.— Because the lower limits of detection using these methods was determined to be ∼5% (see the Patients and Methods section), we could exclude the possibility of >5% somatic mosaicism in the mothers in families H and I.
The order is meant to highlight the finding that those with ⩽22% mutation burden are clinically unaffected, whereas those with ⩾31% mutation burden are clinically affected.
Germline DCX Mutation Mosaicism in Unaffected Parents of Two Children
In families H and I, the unaffected parents had two children with DC/XLIS, consistent with either maternal germline or somatic mosaicism. Both families have demonstrable DCX mutations in both affected children. Since the father did not transmit an X chromosome to his son, each mother must carry the DCX mutation in her germline. However, the maternal genotype of lymphocytes in both families is normal on the basis of SSCP analysis, suggesting that each mother is a germline mosaic for the DCX mutation (fig. 1). We could not exclude the possibility (1) that either unaffected mother carries a subtle somatic mutation at levels that, in peripheral blood lymphocytes, are too low to be detected by SSCP or (2) that, in additional areas, which are not reflected in lymphocyte-based PCR analysis, either unaffected mother is a somatic mosaic.
Figure 1.
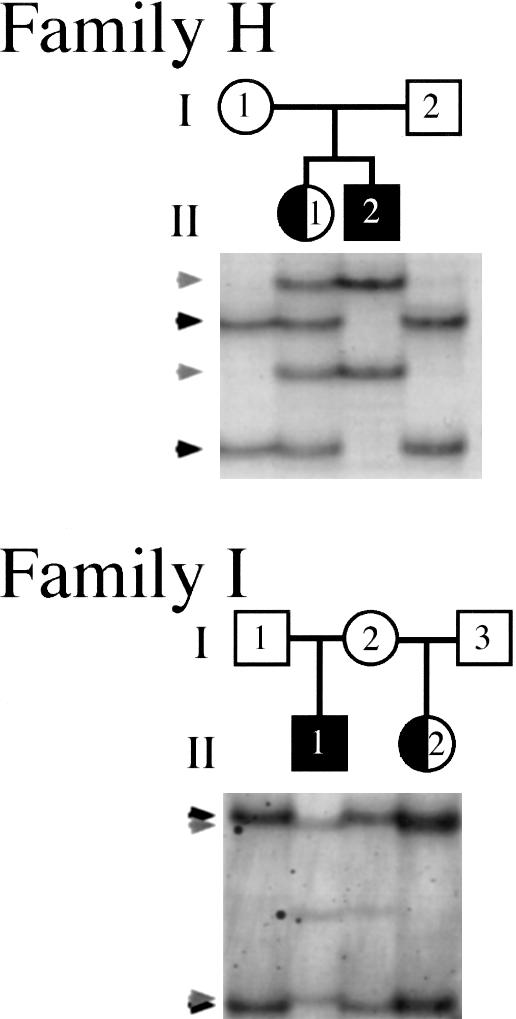
Germline DCX mutation mosaicism in unaffected parents of two affected children. In both families, an unaffected mother has a girl with DC (half-blackened circle) and a boy with XLIS (blackened square), and the maternal genotype appears to be normal. SSCP gel analysis of both families demonstrates two normal bands in each parent (black arrows), two mutant bands from males with lissencephaly (gray arrows), and four bands (reflecting a normal allele and a mutant allele) from females with DC. In family I, other faint bands are visible in individuals II-1 and I-2, likely representing either false priming or minor conformers of the PCR product.
Somatic DCX–Mutation Mosaicism in the Unaffected Parents of a Patient with Sporadic DC
On the basis of these results, we considered that some unaffected parents of patients with sporadic DC might carry a somatic DCX mutation, so we tested for somatic mosaicism in each of the parents of our cohort of six patients with sporadic DC from whom DNA was available. The unaffected mother of DC patient 7 showed evidence of somatic mosaicism, in peripheral blood lymphocytes, of ∼22% (fig. 2A), whereas, on the basis of SSCP analysis, both parents from each of the other families had a normal genotype. Direct sequencing of the genomic PCR product showed the wild-type sequence and the frameshift mutation, with the mutation represented at a level below that of the wild-type, supporting the SSCP-based finding of mosaicism (fig. 2B). To be certain that the difference in intensity between the wild-type and mutant allele was not the result of selective PCR amplification of the wild-type allele, the two alleles were separated by SSCP and cloning and were individually reamplified by use of the same primer pair. Selective amplification of one allele over the other was not detected by either method (data not shown), suggesting that the difference, in band intensity, that genomic PCR detected between the wild-type and the mutant allele is the result of mosaicism.
Figure 2.
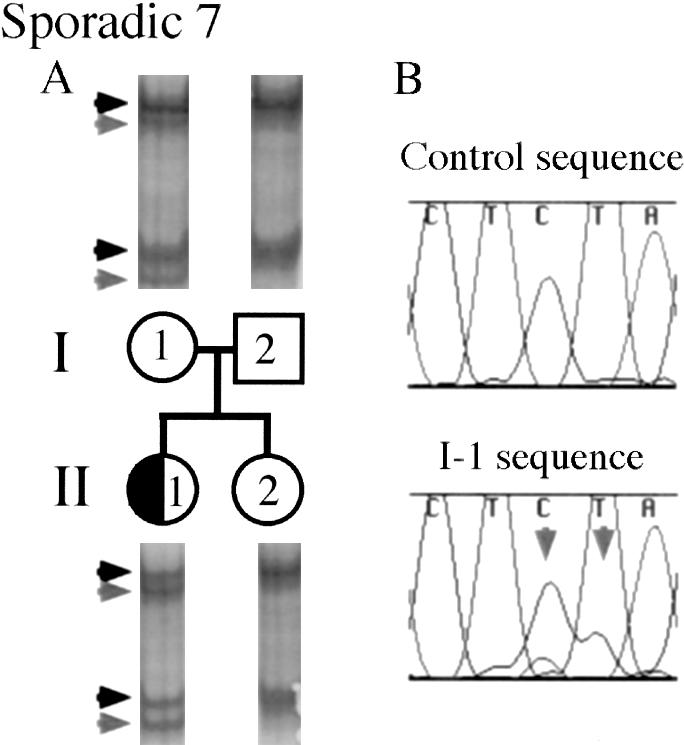
Somatic DCX–mutation mosaicism in the unaffected parents of an affected child. An unaffected mother has a daughter with DC (half-blackened circle). As is shown in panel A, this mother (I-1) was found to carry the same DCX mutation as was seen in her daughter, but the mutant band (rightward-pointing gray arrowheads) is of lower intensity than the normal band (rightward-pointing black arrowheads) (i.e., 22% of the intensity of the wild-type band), suggesting that the mother is a somatic mosaic for the DCX mutation. The mother has normal intelligence and the results of a brain MRI were normal, suggesting that she is clinically and subclinically unaffected. As is shown in panel B, the somatic DCX mutation is also visible on the basis of direct sequencing of genomic DNA in I-1 compared with that in a normal control. In I-1 sequence, the mutant sequence is seen as lower-height peaks that underlie the normal peaks. Because the mutation in I-1 is a CT deletion, the lower-height peaks in this figure are the bases AC, which are indicated by two downward-pointing gray arrows.
Because DC can have a very subtle phenotype (Pinard et al. 1994), we questioned whether the mother of patient 7 could be a very subtly affected somatic mosaic. Thus, she was studied by brain MRI, currently the most sensitive test for DC. The results of her brain MRI were normal, and she has normal intelligence and no history of seizures, making it very unlikely that she is affected clinically or subclinically. Therefore, her somatic DCX mutation may have arisen during development, after ectoderm (brain) differentiated from mesoderm (lymphocyte and germ cells). Another possibility is that the mutation is present in all three of her somatic cell lines but that, because favorable X inactivation hinders expression of the mutant DCX in her nervous system, she does not display DC. X-inactivation studies using lymphocyte-derived DNA from this individual do not show significantly skewed X inactivation (data not shown), but this finding does not fully exclude this possibility. Analysis of skin-biopsy results could help to distinguish between these two possibilities, but this procedure was not performed in this study.
Somatic DCX–Mutation Mosaicism in Affected Females with Sporadic DC
On the basis of these results, we considered that some of the patients with sporadic DC and previously identified mutations might, in fact, carry a somatic DCX mutation, rather than a germline mutation. Therefore, we analyzed our cohort of 16 females with sporadic DC, to investigate this possibility. Two of the 16 females with sporadic DC showed evidence of somatic mutations, involving either 31% or 43% of lymphocyte (somatic) cells (fig. 3A and B). Direct sequencing of the genomic PCR product from patient 64 with sporadic DC showed the wild-type sequence and the frameshift mutation, with the mutation represented at a level below that of the wild-type, supporting the SSCP-based finding of mosaicism (fig. 3A). Again, mutant and wild-type alleles were separated and were individually reamplified; and no selective amplification of one allele over the other was detected (data not shown), supporting the finding of mutation mosaicism in these patients. The unaffected parents do not show, on the basis of SSCP, evidence of the mutation, suggesting that the mutation arose sometime during embryogenesis of the affected patient. The phenotype of one of these patients (patient 17) is clinically indistinguishable from that of other females with sporadic DC (Gleeson et al. 2000), and the phenotype of the other mosaic patient with sporadic DC is also typical of DC (W.B.D. and M.E.R., unpublished data), suggesting that patients with somatic DCX mutations are at risk for the full clinical severity of disease.
Figure 3.
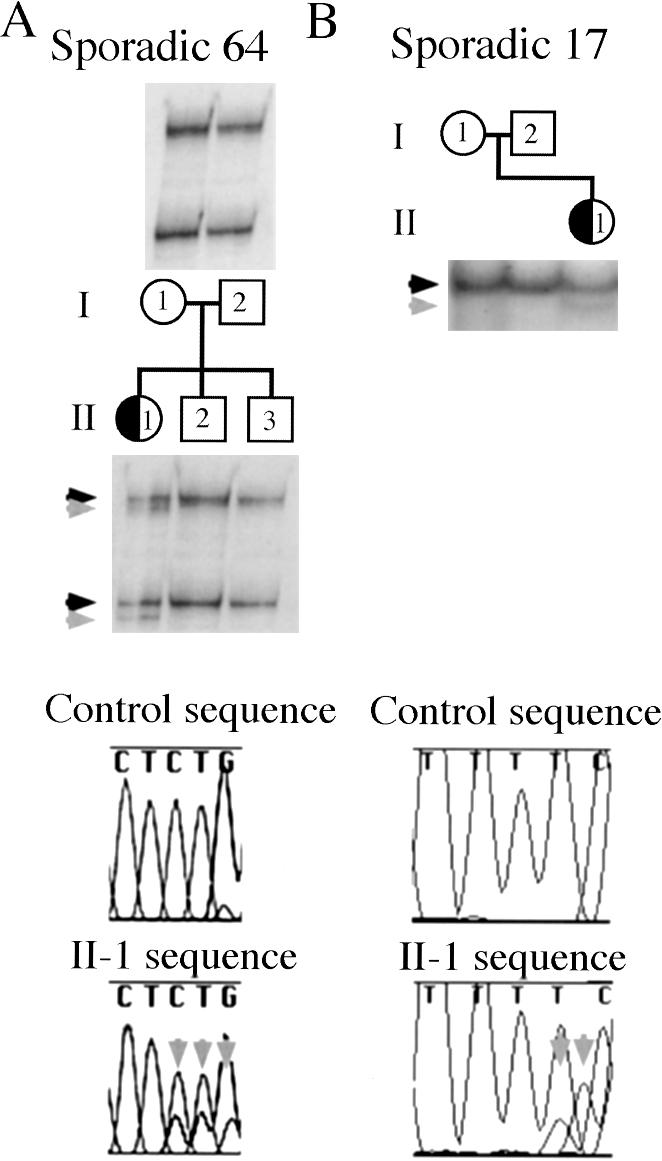
Somatic DCX–mutation mosaicism in affected females with sporadic DC (half-blackened circles). These two females with DC—Sporadic 64 (II-1) and Sporadic 17 (II-1)—have de novo DCX mutations (rightward-pointing gray arrowheads) that are, respectively, 31% and 43% of the intensity of the wild-type band (rightward-pointing black arrowheads), suggesting that they are somatic mosaics for the DCX mutation. The somatic DCX mutations are also visible on the basis of direct sequencing of genomic DNA in both patients, compared with that in a normal control. For Sporadic 64, the mutant sequence is seen as lower-height peaks that underlie the normal peaks. Because the mutation in Sporadic 64 is a CT deletion, the lower-height peaks in this figure are the bases GGA, which are indicated by three downward-pointing gray arrows. For Sporadic 17, the lower-height peaks are the bases CA, which are indicated by two downward-pointing gray arrows. The sequence for Sporadic 17 was obtained with the reverse primer, and thus the A deletion is seen as a T deletion, with a resultant leftward shift of the bases of the mutant allele. Because of an artifact of the ABI 310 sequencer, the mutant A peak, which is indicated by the second downward-pointing gray arrow, is shifted slightly leftward relative to the wild-type C peak.
Somatic DCX Mutation–Mosaicism in an Affected Male with Sporadic DC
We considered the possibility that males with DC could display somatic DCX mutations, as an explanation for why these males display the heterozygous (female) phenotype, as opposed to the more typical hemizygous (male) phenotype, of lissencephaly. In our cohort of patients with DC, there were two males with sporadic DC on whom DCX mutation analysis was performed for this study. One of these males (male 1) displayed a de novo G628T DCX mutation, predicted to lead to a V210F amino acid substitution. This male displayed two alleles for this exon (fig. 4A), raising the possibility that he either has a duplication of part or all of the X chromosome or carries a somatic DCX mutation. We used karyotype analysis to investigate the possibility of an XXY genotype, and we used Southern blot analysis with a full-length DCX probe to search for a duplication; results of both analyses were normal (data not shown). On the basis of SSCP analysis, the intensity of the mutant band was ∼31% of that of the wild-type band, suggesting that the majority of the patient's cells have the wild-type DCX allele. Direct sequencing of the genomic PCR product showed the wild-type sequence and the base-substitution mutation, with the mutation represented at a level below that of the wild-type sequence, supporting the SSCP-based finding of mosaicism (fig. 4B). Again, mutant and wild-type alleles were separated and individually reamplified; and no selective amplification of one allele over the other was detected (data not shown), supporting the finding of mutation mosaicism in this patient. Therefore, this male most likely has in DCX a somatic mutation leading to a mosaic state and, because of his mosaic mutation, is spared from the more severe phenotype of lissencephaly.
Figure 4.
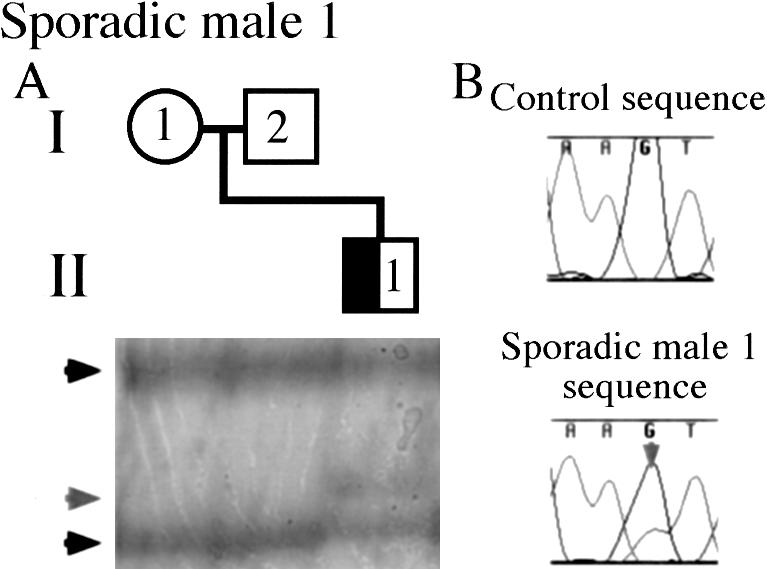
Somatic DCX–mutation mosaicism in an affected male with sporadic DC (half-blackened square). As is shown in panel A, this male was found to carry a de novo DCX mutation allele (rightward-pointing gray arrowhead) and a wild-type DCX allele (rightward-pointing black arrowheads). Males should have just one DCX allele, suggesting that this male is a somatic mosaic for the DCX mutation. As is shown in panel B, the somatic DCX mutation is also visible on the basis of direct sequencing of genomic DNA in male 1 with sporadic DC (i.e., II-1), compared with that in a normal control. The mutant sequence is shown as a lower-height T peak that underlies the normal G peak, which is indicated by the downward-pointing gray arrow.
Discussion
We demonstrate that germline and somatic mosaicism for DCX mutations is not infrequent and may take one of many presentations. Germline mosaicism for DCX was identified in two unaffected mothers who transmitted the mutation to two of their children. Somatic mosaicism for DCX was identified in an unaffected mother who transmitted the mutation to her child, in two affected females with sporadic DC, and in one male who displayed the female phenotype. These results further support the hypothesis that the DC phenotype results from a mosaic state in which some neurons express a normal copy of DCX and some neurons express a mutant copy of DCX. The mosaic state in a female with DC is typically a result of X inactivation, but other forms of mosaicism can produce the same mosaic state. These results are based on analysis of lymphocyte-derived DNA and will need to be verified by use of tissue derived from a second source (e.g., by skin biopsy). Nevertheless, these finding suggest that multiple forms of mosaicism (X-inactivation mosaicism and somatic mosaicism) may be present in a single patient with DC.
des Portes et al. (1998a) have reported a family with two affected daughters and a less severely affected mother with a likely somatic DCX mutation. In this family, the mother had a thin frontal band and displayed mental retardation less severe than that in her two affected daughters, suggesting that her mosaicism may have spared her from the more severe phenotype associated with the germline mutation and displayed by her daughters. Although our study identified a mother with a somatic mosaic DCX mutation as well, the mother in our study, unlike the mother reported by des Portes et al., was clinically and radiographically unaffected. The band intensity of the wild-type allele and the mutant allele was not quantitated in the report by de Portes et al., so it is not possible to correlate their results with ours, but their data support our finding that mosaicism among patients with a DCX mutation is not an infrequent occurrence.
Although DC in males is infrequent, it appears that both mutations in DCX and mutations in LIS1 can lead to DC in males. Because neither mutations in DCX nor mutations in LIS1 were predicted to lead to DC in males, the genetic etiology of DC in males was a mystery. Pilz et al. (1999) recently described a male with DC and a de novo LIS1 mutation, as well as two males with DC and DCX mutations. Because DC apparently is a mosaic phenotype, it was initially suspected that perhaps these DC males would display mosaic mutations; however, the data suggested that each of these mutations was germline in origin. Although the mechanism through which these germline LIS1 and DCX mutations lead to DC is unclear, Pilz et al. suggested that perhaps the mutations were hypomorphic alleles, rather than complete null alleles, accounting for the less severe phenotype of DC rather than lissencephaly. We could not exclude a similar mechanism in the male with sporadic DC who is presented here, but our findings demonstrating a male with a mosaic DCX mutation who displays the female phenotype fit well with the idea that DCX may function in a cell-autonomous manner during neuronal migration (Francis et al. 1999; Gleeson et al. 1999a).
On the basis of these results, we hypothesize that at least some patients with focal cortical dysplasia may have somatic DCX mutations. In a previous study, we described three individuals with focal subcortical band heterotopia in whom no DCX mutation could be identified (Gleeson et al. 2000); we hypothesized that somatic mosaicism could underlie the disease in these individuals. The current findings support the possibility that focal subcortical heterotopia may be due, at least in part, to somatic mutations in DCX. It would be interesting to examine cortical resections from a large cohort of patients with focal cortical dysplasias, for evidence of mosaic DCX mutations at the site of dysplasia.
In the present study, individuals displaying <30% mosaicism were clinically unaffected, whereas individuals with >30% mosaicism were clinically affected with DC, suggesting that individuals with a higher mutation load are more likely to be clinically and radiographically affected. For example, the unaffected mothers in families I and H and the mother of female 7 with sporadic DC each display ⩽22% mosaicism for the DCX mutation and are unaffected, whereas females 17 and 63 with sporadic DC and male 1 with sporadic DC each display ⩾31% mosaicism for the DCX mutation. The small number of patients in the present study precludes a meaningful statistical analysis, and further refinement of this lower limit required for phenotypic expression (>30% mosaicism) awaits analysis of a larger patient group. Nevertheless, these data suggest that there may be a critical percentage of mosaicism in peripheral blood lymphocytes that is associated with clinical and radiographic features of DC.
Our findings may have bearing on genetic counseling regarding the risk of recurrence after the first child is diagnosed with either DC or XLIS. The risk of recurrence of either DC or XLIS after the birth of a first affected child has been believed to be low, especially if results of brain MRI of the mother are normal (Clark and Noebels 1999). However, we have presented three unaffected females with germline or somatic mosaicism for DCX, each with presumably a 50% chance of recurrence of the disease in future children. It is not possible to predict at this time the recurrence risk after the birth of a single child affected with either DC or XLIS, but analysis of a larger cohort of patients with sporadic DC and their parents may help to clarify this issue. Our experience with a limited cohort suggests that somatic and germline mosaicism in unaffected mothers with either DC or XLIS is seen in ∼10% of cases, a finding that is in keeping with those for autosomal dominant disorders such as osteogenesis imperfecta and neurofibromatosis (Hall 1988; Zlotogora 1998). Geneticists must continue to take the risk of somatic and germline mosaicism into account when the parents of a patient with either sporadic DC or XLIS are counseled about the risk of recurrence (Sippel et al. 1998), and DNA testing for DCX mutations in this clinical setting may be helpful. For example, the mother in family I (I-2) recently became pregnant, and amniocentesis was performed to determine whether the DCX mutation was present. Fortunately, the fetus did not carry the mutation, and the woman gave birth to a normal child (I.A.Y., J.G.G., and C.A.W., unpublished data).
Genetic counselors may also need to take these results into consideration when determining the recurrence risk for lissencephaly in males, since mutations in either DCX or the chromosome 17 gene LIS1 (MIM 247200) lead to nearly indistinguishable phenotypes, although the inheritance and recurrence risk are very distinct. LIS1 mutations appear to be exclusively germline, and the only documented recurrences have been in the setting of a balanced translocation in one parent that became unbalanced on transmission to the affected children (Goutieres et al. 1987; Alvarado et al. 1993; Pollin et al. 1999). Therefore, if a LIS1 mutation is identified in the absence of a translocation, there is very little risk of recurrence. However, on the basis of these results, if a DCX mutation is identified, there appears to be a significant (up to 10%) risk of recurrence, on the basis of the possibility of mosaicism in the mother. In the absence of demonstrable mutations in either gene, the risk of recurrence is indeterminate. Therefore, mutation analysis can be very helpful in appropriate situations.
Acknowledgments
We are deeply indebted to the patients who participated in these studies and to the clinicians who have supplied patient samples not used directly in this work. Thanks to David Ledbetter and Naomichi Matsumoto for critical review of the manuscript. J.G.G. was supported by NSADA fellowship 5K12NS01701-05 from the NINDS, by an Epilepsy Foundation Junior Investigator Award, and by the John Merck Scholars Program in the Biology of Developmental Disabilities in Children. This work was also supported by NINDS grants RO1-NS 35129 (to C.A.W.), R01-NS35515 (to M.E.R.), and P01-NS38289 (to W.B.D., M.E.R., and C.A.W.) and by the Mental Retardation Research Center at Children’s Hospital.
Electronic-Database Information
Accession numbers and the URL for data in this article are as follows:
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for DC [MIM 600348 and MIM 300067], XLIS [MIM 300067], DCX [MIM 300121], and LIS1 [MIM 247200])
References
- Alvarado M, Bass HN, Caldwell S, Jamehdor M, Miller AA, Jacob P (1993) Miller-Dieker syndrome: detection of a cryptic chromosome translocation using in situ hybridization in a family with multiple affected offspring. Am J Dis Child 147:1291–1294 [DOI] [PubMed] [Google Scholar]
- Berg MJ, Schifitto G, Powers JM, Martinez-Capolino C, Fong CT, Myers GJ, Epstein LG, Walsh CA (1998) X-linked female band heterotopia-male lissencephaly syndrome. Neurology 50:1143–1146 [DOI] [PubMed] [Google Scholar]
- Clark GD, Noebels JL (1999) Cortin disaster: lissencephaly genes spell double trouble for the developing brain. Ann Neurol 45:141–142 [DOI] [PubMed] [Google Scholar]
- des Portes V, Francis F, Pinard JM, Desguerre I, Moutard ML, Snoeck I, Meiners LC, Capron F, Cusmai R, Ricci S, Motte J, Echenne B, Ponsot G, Dulac O, Chelly J, Beldjord C (1998a) doublecortin is the major gene causing X-linked subcortical laminar heterotopia (SCLH). Hum Mol Genet 7:1063–1070 [DOI] [PubMed] [Google Scholar]
- des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J (1998b) A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell 92:51–61 [DOI] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC, Friocourt G, McDonnell N, Reiner O, Kahn A, McConnell SK, Berwald-Netter Y, Denoulet P, Chelly J (1999) Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 23:247–256 [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA (1998) doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92:63–72 [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA (1999a) Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23:257–271 [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Luo RF, Grant PE, Guerrini R, Huttenlocher PR, Berg MJ, Ricci S, Cusmai R, Wheless JW, Berkovic S, Scheffer I, Dobyns WB, Walsh CA (2000) Genetic and neuroradiological heterogeneity of double cortex syndrome. Ann Neurol 47:265–269 [PubMed] [Google Scholar]
- Gleeson JG, Minnerath SR, Fox JW, Allen KM, Luo RF, Hong SE, Berg MJ, Kuzniecky R, Reitnauer PJ, Borgatti R, Mira AP, Guerrini R, Holmes GL, Rooney CM, Berkovic S, Scheffer I, Cooper EC, Ricci S, Cusmai R, Crawford TO, Leroy R, Andermann E, Wheless JW, Dobyns WB, Ross ME, Walsh CA, et al (1999b) Characterization of mutations in the gene doublecortin in patients with double cortex syndrome. Ann Neurol 45:146–153 [DOI] [PubMed] [Google Scholar]
- Goutieres F, Aicardi J, Rethore MO, Prieur M, Lejeune J (1987) Familial Miller-Dieker syndrome and (15;17) chromosome translocation (in French). Arch Fr Pediatr 44:501–504 [PubMed] [Google Scholar]
- Hall JG (1988) Review and hypotheses: somatic mosaicism: observations related to clinical genetics. Am J Hum Genet 43:355–363 [PMC free article] [PubMed] [Google Scholar]
- Pilz DT, Kuc J, Matsumoto N, Bodurtha J, Bernadi B, Tassinari CA, Dobyns WB, Ledbetter DH (1999) Subcortical band heterotopia in rare affected males can be caused by missense mutations in DCX (XLIS) or LIS1. Hum Mol Genet 8:1757–1760 [DOI] [PubMed] [Google Scholar]
- Pinard JM, Motte J, Chiron C, Brian R, Andermann E, Dulac O (1994) Subcortical laminar heterotopia and lissencephaly in two families: a single X linked dominant gene. J Neurol Neurosurg Psychiatry 57:914–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollin TI, Dobyns WB, Crowe CA, Ledbetter DH, Bailey-Wilson JE, Smith AC (1999) Risk of abnormal pregnancy outcome in carriers of balanced reciprocal translocations involving the Miller-Dieker syndrome (MDS) critical region in chromosome 17p13.3. Am J Med Genet 85:369–375 [PubMed] [Google Scholar]
- Sippel KC, Fraioli RE, Smith GD, Schalkoff ME, Sutherland J, Gallie BL, Dryja TP (1998) Frequency of somatic and germ-line mosaicism in retinoblastoma: implications for genetic counseling. Am J Hum Genet 62:610–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotogora J (1998) Germ line mosaicism. Hum Genet 102:381–386 [DOI] [PubMed] [Google Scholar]