

Abstract
A variety of mtDNA mutations responsible for human diseases have been associated with molecular defects in the OXPHOS system. It has been proposed that mtDNA genetic alterations can also be responsible for sperm dysfunction. In addition, it was suggested that if sperm dysfunction is the main phenotypic consequence, these mutations could be fixed as stable mtDNA variants, because mtDNA is maternally inherited. To test this possibility, we have performed an extensive analysis of the distribution of mtDNA haplogroups in white men having fertility problems. We have found that asthenozoospermia, but not oligozoospermia, is associated with mtDNA haplogroups in whites. Thus, haplogroups H and T are significantly more abundant in nonasthenozoospermic and asthenozoospermic populations, respectively, and show significant differences in their OXPHOS performance.
Introduction
There is increasing evidence showing that sperm motility is strongly dependent on the ATP supplied by the mitochondrial oxidative phosphorylation system (OXPHOS) (Ford and Harrison 1981; Halangk et al. 1985; Pascual et al. 1996; Ruiz-Pesini et al. 1998). Four of the five enzymatic complexes that constitute the OXPHOS system are partially encoded by mitochondrial DNA (mtDNA). Thus, mutations in mtDNA genes that impair the expression of one or more proteins encoded in the mtDNA can promote diseases in humans (Enriquez et al. 1995; Larsson and Clayton 1995; Wallace 1995; Schon et al. 1997; Zeviani and Antozzi 1997). Accordingly, it has been proposed that mutations in the mtDNA affecting the performance of mitochondria ATP production could cause reduced sperm motility and, therefore, asthenozoospermia (Følgero et al. 1993; Kao et al. 1995; Frank and Hurst 1996; St. John et al. 1997; Zeviani and Antozzi 1997). About 10%–13% of couples suffer from infertility, which is caused by male dysfunction in 40% of the cases. Untreatable subfertility caused by poor semen quality accounts for 75% of patients consulting for fertility problems. This is due mainly to a low number of sperm cells (oligozoospermia) or to asthenozoospermia (Baker 1994).
The frequency of the known pathological mtDNA mutations within the population is insufficient to explain a significant proportion of the asthenozoospermic patients. However, it was recently proposed that if sperm dysfunction is the main, or the only phenotypic consequence of a mtDNA mutation, those mutation(s) could be fixed as stable mtDNA haplotype(s) or even haplogroup(s). This could be possible due to the fact that mtDNA is maternally inherited, and would not suffer selective pressure throughout the male lineage (Frank and Hurst 1996). Therefore, these mutations could be accumulated within a population in a relatively high proportion. We have investigated this possibility by analyzing the distribution of Caucasian mtDNA haplogroups (Wallace 1995; Brown et al. 1997; Hofmann et al. 1997b, 1997c; Richards et al. 1996; Torroni et al. 1996, 1997) in men having fertility problems.
Material and Methods
Semen Collection and Analysis
Donors from the area of Madrid and Zaragoza (Spain) were asked to collect semen by masturbation under hygienic conditions, after a period of sexual abstinence of 3–5 d. The samples were analyzed following standard protocols (WHO 1992) within a period of 2 h at hospital fertility facilities. They were allowed to liquefy for 30 min at 37°C. Then the volume of the ejaculate was measured, and the number and percentage of motile spermatozoa was evaluated (WHO 1992). In particular, sperm-motility analysis was performed at room temperature, a minimum of four microscopy fields were systematically scanned and the motility of each spermatozoon graded as “a,” “b,” “c,” or “d,” according to whether it showed rapid progressive motility, slow or sluggish progressive motility, nonprogressive motility, or no motility at all. The percentage of spermatozoa in each category was estimated after ⩾100 spermatozoa were counted.
Spermatozoa morphology was assessed by optic microscopy. The proportion of sperm cells with apparent head defects (large heads, small heads, and tapering heads), midpiece malformations, or tail abnormalities (multiple tails or coiled tails) was estimated. Samples showing sperm cell agglutination, severe flagellar defects, or very high semen viscosity were discarded.
Flagella-Activity Measurements
The activity of several mitochondrial OXPHOS complexes was inhibited by increased concentrations of rotenone (an inhibitor for the NADH dehydrogenase or complex I-EC 1.6.5.3), antimycine A (an inhibitor for the cytochrome c reductase or complex III-EC 1.10.2.2), or cyanide (inhibitor for the cytochrome c oxidase or complex IV- EC 1.9.3.1) and the proportion of sperm cells able to manifest any kind of flagella activity was monitored by microscopy cell count in a Neubauer chamber after 3 min in the presence of the drug. In order to maintain a short and homogenous timing between the different measurements, several microscopic fields in each sample were video recorded for further quantification.
Spermatozoa Vertical Progression
Spermatozoa vertical progression was estimated by vertical introduction of a capillary tube filled with a 4.8% glucose isotonic solution to a semen sample and allowance of the entrance of the spermatozoa for 30 min. After that period of time, the distance (in mm) that the spermatozoa were able to progress along the capillary tube was estimated.
DNA Analysis and Sequencing
Mitochondrial DNA levels were estimated as follows: ∼0.5 μg of total DNA, extracted from semen and previously denaturalized with 0.5 M NaOH, was transferred to a nylon filter by Slot-Blot. Then, filters were hybridized for the first time with a 273-bp mitochondrial probe against the 16S mt-rRNA gene (nt 1621–1894). The same filter was rehybridized, after stripping, with a 431-bp nuclear probe against the 18S n-rRNA gene (nt 757–1188). Probes were labeled by PCR with 32[P]-dTTP (Amersham). Filters were hybridized with 500,000–1,000,000 counts per minute/ml of the appropriate probe. Hybridization was performed in 6× SSC at 68°C and the blots were exposed for fluorography. Autoradiograms were evaluated by a LKB laser densitometer (model ULTROSCAN XL), with GelScan XL software. DNA sequencing was performed by the Thermo Sequenase radiolabeled terminator cycle sequencing Kit (Amersham) as specified by the manufacturer.
Haplogroup Determination
The samples were haplogrouped by PCR amplification of short mtDNA fragments, followed by restriction enzyme analysis, as described elsewhere (Torroni et al. 1996), but with some modifications (table 1table 1). Thus, we have used the nomenclature proposed by the Emory group, with minor modifications (Wallace 1995; Torroni et al. 1996; Brown et al. 1997; Torroni et al. 1997). In particular, since it has been recently proposed that the haplogroup previously designated as “K” belongs to the haplogroup U and should be considered a sub haplogroup of it (Macaulay et al. 1999), we have joined the individuals of K and U haplogroups and designated them as “haplogroup KU.” In addition, we have reduced the number of the polymorphic sites screened in all samples to four specific positions: the 7025 AluI, 10394 DdeI, and 12308 HinfI sites (Torroni et al. 1996) and the 4216 AflIII site.
Table 1.
MtDNA Polymorphic Sites Used to Define the mtDNA Haplogroups[Note]
|
Results at Sitea |
|||||||||||||||
| mtDNAHaplogroup | +7025AluI | +12308HinfI | +4216AflIII | +4914MaeI | +13366BamH1 | +13704MvaI | +8249AvaII | +4529HaeII | +8994HaeIII | +14465AccI | +10394DdeI | +10397HaeIII | +3592HpaI | +1715DdeI | +4577NlaIII |
| H | − | − | −/+ | ND/− | ND/− | ND/+ | ND | ND | ND | ND | − | ND | ND | ND | ND |
| V | + | − | − | ND | ND | ND | − | ND | ND | ND | − | ND | ND | + | − |
| KU | + | + | − | ND | ND | ND | ND | ND | ND | ND | ± | ND | ND | ND | ND |
| J | + | − | + | − | − | − | ND | ND | ND | ND | ± | ND | ND | ND | ND |
| T | + | − | + | + | + | + | ND | ND | ND | ND | ± | ND | ND | ND | ND |
| I | + | − | − | ND | ND | + | + | − | ND | ND | + | ND | ND | ND | ND |
| W | + | − | − | ND | ND | ND | + | + | − | ND | − | ND | ND | ND | ND |
| X | + | − | − | ND | ND | ND | − | + | + | + | − | ND | ND | − | + |
| M | + | − | − | ND | ND | + | − | ND | ND | ND | + | + | ND | ND | ND |
| L | + | − | − | ND | ND | + | − | + | + | − | + | − | + | ND | ND |
| O | + | − | − | ND | ND | + | − | + | + | − | +/− | −/ND | − | + | ND/+ |
Note.— The presence of the indicated restriction-enzyme polymorphic sites was investigated in short PCR fragments amplified from mtDNA. The plus sign (+) indicates that the enzyme is able to cut the amplified PCR fragment, and the minus sign (−) indicates that it is not. The plus/minus sign (±) indicates that there are members of these haplogroups that could, or could not, be cut by the enzyme. Results following a slash (/) apply to only a few individuals. The names of the haplogroups are as described elsewhere (Macaulay et al. 1999), except for O, which corresponds to a set of unassigned individuals.
nd = not determined.
Table 1.
MtDNA Polymorphic Sites Used to Define the mtDNA Haplogroups[Note]
|
Results at Sitea |
|||||||
| mtDNAHaplogroup | +7025AluI | +12308HinfI | +4216AflIII | +4914MaeI | +13366BamH1 | +13704MvaI | +8249AvaII |
| H | − | − | −/+ | ND/− | ND/− | ND/+ | ND |
| V | + | − | − | ND | ND | ND | − |
| KU | + | + | − | ND | ND | ND | ND |
| J | + | − | + | − | − | − | ND |
| T | + | − | + | + | + | + | ND |
| I | + | − | − | ND | ND | + | + |
| W | + | − | − | ND | ND | ND | + |
| X | + | − | − | ND | ND | ND | − |
| M | + | − | − | ND | ND | + | − |
| L | + | − | − | ND | ND | + | − |
| O | + | − | − | ND | ND | + | − |
Note.— The presence of the indicated restriction-enzyme polymorphic sites was investigated in short PCR fragments amplified from mtDNA. The plus sign (+) indicates that the enzyme is able to cut the amplified PCR fragment, and the minus sign (−) indicates that it is not. The plus/minus sign (±) indicates that there are members of these haplogroups that could, or could not, be cut by the enzyme. Results following a slash (/) apply to only a few individuals. The names of the haplogroups are as described elsewhere (Macaulay et al. 1999), except for O, which corresponds to a set of unassigned individuals.
nd = not determined.
Table 1.
MtDNA Polymorphic Sites Used to Define the mtDNA Haplogroups
| Results at Sitea | |||||||
| +4529HaeII | +8994HaeIII | +14465AccI | +10394DdeI | +10397HaeIII | +3592HpaI | +1715DdeI | +4577NlaIII |
| ND | ND | ND | − | ND | ND | ND | ND |
| ND | ND | ND | − | ND | ND | + | − |
| ND | ND | ND | ± | ND | ND | ND | ND |
| ND | ND | ND | ± | ND | ND | ND | ND |
| ND | ND | ND | ± | ND | ND | ND | ND |
| − | ND | ND | + | ND | ND | ND | ND |
| + | − | ND | − | ND | ND | ND | ND |
| + | + | + | − | ND | ND | − | + |
| ND | ND | ND | + | + | ND | ND | ND |
| + | + | − | + | − | + | ND | ND |
| + | + | − | +/− | −/ND | − | + | ND/+ |
nd = not determined.
These four polymorphic sites allow the classification of individuals into four groups: those who belong to haplogroups H, KU, and T–J, and those who belong to haplogroups L, M, I, W, X, and V (Torroni et al. 1996, 1997). The identity of the haplogroups T and J was further established by the analysis of additional polymorphic sites as indicated intable 1table 1. Because of the low ratio of mtDNA/nDNA present in spermatozoa, PCR conditions were optimized in order to avoid amplification of mitochondrial pseudogenes. Screening for the T10463C and the G15928A polymorphisms was performed by RFLP analysis, as described elsewhere (Houshmand et al. 1994). The G15928A polymorphism was confirmed by DNA sequencing. A full description of the oligodeoxynucleotides utilized and the PCR amplification conditions are available upon request.
Biochemical Determinations
Semen samples (0.5–2 ml) were centrifuged for 10 min at 600 × g at room temperature. Seminal plasma was collected for the biochemical determinations. Fructose and citric acid concentration were determined by spectrophotometry in an Hitachi autoanalyzer, using standard kits (Boehringer Mannheim catalogue numbers 139106 and 139076) according to the supplier's specifications. Carnitine concentration was estimated as previously described (Rodriguez-Segade et al. 1985). The concentration of different metals (calcium, zinc, and magnesium) was estimated at the hospital facilities by atomic absorption.
Enzyme Activity Analysis
At room temperature, 0.5–2 ml of semen was centrifuged for 10 min at 600 × g. Seminal plasma was eliminated and the pellet was washed with saline solution. Samples were centrifuged again for 10 min at 600 × g, supernatants were discarded, and the spermatozoa were resuspended in the required volume of 20 mM potassium phosphate (pH 7.0) to reach a final concentration of 2 × 105 cells/μl. Then samples were homogenized by freeze thawing before analysis, as described elsewhere (Ruiz-Pesini et al. 1998). The activities of NADH dehydrogenase (NADH DH, complex I), succinate dehydrogenase (complex II- EC 1.3.99.1), cytochrome c oxidase (COX, complex IV), and citrate synthase (CS-EC 4.1.3.7) in spermatozoa homogenates were measured spectrophotometrically in a Beckman DU-650 spectrophotometer (Beckman Instruments) by monitoring the reduction of ferricyanide (complex I), 2,6 diclorophenolindophenol (DCPIP) (complex II), the oxidation of cytochrome c (complex IV) or the condensation of ditionitrobenzoate (DTNB) with free Coenzyme A, as previously described (Ruiz-Pesini et al. 1998). Protein concentration was measured by the Lowry's method. Specific activities were expressed as nmol × min−1 × mg protein−1. All chemicals were from Boehringer Mannheim and Sigma Chemicals.
Statistical Analysis
The differences in haplogroup frequency distribution between populations were assessed by the χ2 test from contingency tables. The contribution of individual table cells to the χ2 was done by post hoc analysis by the following statistic:
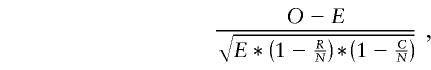 |
where O is the observed frequency, C the column total, R the row total, N the grand total and E is (CR)/N. This form of standardized residual follows a standard normal distribution.
Additionally, probabilities for the distribution of individual haplogroups in asthenozoospermic and nonasthenozoospermic populations was estimated by means of Fisher's exact test from 2 × 2 contingency tables, where the frequency of all haplogroups but the one to be tested was added. The differences between haplogroups in seminogram parameters, spermatozoa vertical progression, specific activity of NADH dehydrogenase (complex I), succinate dehydrogenase (complex II), cytochrome c oxidase (complex IV), and citrate synthase were assessed by analysis of variance (ANOVA). Paired haplogroup differences were assessed by the post hoc Fisher’s protected least significant difference test (PLSD). All tests and calculations were done with the statistical package StatView 5.0 for Macintosh (SAS Institute, Inc.).
Results
OXPHOS and Spermatozoa Motility
Our primary goal was the investigation of fixed mtDNA variants with potential phenotypic consequences in human sperm quality. Therefore, we focused on the potential association between mtDNA and one of the more relevant parameters of the spermiogram, the motility of the spermatozoa. The spermatozoa have very specialized mitochondria that are exclusively located in the midpiece of the cell at the position where the flagellum is inserted. This localization strongly suggests that ATP production by mitochondria can play a relevant role in sperm motility. However, a surprising absence of consensus about the relevance of mitochondrial OXPHOS in sperm motility (Cummins et al. 1998) prompted us to investigate this aspect first. Figure 1 shows how the inhibition, by specific drugs, of the activity of three OXPHOS complexes that are partially encoded by mtDNA progressively impairs the quality of sperm motility and finally blocks any flagellar movement. These results demonstrate that the motility of human spermatozoa is fully dependent on the OXPHOS functionality and confirm previous observations pointing in the same direction (Ford and Harrison 1981; Halangk et al. 1985; Pascual et al. 1996; Ruiz-Pesini et al. 1998).
Figure 1.

Dependence of the sperm flagella movement on the activity of the mitochondrial oxidative phosphorylation complexes. The activity of several mitochondrial OXPHOS complexes was inhibited by increasing concentrations of the indicated drug, and the proportion of sperm cells able to move following a quick and straight trajectory (open circles) or that manifest any kind of flagella activity (close circles) was evaluated after 3 min in the presence of the drug by microscopy count in a Neubauer chamber. Data represent average ± standard error of the mean (SEM) of four independent experiments.
mtDNA Haplogroup and Sperm Motility Phenotype
Sperm samples from 545 donors were collected and analyzed by standard procedures (see Methods). Then, total DNA extraction and mtDNA PCR amplification, followed by restriction enzyme analysis, were performed to ascribe each donor to one of the different haplogroups previously described (table 1table 1). In this study, we have used the nomenclature proposed by the Emory group (Wallace 1995; Torroni et al. 1996, 1997; Brown et al. 1997) and recently updated (Macaulay et al. 1999). In summary, and in good agreement with previous descriptions of other white populations, >90% of the individuals belong to one of the previously known haplogroups (table 2), and their distribution was similar to those described elsewhere (Richards et al. 1996; Torroni et al. 1996, 1997; Brown et al. 1997; Hofmann et al. 1997b, 1997c).
Table 2.
Observed Frequencies for Each Haplogroup in the Whole Sample and in Asthenozoospermic versus Non-AP Populations
|
Results in Populationa |
||||||
| Whole Sample |
Non-AP |
AP |
||||
| mtDNAHaplogroup | n | f | n | f | n | f |
| Hb | 271 | 49.72 | 107 | 59.44 | 164 | 44.93 |
| I | 8 | 1.47 | 2 | 1.11 | 6 | 1.64 |
| J | 38 | 6.97 | 11 | 6.11 | 27 | 7.40 |
| KU | 106 | 19.45 | 31 | 17.22 | 75 | 20.55 |
| L | 7 | 1.28 | 2 | 1.11 | 5 | 1.37 |
| M | 8 | 1.47 | 1 | 0.56 | 7 | 1.92 |
| O | 39 | 7.16 | 10 | 5.56 | 29 | 7.95 |
| Tc | 31 | 5.69 | 4 | 2.22 | 27 | 7.40 |
| V | 21 | 3.85 | 7 | 3.89 | 14 | 3.84 |
| W | 6 | 1.10 | 2 | 1.11 | 4 | 1.10 |
| X | 10 | 1.83 | 3 | 1.67 | 7 | 1.92 |
| Total | 545 | 100.00 | 180 | 100.00 | 365 | 100.00 |
| IWXd | 24 | 4.40 | 7 | 3.89 | 17 | 4.66 |
| LMOd | 54 | 9.91 | 13 | 7.23 | 41 | 11.24 |
Samples were considered as asthenozoospermic phenotype (AP) when showing <50 % of progressive motile spermatozoa (WHO 1992). χ2 = 13.873 for haplogroup distribution vs. AP or Non-AP (6 df; P=.0311). n = indicates the number of individuals; f = the frequency expressed as percentage.
Haplogroup H contribution to χ2=5.106 (Fisher’s exact test P=.0019). Difference in frequency distribution from the expected values for haplogroup H in non-AP population: P=.0014
Haplogroup T contribution to χ2=5.676 (Fisher’s exact P=.0168). Difference in frequency distribution from the expected values for haplogroup T in the AP population: P=.0142.
IWX and LMO represent the added frequencies of I, W, and X and of L, M, and O, respectively.
To investigate the potential genetic association between mtDNA variants and asthenozoospermia, we divided our sample in two groups according to the motility of the sperm cells: Asthenozoospermic (AP), individuals with low sperm motility (< 50% of progressive spermatozoa) and Non-AP, individuals with normal sperm motility (⩾ 50%) (WHO 1992), and we estimated the frequency of each haplogroup in the non-AP and AP populations (table 2). Since the expected frequencies for several haplogroups were <5, we have grouped the low-represented ones (W, X, and I) according to their proposed evolutionary proximity (Macaulay et al. 1999). Furthermore, we have also grouped those of nonwhite origin, haplogroups L (African) and M (Asian), with the samples that we could not ascribe to any of the known haplogroups (O). In this way, the minimum expected frequency to ensure the accuracy in the χ2 independence test is reached (table 2). Contingency table analysis suggests that the asthenozoospermic character may not be independent on the mtDNA haplogroup (χ2=13.873, P=.0311, 6 df). When the contribution of the different haplogroups to the χ2 was examined, it could be appreciated that T (χ2 contribution 5.676) and H (χ2 contribution 5.106) mtDNAs were the main responsible for the high χ2. Furthermore, Fisher's exact test also suggests a significant deviation from the expected frequencies for H (P=.0019) and T (P=.0168) mtDNA haplogroups. In fact, a post hoc analysis on the cell contribution to the χ2 revealed that a significant deviation from the expected frequency could be observed for the presence of mtDNA haplogroup H in normal individuals (post hoc cell contribution 3.187, P=.0014). This was also the case for the accumulation of T mtDNA haplogroup within the asthenozoospermic population (post hoc cell contribution 2.453, P=.0142). None of the other haplogroups showed a frequency distribution in AP and non-AP populations significantly deviated from the expected values when a hypothesis of independence was assumed.
We reevaluated the association between mtDNA haplogroups and motility phenotype by a semiquantitative approach. Thus, we divided our samples by calculating the frequency of the different haplogroups in three categories: nonasthenozoospermic (Non-AP; ⩾ 50% of progressive spermatozoa), moderate asthenozoospermic phenotype (MAP; ⩾25% and <50% of progressive spermatozoa) and severe asthenozoospermic phenotype (SAP; < 25% of progressive spermatozoa) (table 3). Contingency table analysis by the χ2 test confirmed that the degree of asthenozoospermia was not independent on the mtDNA of the probands (χ2=29.104, P=.0038, 12 df). Again, T and H mtDNA haplogroups are the two major contributors to the χ2 (T haplogroup contribution to χ2=14.955; H haplogroup contribution to χ2=5.165). Moreover, a post hoc analysis on the cell contribution to the χ2 shows that haplogroup H was, in fact, significantly overrepresented in non-AP individuals (post hoc cell contribution 3.187, P=.0014), whereas haplogroup T is very much accumulated in the MAP group (post hoc cell contribution 3.977, P=.00007). None of the other haplogroups showed an association with any of the motility phenotypes.
Table 3.
Observed Frequencies for mtDNA Haplogroups in SAP, MAP, and Non-AP
|
Results in Phenotypea |
||||||||
| Non-AP |
MAP |
SAP |
All |
|||||
| mtDNAHaplogroup | n | f | n | f | n | f | n | f |
| Hb | 107 | 59.44 | 100 | 45.66 | 64 | 43.84 | 271 | 49.72 |
| IWX | 7 | 3.89 | 10 | 4.57 | 7 | 4.79 | 24 | 4.40 |
| J | 11 | 6.11 | 19 | 8.68 | 8 | 5.48 | 38 | 6.97 |
| KU | 31 | 17.22 | 39 | 17.81 | 36 | 24.66 | 106 | 19.45 |
| LMO | 13 | 7.22 | 22 | 10.05 | 19 | 13.01 | 54 | 9.91 |
| Tc | 4 | 2.22 | 23 | 10.50 | 4 | 2.74 | 31 | 5.69 |
| V | 7 | 3.89 | 6 | 2.74 | 8 | 5.48 | 21 | 3.85 |
| Total | 180 | 100.00 | 219 | 100.00 | 146 | 100.00 | 545 | 100.00 |
Samples were considered as MAP when they showed <50% and ⩾25% of progressive motile spermatozoa, and they were considered as SAP when they showed <25% of progressive motile spermatozoa. χ2=29.104 for haplogroup distribution vs. SAP, MAP, or non-AP (12 df; P=.0038). n = the number of individuals; f = the frequency expressed as percentage.
Haplogroup H contribution to χ2=5.165. Difference in frequency distribution from the expected values for haplogroup H in non-AP population: P=.0014.
Haplogroup T contribution to χ2=14.955. Difference in frequency distribution from the expected values for haplogroup T in AP population: P=.00007.
As an additional control of the observed association between the motility phenotype and the mtDNA haplogroup, we divided our sperm-donors panel according to a different parameter of the spermiogram, the concentration of sperm cells: oligozoospermic (OP), individuals showing reduced cell concentration (<20 million/ml); and non-OP, individuals showing normal sperm concentration (⩾20 million/ml) (WHO 1992). A similar statistical approach determined that the oligozoospermic character should be considered independent of the mtDNA haplogroup (χ2=4.771, P=.5736; 6 df), and none of the individual haplogroups showed a significant association with OP or non-OP populations. Moreover, when we reanalyzed our data for any potential relationship between severe oligozoospermia (<5 million sperm cells/ml) and mtDNA haplogroup, no association was observed (χ2=6.065; P=.6400; 8 df).
The combination of the two characters of the spermiogram, motility and concentration of sperm cells, define four distinct phenotypes: 241 individuals were pure asthenozoospermic A (pA), 25 were pure oligozoospermic (pO), 123 were asthenooligozoospermic (AO), and 155 did not show any deficient phenotype and were designated controls (C). Therefore, to confirm that the asthenozoospermic character alone is responsible for the observed associations, the distribution of the mtDNA haplogroups in the control subpopulation was compared to that in the pure asthenozoospermic one (table 4). In order to reach the minimal number of expected individuals for a confident χ2 analysis, the reduction in the number of samples forced us to consider several haplogroups of lower interest together (IWX, LMO, and V) in one that was called “Rest.” Despite of that, the χ2-independence test supports again the interpretation proposed above, i.e., the non-random distribution of mtDNA haplogroups with respect to the motility phenotype (χ2=16.99; P=.0302; 8 df). Thus, a significant accumulation of H mtDNA haplotypes within the control population (post hoc test 3.282; P=.001) and of T mtDNA haplotypes within the MAP population (post hoc test 2.650; P=.008) was observed.
Table 4.
Observed Frequencies for mtDNA Haplogroups in Non-AP, Pure SAP, and Pure MAP
|
Results in Populationa |
||||||||
| Non-AP |
Pure MAP |
Pure SAP |
All |
|||||
| mtDNAHaplogroup | n | f | n | f | n | f | n | f |
| Hb | 95 | 61.29 | 70 | 42.94 | 37 | 47.44 | 202 | 51.01 |
| J | 8 | 5.16 | 16 | 9.82 | 5 | 6.41 | 29 | 7.32 |
| KU | 25 | 16.13 | 32 | 19.63 | 16 | 20.51 | 73 | 18.43 |
| Rest | 23 | 14.84 | 30 | 18.40 | 17 | 21.79 | 70 | 17.68 |
| Tc | 4 | 2.58 | 15 | 9.20 | 3 | 3.85 | 22 | 5.56 |
| Total | 155 | 100.00 | 163 | 100.00 | 78 | 100.00 | 396 | 100.00 |
Samples were considered to be pure MAP when they show <50% and ⩾25% of progressive motile spermatozoa and ⩾20 × 106 spermatozoa per milliliter. Samples were considered to be pure SAP when they showed <25% of progressive motile spermatozoa and ⩾20 × 106 spermatozoa per milliliter. χ2=16.988 for haplogroup distribution vs. pure SAP, pure MAP, or Control (8 df; P=.0302). n = indicates the number of individuals; f = frequency, expressed as a percentage.
Haplogroup H contribution to χ2=5.49. Difference in frequency distribution from the expected values for haplogroup H in non-AP: P=.001.
Haplogroup T contribution to χ2=6.78. Difference in frequency distribution from the expected values for haplogroup T in AP: P=.008.
The observed accumulation of H and T mtDNAs within the non-AP and the MAP motility groups, respectively, could be a secondary association determined by an additional parameter of the seminogram. To investigate this possibility, we tested the potential variation between haplogroups of several other parameters of the seminogram. These evaluate a potential bias in the sampling process (age of the donors, time between sample collection, and analysis), differences in ejaculated volume, or pH as well as in the cellular composition of the semen (spermatozoa concentration, spermatozoa vitality, proportion of abnormal spermatozoa morphology, and concentration of round cells). In addition, several biochemical parameters that are used as markers of the prostatic functionality (citric acid, zinc, magnesium, and calcium concentration), seminal vesicle functionality (fructose concentration) and epididimal functionality (carnitine concentration) were evaluated. The number of individual samples utilized for these analyses was variable and dependent on their availability. Therefore we also estimated if the observed accumulation of haplogroups T and H within the MAP and, respectively, the non-AP population, was maintained in the subpopulation utilized for the analysis of each parameter. Table 5 shows a summary of the results obtained and indicates that none of the investigated parameters but motility phenotype is significantly different between haplogroups H and T.
Table 5.
Differences in Seminal Parameters between H and T mtDNA Carrier Populations
|
n |
Haplogroup |
||||||||
| Parameter | All | H | T | Whole Population | H | T | T versus Ha | T in MAPb | H in Non-APb |
| Age (years) | 360 | 169 | 17 | 32.6 ± 6.0 | 32.6 ± 6.3 | 33.6 ± 4.8 | .4778 | <.001 | <.001 |
| Preanalytic time (min) | 235 | 109 | 12 | 45.9 ± 14.7 | 44.1 ± 12.9 | 47.9 ± 18.8 | .3933 | .002 | .002 |
| Seminal volume (ml) | 513 | 252 | 31 | 3.3 ± 1.7 | 3.3 ± 1.7 | 3.4 ± 1.9 | .7039 | <.001 | <.001 |
| Seminal pH | 317 | 156 | 18 | 7.7 ± .3 | 7.8 ± .3 | 7.8 ± .4 | .4213 | .004 | .002 |
| Prostatic markers: | |||||||||
| Citric acid (mg/dl) | 202 | 95 | 11 | 533 ± 300 | 570 ± 343 | 550 ± 244 | .8329 | <.001 | <.001 |
| Zn (mg/dl) | 201 | 95 | 11 | 10.8 ± 6.3 | 11.3 ± 6.4 | 11.9 ± 6.2 | .8082 | <.001 | <.001 |
| Mg (mg/dl) | 202 | 95 | 11 | 8.2 ± 4.4 | 8.5 ± 4.6 | 7.5 ± 4.2 | .8080 | <.001 | <.001 |
| Ca (mg/dl) | 202 | 95 | 11 | 35.4 ± 14.6 | 37.5 ± 15.6 | 33.6 ± 12.6 | .4035 | <.001 | <.001 |
| Seminal vesicle marker: | |||||||||
| Fructose (mg/dl) | 202 | 95 | 11 | 295 ± 132 | 281 ± 142 | 344 ± 164 | .1383 | <.001 | <.001 |
| Epididimal marker: | |||||||||
| Carnitine (μM) | 197 | 93 | 11 | 485 ± 231 | 497 ± 234 | 446 ± 211 | .4877 | <.001 | <.001 |
| Cellular parameters: | |||||||||
| Sperm concentration (million cells/ml) | 544 | 270 | 31 | 48.4 ± 44.1 | 51.4 ± 46.3 | 40.9 ± 30.6 | .2084 | <.001 | <.001 |
| Vital spermatozoa (%) | 211 | 100 | 17 | 49.6 ± 16.7 | 49.5 ± 17.8 | 47.2 ± 13.8 | .6577 | .006 | .006 |
| Abnormal spermatozoa (%) | 238 | 119 | 17 | 50.7 ± 17.0 | 48.4 ± 16.8 | 52.1 ± 14.9 | .4082 | <.001 | <.001 |
| Round cells (million cells/ml) | 251 | 119 | 15 | 4.3 ± 3.5 | 4.0 ± 3.2 | 3.8 ± 3.2 | .7807 | <.001 | <.001 |
Estimation of the potential signification (Fisher's PLSD test) for the difference between the mean values of the indicated parameter for H and T mtDNA carriers.
Significance of the association of T and H individuals with the MAP and non-AP populations, respectively, for the subset of samples included in the analysis of each seminal parameter.
For a large set of the samples (n=223), we could perform an additional motility test consisting in the measurement of the maximum distance that the spermatozoa population from different individuals are able to swim, in 30 min; in a capillary tube introduced vertically into the semen sample (vertical progression). This analysis was performed before the mtDNA haplogroup of the samples was determined and without knowing the spermiogram analysis. As shown in figure 2, the vertical progression of spermatozoa was variable between haplogroups. Interestingly, it was maximum for haplogroup H and minimum for haplogroup T being significantly different (P=.029, Fisher PLSD test). The rest of the haplogroups showed an intermediate capacity of vertical progression although the differences between them were not significant.
Figure 2.

Vertical-progression analysis. Differences in the ability of spermatozoa carrying different mtDNAs to swim from the semen into a capillar tube in a period of 30 min. n is the number of samples, and the asterisk (*) indicates significant differences between the distance of vertical progression for haplogroup T versus haplogroup H carrying mtDNA samples (P=.029; Fisher’s PLSD test).
Confirmation that the vertical progression is also a good quantitative estimation of the degree of asthenozoospermia came from the fact that those individuals classified as nonasthenozoospermic showed an average spermatozoa vertical progression of 6.87 ± 1.74 mm (n=70), whereas in moderate asthenozoospermic was 4.89 ± 1.67 mm (n=103) and severe asthenozoospermic was 2.380 ± 1.74 mm (n=50). As expected, there is a strong association between vertical progression and the motility phenotype (SAP, MAP, and non-AP) established by the percentage of motile spermatozoa (F=101.086; 2 df; P<.0001; ANOVA). Furthermore, differences in vertical progression between paired groups were always significant as estimated by the Fisher's PLSD post hoc test (P<.0001 in all cases).
Finally, to discard any bias in the sampling of our haplogroup T individuals, the hypervariable region I (position 16083–16378 in the noncoding region of mtDNA) of 29 of our 31 Ts was sequenced. Thus, we were able to identify several main sublineages with 20 different haplotypes (fig. 3). Interestingly, they include all the T branches described for the German population (Hofmann et al. 1997c), showing that our T population is truly representative of the white T haplogroup. In addition, the non-AP as well as the SAP individuals were scattered among the different branches of the T haplotype tree. These results strongly suggest that the accumulation of T in the MAP population is due to the haplogroup itself rather than to a particular sublineage within the haplogroup T.
Figure 3.

Sequence analysis of mtDNA haplogroup T carriers. Nucleotide differences at the hypervariable region I (HVRI) of the noncoding region of mtDNA that define 20 haplotypes in 29 individuals harboring the T haplogroup. The phenotype of the different individuals is indicated as SAP (blackened circles) or non-AP (unblackened circles); all the other samples showed MAP (striped circles). Each individual is indicated by a letter/number code.
Different OXPHOS Performance between Sperm Samples Carrying H or T mtDNAs
In order to get some insights on the molecular mechanism underlying the observed association between mtDNA haplogroups and spermatozoa motility, we first tested the OXPHOS performance in sperm homogenate of the 136 individuals from whom we had enough available sample. Those 136 samples represented a mix of SAP, MAP, and non-AP (39, 58, and 39 samples, respectively) and a variety of haplogroups H, J, KU, and T (60, 17, 27, and 5, respectively). Samples belonging to the remaining haplogroups (I, X, V, W, L, M, and O; 5, 3, 3, 1, 4, 4, and 7) were grouped together (n=27) and were called “Rest” for the purpose of statistical analysis. As shown in figure 4, complex I–specific activity measured in spermatozoa homogenates was variable between different haplogroups, being, on average, 22% lower in samples carrying T mtDNAs than in those carrying H mtDNAs. However, this variability was not significant enough to reject the independence hypothesis between the mtDNA haplogroups and NADH DH activity (ANOVA P=.593), and no significant differences between paired haplogroups were observed. Since all T samples fall in the MAP category, the difference in NADH DH activity considering only MAP individuals was also investigated. Again, a nonsignificant reduction of the complex I activity of T individuals versus that in H individuals could be observed (ANOVA P=.873). On the contrary, ANOVA analysis indicated that complex IV–specific activity showed a significant variability between the different haplogroups regardless of the motility phenotype in the subpopulations included in the analysis (ANOVA P=.0031 for total sample and P=.0464 for MAP individuals; fig. 4). Thus, T haplogroup showed the lowest COX activity when compared with H mtDNA haplogroup. This difference was significant by the Fisher’s PLSD post hoc test (P=.0184 for total sample and P=.0159 for MAP individuals). The other haplogroups showed intermediate COX activity that were not (J and KU) or were significantly different (Rest, P=.0016 for total sample and P=.0116, for MAP) from that estimated for the H haplogroup (fig. 4). In addition, small and nonsignificant differences between haplogroups in the specific activity of complex II (the only respiratory complex with all subunits encoded by nuclear genes) and citrate synthase (a mitochondrial matrix located enzyme) could be observed (fig. 4).
Figure 4.

OXPHOS-activity analysis. Bar diagrams showing the differences in activity of two fully nuclear-encoded mitochondrial enzymes, citrate synthase (CS, a mitochondrial-matrix enzyme) and succinate dehydrogenase (complex II) and two enzymatic complexes partially encoded by mtDNA: NADH dehydrogenase (complex I) and cytochrome c oxidase (COX or complex IV). Bars represent specific enzyme activity (mean ± SEM) for the different haplogroups when either all (gray) or only MAP (crossed) individuals were considered. ANOVA test indicate the absence of significant differences between haplogroups for CS, complex II, and complex I, as well as a significant difference for COX activity when all (P=.0031) or only MAP (P=.0464) individuals were considered. Post hoc analysis by Fisher’s PLSD test reveals that T and Rest showed a significantly lower COX activity when compared with H, regardless of whether all or only MAP individuals were considered (P<.05).
Furthermore, when the activity of the different respiratory complexes was normalized to the activity of the citrate synthase, an interesting figure arose (fig. 5). Both complex I and complex IV showed high relative activity in samples carrying the H mtDNA haplogroup and the lowest relative activity in samples harboring the T mtDNA. Thus, T samples showed 23% and 29% reductions, respectively, in complexes I and IV, with respect to H samples. However, these differences were not statistically significant by the ANOVA test. Moreover, the activity of complex II, when normalized by citrate synthase, was virtually identical between the different haplogroups, and, in particular, between H and T. Despite the absence of statistical significance, it should be remarked that those complexes that are partially encoded by mtDNA (complex I and IV) showed a similar reduction in their relative activity per mitochondria between haplogroups H and T. On the contrary, the complex that has no subunits encoded by mtDNA showed similar activity in all haplogroups. Therefore, samples of individuals belonging to the T and H mtDNA haplogroups seem to present the largest difference in sperm OXPHOS performance between haplogroups.
Figure 5.
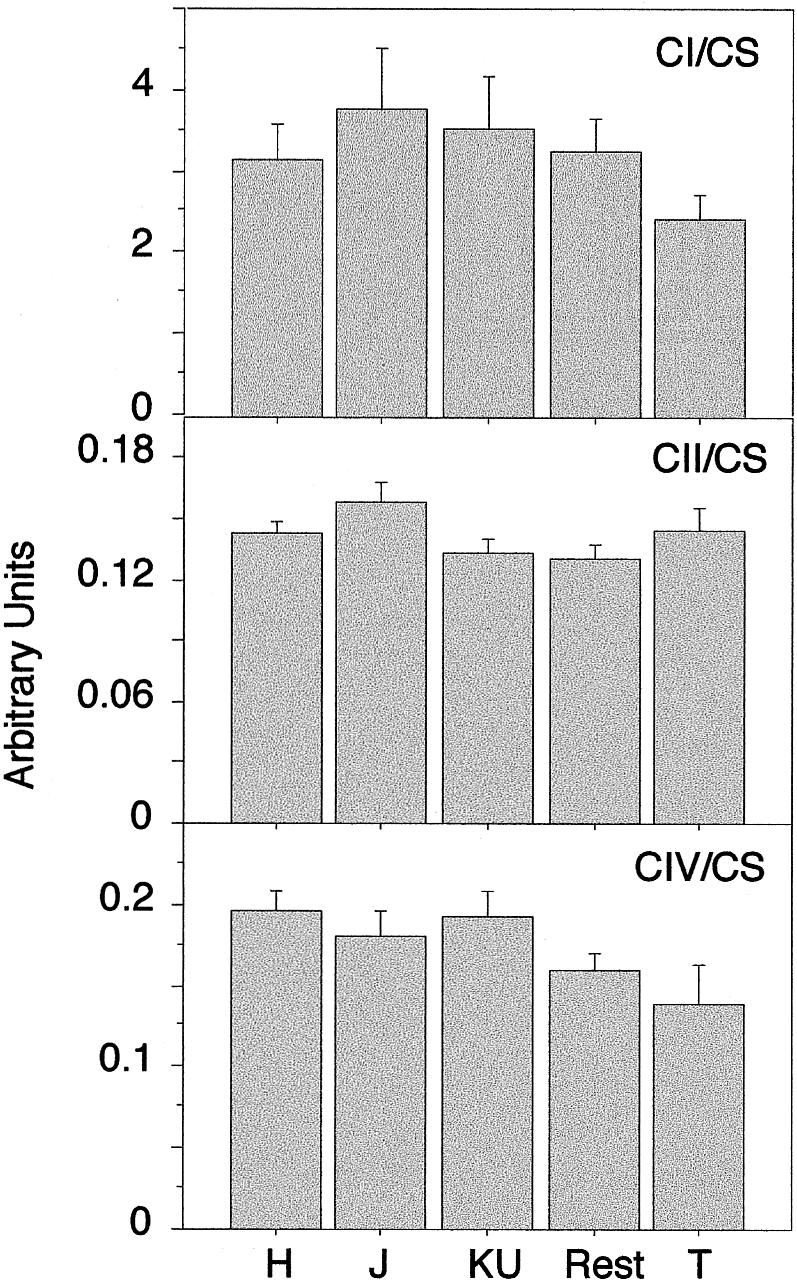
Bar diagrams showing the relative activity of three respiratory complexes (complexes I and IV, partially encoded by mtDNA, and complex II, fully encoded by nuclear genes) in each haplogroup, when they are normalized by the activity of citrate synthase. Data are given as mean ± SEM in arbitrary units.
Additional Haplogroup T–Specific mtDNA Polymorphisms
The differences in OXPHOS performance between haplogroups were not a consequence of haplogroup-specific differences in mtDNA level as estimated by ANOVA analysis, since the proportion of mtDNA/nDNA between the different haplogroups was very similar (data not shown). Several polymorphic markers that differentiate the T haplogroup are located at genes that encode complex I subunits (T4216C at ND1, A 4917G at ND2, and G13368A at ND5) which promote either amino acid substitutions (Y→H at ND1 and D→N at ND2) or no changes (ND5). These changes could be invoked to explain the apparently lower performance of complex I in T mtDNA carrying individuals. However, these could not explain the more significant reduction in complex IV activity observed in T individuals. Therefore, to investigate the molecular reason why COX activity was reduced in T samples, we sequenced the full set of COX genes encoded in the mtDNA for one T individual. Three alterations from the Cambridge sequence were detected. One, the expected C7028T transition, was located in the CO I gene (which is common to all white haplogroups, with the exception of H) and is a synonymous mutation. The second was located in the CO II gene and is also a synonymous C7891T transition not previously described. Finally, the third mutation, a G9559C transversion, was located in the CO III gene, and it implies a variation from proline to arginine. This last mutation has been recently reported as an error of the Cambridge sequence (Andrews et al. 1999) and is not specific for haplogroup T. Therefore, none of these polymorphisms could justify the reduced COX activity associated with T mtDNA.
In addition, the accumulation of T mtDNA within the asthenozoospermic population could be due to an inefficient mitochondrial ATP synthase activity (complex V). Since the evaluation of the activity of complex V in spermatozoa is very much compromised by the heavy Dinein ATPase activity, we sequenced the mtDNA encoded ATP synthase subunits of one individual to search for any T-specific polymorphic substitutions. Three mutations were detected, all in the ATPase 6 gene. One, a synonymous G8697A transition, was previously described in another T individual and seems to be a haplogroup-specific polymorphism (Ohlenbusch et al. 1998). The second, an A8860G transition, was described previously in other haplogroups, including H (Marzuki et al. 1991; Ohlenbusch et al. 1998; Andrews et al. 1999). The third mutation was an A9037G transition not described previously but not present in other T individuals.
Mild reduction in OXPHOS activity also can be a consequence of mutations in the mtDNA genes that encode for rRNAs or tRNAs. Thus, an homoplasmic mutation at nt 7445 has been described that decreases the amount of mitochondrial tRNASerUCN (Reid et al. 1994; Fischel-Ghodsian et al. 1995; Reid et al. 1997; Guan et al. 1998). In cultured cells, this mutation is associated with a moderate reduction in mitochondrial protein synthesis and a relatively mild phenotype (Guan et al. 1998). The fact that activity of complex I, as well as that of complex IV, apparently was reduced in T samples also suggests that they could be the consequence of a reduced performance in protein synthesis caused by a haplogroup-associated mutation in tRNA genes. To test this hypothesis, we sequenced the 22 tRNAs of one T individual. Two mutations were detected: a T10463C transition in the tRNA Arg gene within an uncommon U/U base pair at the bottom of the acceptor stem that becomes here U/C (fig. 6) and a G15928A transition in the tRNA Thr gene that transforms the first base pair at the anticodon stem into a non–standard Watson and Crick base pair C/A (fig. 6). Both tRNA mutations were previously described (MITOMAP), and it was proposed that are haplogroup T–specific polymorphic sites (Macaulay et al. 1999). In confirmation of that, they were detected in all our T individuals. In addition, the G15928A transition at the tRNA Thr gene was not present in any of the 495 non-T individuals investigated, as determined by RFLP analysis (see Material and Methods) and as confirmed by sequencing of the only three samples that showed alterations in their RFLP pattern. Further molecular analysis will be needed to determine if any of these tRNA variants, or both synergically, influence mitochondrial protein synthesis in a way that could justify the lower COX activity found in T mtDNA–carrying organelles.
Figure 6.

Haplogroup T–specific tRNA mutations. Sequencing of all the tRNA genes of one individual showed, in two tRNAs, mutations that were specific for the haplogroup T individuals: left, A15928G transition at the top of anticodon stem of the tRNA Thr; right, U10463C transition at the bottom of the acceptor stem of the tRNA Arg.
Discussion
Two major conclusions can be drawn from the work presented in this report. First, we have unequivocally demonstrated that human spermatozoa motility is fully dependent on the functionality of the OXPHOS system. Second, we have obtained compelling evidence indicating that the spermatozoa-motility phenotype is conditioned by the mtDNA haplogroup.
In particular, those probands carrying the H haplogroup are significantly more frequent in populations selected by their high sperm motility, and those carrying the T haplogroup are accumulated in populations selected by their reduced sperm motility. Haplogroup H is the most common variant in white mtDNA populations (Richards et al. 1996; Torroni et al. 1996; Brown et al. 1997; Hofmann et al. 1997a, 1997c; Torroni et al. 1997). Therefore, in light of the direct dependence of spermatozoa motility on the OXPHOS functionality (Halangk et al. 1985; Pascual et al. 1996; Ruiz-Pesini et al. 1998), it is reasonable to interpret the accumulation of the H mtDNA haplogroup in the non-AP population as an indication of a robust genetic background preventing against some OXPHOS weakness in spermatozoa. More interesting seems to be the increased frequency of haplogroup T in the MAP population. Haplogroup T is widely distributed among white populations. Its frequency is very variable, ranging from 22% in Swedes to 12% in central Germans to 4.4% in the Druze (Torroni et al. 1996, 1997; Hofmann et al. 1997c). From our results, it could be inferred that haplogroup T represents a weak genetic background that can predispose to asthenozoospermia.
In support of this interpretation, a difference in sperm COX-specific activity between samples harboring H and T mtDNAs also has been observed. The interpretation of the OXPHOS performance analysis should be done with care. Several facts suggest that no dramatic difference should be expected between the activity of the different respiratory complexes under each particular mtDNA haplogroup. (1) The association of T with MAP and H with non-AP subpopulations, and, more quantitatively, the 27% reduction in vertical progression of T versus H spermatozoa, suggests that the phenotypic effect induced by the type of mtDNA, although significant, is mild. (2) A severe OXPHOS defect caused only by an intrinsic characteristic of a particular mtDNA should imply a very severe loss of motility and would likely have phenotypic effects in other tissues. To our knowledge, the only potentially relevant health problem reported by a portion of the individuals included in this study is the difficulty of the couple to get pregnant and, in a significant portion of these couples, the origin of the problems should be attributed to the female partner. On the contrary, if specific particularities of the sperm OXPHOS system biogenesis were the main factor responsible for the association between low sperm motility and haplogroup T, either severe or moderate asthenozoospermic phenotype could be expected without compromising the OXPHOS performance in other tissues. (3) However, the relatively high frequency of the T mtDNA in white populations strongly suggests that the effect of this mtDNA in male fertility should not be dramatic. This is true since the lack of selective pressure against male mtDNA is not absolute because populations with a high proportion of otherwise healthy infertile males, will reduce the successful pregnancy rate, and the survival of the population would be compromised. It should be mentioned here that MAP men must be considered subfertile individuals, rather than infertile ones (Baker 1994). Therefore, it is reasonable to expect only mild OXPHOS differences between H and T mtDNAs.
The determination of consistent small differences in the activity of OXPHOS complexes between samples is always compromised by the high intrinsic variability of the enzymatic determinations. In fact, this has been also difficult to establish in well-characterized pathogenic homoplasmic mtDNA mutations—that is, those causing nonsyndromic deafness (Prezant et al. 1993; Reid et al. 1994; Fischel-Ghodsian et al. 1995) or Leber's optic neuropathy (LHON) (Howell 1999) (see below). In spite of that, in this work, we were able to detect statistically significant differences in the COX activity between individuals harboring the H mtDNA and those harboring the T mtDNA. The difference is maintained when the activity is normalized by a mitochondrial-matrix enzyme activity (citrate synthase), although the statistical significance is lost. Nevertheless, there are additional indications that made us to propose that the COX activity—and probably the NADH DH activity as well—are truly different between spermatozoa harboring H and T mtDNAs. First, there is a coherent relationship between the accumulation of H or T individuals within the non-AP and AP populations, respectively, and the OXPHOS figures. Second, and more importantly, when normalized by citrate synthase, there is a positive correlation between the activity of the complexes partially encoded by mtDNA (complex I, 23% decreased in T vs. H; complex IV, 29% decreased in T vs. H). On the contrary, this correlation is not maintained with the only complex that has all its subunits encoded by nuclear genes (complex II, 1% increased in T vs. H). Very interestingly, the amount of reduction in complex I or complex IV needed to affect overall cell respiration in cultured human cells, and, therefore, ATP production is 20% for COX (Villani and Attardi 1997) and ∼20% for NADH DH (Bai et al. 2000). Thus, the estimated reduction in OXPHOS performance in T versus H mtDNA backgrounds seems to be sufficient to induce differences in overall respiration rate and ATP production. Then, the relative lower ATP production capacity of spermatozoa harboring T mtDNA, in a situation of high energy demands (motile spermatozoa), would be the cause of the observed differences in the motility of the spermatozoa. Therefore, although we still must be cautious in the interpretation of this data, we strongly believe that they indicate that a minor, but consistent, general difference between the OXPHOS performance of spermatozoa harboring H or T mtDNAs explains their association with populations showing variable degree of sperm-motility capacity.
If this interpretation is correct, it is still necessary to understand how established mtDNA haplogroups (like T) can favor asthenozoospermia and not obviously involve other tissues. It could be that some particular characteristics of spermatozoa (e.g., nuclear-encoded sperm-specific OXPHOS subunits, etc.) make them especially sensitive to particular haplogroups. Therefore, this sensitivity would be not only an intrinsic characteristic of a particular mtDNA, but also a consequence of the specific characteristics of the biogenesis of the sperm OXPHOS system. Alternatively, it could happen that, in fact, some haplogroups promote a reduction in OXPHOS performance, albeit a very mild one in normal circumstances. Thus, only in the presence of environmental insults and/or certain nuclear backgrounds will the OXPHOS activity associated with a particularly inefficient mtDNA haplogroup drop below the pathogenic threshold. On the contrary, a more robust haplogroup will keep the OXPHOS activity over such a threshold in similar circumstances. It is, then, a plausible hypothesis that one of the more demanding OXPHOS activities (e.g., flagellar movement) will be affected first. Moreover, this would also imply that, under certain circumstances, this lower performance could be further amplified and could promote defects other than moderated asthenozoospermia.
Tissue specificity and variable degree of penetrance are hallmarks of mtDNA mutation–dependent disorders, most typically of those associated with homoplasmic mtDNA mutations that cause nonsyndromic deafness (Prezant et al. 1993; Reid et al. 1994; Fischel-Ghodsian et al. 1995) or LHON (for review, see Howell [1999]). The use of cellular models for the investigation of an homoplasmic mtDNA mutation at position 7445, affecting tRNA Ser (UCN) precursor processing, a nonsyndromic deafness–causing mutation, has revealed a mild biochemical phenotype (Guan et al. 1998) or a complete absence of one (Reid et al. 1997). However, a clear defective molecular phenotype could be observed in all cases: a severe reduction in the level of tRNA Ser (UCN) (Reid et al. 1997; Guan et al. 1998). On the other hand, a potential influence of the nuclear context in the expression of the biochemical defects associated with a mitochondrial 12S rRNA mutation causing also nonsyndromic deafness has been proposed (Guan et al. 1996). Interestingly enough, it has also been proposed that, at least in some families, the latest mutation confers susceptibility to aminoglycoside-induced ototoxicity (Prezant et al. 1993). Therefore, the molecular consequence of a mild (and homoplasmic) mtDNA mutation is transformed into a biochemical defect only after environmental insults, in susceptible nuclear backgrounds, and/or in specific cellular types. Despite that, the primary molecular cause of the disease is the mtDNA mutation. Thus, analysis of the expression of well-characterized homoplasmic pathogenic mtDNA mutations can illustrate the potential phenotypic consequences, although milder than those of such mutations, of some mtDNA haplogroups.
Additional observations give support to the interpretation that mtDNA haplogroup T is intrinsically prone to development of OXPHOS defects. DIDMOAD is a rare human disease characterized by diabetes insipidus, diabetes mellitus, optic atrophy and deafness, and it can be caused by defects in both nuclear and mitochondrial genomes (Rötig et al. 1993; Polymeropoulos et al. 1994). Very interestingly, a group of eight DIDMOAD patients were found to be preferentially associated with haplogroup T (Hofmann et al. 1997b, 1997c). In addition, an increased frequency of T mtDNA among multiple sclerosis (MS) patients has been described (Kalman et al. 1995; Mayr-Wohlfart et al. 1996).
The demonstration of negative (H) as well as positive (T) association of mtDNA haplogroups with asthenozoospermia strongly supports a model in which mtDNA haplogroups could contribute differentially to the performance of the OXPHOS system. In fact, it is intriguing that other mtDNA haplogroups were asymmetrically distributed between AP and non-AP populations and showed differences in COX-specific activity. It is also plausible that only certain sublineages within a particular haplogroup may be phenotypically relevant. Therefore, further investigation, with a more extended sample, will be needed to establish definitively the potential influence of other white mtDNA haplogroups. Nevertheless, a tendency has been observed suggesting that mtDNA haplogroups, at first thought to be phenotypically indistinguishable, could have OXPHOS phenotypic consequences and be involved in human pathology.
In this respect, the existence of tissue-specific selective pressures for different polymorphic mouse mtDNA variants has been recently proposed (Jenuth et al. 1997). There is also an increasing amount of evidence suggesting that the penetrance of two of the three common primary LHON mutations is significantly increased in haplogroup J (Brown et al. 1997; Hofmann et al. 1997b, 1997c; Torroni et al. 1997). In addition, the positive and negative associations of haplogroups T and H with asthenozoospermia found in this report, the prevalence of haplogroup T in DIDMOAD and MS patients mentioned above (Kalman et al. 1995; Mayr-Wohlfart et al. 1996; Hofmann et al. 1997b, 1997c), the accumulation of haplogroup U in occipital stroke (Majamaa et al. 1998), and the J haplogroup in human centenarians (De Benedictis et al. 1999) also point in the same direction.
Asthenozoospermia is considered a major cause of untreatable male subfertility (Baker 1994). The role of haplogroup T in promoting asthenozoospermia is probably related to an intrinsic higher sensitivity of the OXPHOS energy production to a variety of environmental or genetic factors. Thus, this mtDNA background could be a secondary, but relevant, contributing factor in at least 7%–10% of the asthenozoospermic males. Moreover, it is very likely that future studies will extend further the implication of both mtDNA- and nuclear-encoded OXPHOS genes as a major cause of sperm dysfunction (Følgero et al. 1993; Frank and Hurst 1996; Kao et al. 1995; St. John et al. 1997; Zeviani and Antozzi 1997; Cummins et al. 1998; Ruiz-Pesini et al. 1998). Then, the isolation of well-defined genetic backgrounds that are positively or negatively associated with asthenozoospermia can contribute to understanding of the causes of male infertility and can help to better define therapeutic strategies.
Acknowledgments
These investigations were supported by the Spanish Fondo de Investigaciones Sanitarias (FIS95-1783 and FIS98-1033) and by the Ayuntamiento de Zaragoza. We are very grateful to Drs. F. Martínez-Azorín, P. Fernández-Sílva, J. Arenas, L. Rodríguez-Vela, C. Leal-Cariñena, F. Aisa, J. A. Duque and J. Sanchez-Rubio for help in various phases of the work. The technical assistance of Santiago Morales and Carsten Krantz is gratefully acknowledged.
Electronic-Database Information
The URL for data in this article is as follows:
- MITOMAP: A Human Mitochondrial Genome Database, http://www.gen.emory.edu/mitomap.html
References
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23:147 [DOI] [PubMed] [Google Scholar]
- Bai Y, Shakeley RM, Attardi G (2000) Tight control of respiration by NADH dehydrogenase ND5 subunit gene expression in mouse mitochondria. Mol Cell Biol 20:805–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker HW (1994) Clinical male infertility. II. Critical evaluation of the prospects for therapy. Reprod Fertil Dev 6:9–12 [DOI] [PubMed] [Google Scholar]
- Brown MD, Sun F, Wallace DC (1997) Clustering of Caucasian Leber hereditary optic neuropathy patients containing the 11778 or 14484 mutations on an mtDNA lineage. Am J Hum Genet 60:381–387 [PMC free article] [PubMed] [Google Scholar]
- Cummins JM, Jequier AM, Martin R, Mehmet D, Goldblatt J (1998) Semen levels of mitochondrial DNA deletions in men attending an infertility clinic do not correlate with phenotype. Int J Androl 21:47–52 [DOI] [PubMed] [Google Scholar]
- De Benedictis G, Rose G, Carrieri G, De Luca M, Falcone E, Passarino G, Bonafe M, Monti D, Baggio G, Bertolini S, Mari D, Mattace R, Franceschi C (1999) Mitochondrial DNA inherited variants are associated with successful aging and longevity in humans. Faseb J 13:1532–1536 [DOI] [PubMed] [Google Scholar]
- Enriquez JA, Chomyn A, Attardi G (1995) MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNA(Lys) and premature translation termination. Nat Genet 10:47–55 [DOI] [PubMed] [Google Scholar]
- Fischel-Ghodsian N, Prezant TR, Fournier P, Stewart IA, Maw M (1995) Mitochondrial mutation associated with nonsyndromic deafness. Am J Otolaryngol 16:403–408 [DOI] [PubMed] [Google Scholar]
- Følgero T, Bertheussen K, Lindal S, Torbergsen T, Oian P (1993) Mitochondrial Disease and Reduced Sperm Motility. Hum Reprod 8:1863–1868 [DOI] [PubMed] [Google Scholar]
- Ford WC, Harrison A (1981) The role of oxidative phosphorylation in the generation of ATP in human spermatozoa. J Reprod Fertil 63:271–278 [DOI] [PubMed] [Google Scholar]
- Frank SA, Hurst LD (1996) Mitochondria and male disease. Nature 383:224 [DOI] [PubMed] [Google Scholar]
- Guan MX, Enriquez JA, Fischel-Ghodsian N, Puranam RS, Lin CP, Maw MA, Attardi G (1998) The deafness-associated mitochondrial DNA mutation at position 7445, which affects tRNASer(UCN) precursor processing, has long-range effects on NADH dehydrogenase subunit ND6 gene expression. Mol Cell Biol 18:5868–5879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan MX, Fischel-Ghodsian N, Attardi G (1996) Biochemical evidence for nuclear gene involvement in phenotype of non-syndromic deafness associated with mitochondrial 12S rRNA mutation. Hum Mol Genet 5:963–971 [DOI] [PubMed] [Google Scholar]
- Halangk W, Bohnensack R, Kunz W (1985) Interdependence of mitochondrial ATP production and extramitochondrial ATP utilization in intact spermatozoa. Biochim Biophys Acta 808:316–322 [DOI] [PubMed] [Google Scholar]
- Hofmann S, Bezold R, Jaksch M, Kaufhold P, Obermaier-Kusser B, Gerbitz KD (1997a) Analysis of the mitochondrial DNA from patients with Wolfram (DIDMOAD) syndrome. Mol Cell Biochem 174:209–213 [PubMed] [Google Scholar]
- Hofmann S, Bezold R, Jaksch M, Obermaier-Kusser B, Mertens S, Kaufhold P, Rabl W, Hecker W, Gerbitz KD (1997b) Wolfram (DIDMOAD) syndrome and Leber hereditary optic neuropathy (LHON) are associated with distinct mitochondrial DNA haplotypes. Genomics 39:8–18 [DOI] [PubMed] [Google Scholar]
- Hofmann S, Jaksch M, Bezold R, Mertens S, Aholt S, Paprotta A, Gerbitz KD (1997c) Population genetics and disease susceptibility: characterization of central European haplogroups by mtDNA gene mutations, correlation with D loop variants and association with disease. Hum Mol Genet 6:1835–1846 [DOI] [PubMed] [Google Scholar]
- Houshmand M, Larsson NG, Holme E, Oldfors A, Tulinius MH, Andersen O (1994) Automatic sequencing of mitochondrial tRNA genes in patients with mitochondrial encephalomyopathy. Biochim Biophys Acta 1226:49–55 [DOI] [PubMed] [Google Scholar]
- Howell N (1999) Human mitochondrial diseases: answering questions and questioning answers. Int Rev Cytol 186:49–116 [DOI] [PubMed] [Google Scholar]
- Jenuth JP, Peterson AC, Shoubridge EA (1997) Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat Genet 16:93–95 [DOI] [PubMed] [Google Scholar]
- Kalman B, Lublin FD, Alder H (1995) Mitochondrial DNA mutations in multiple sclerosis. Mult Scler 1:32–36 [DOI] [PubMed] [Google Scholar]
- Kao SH, Chao HT, Wei YH (1995) Mitochondrial deoxyribonucleic acid 4977-bp deletion is associated with diminished fertility and motility of human sperm. Biol Reprod 52:729–736 [DOI] [PubMed] [Google Scholar]
- Larsson NG, Clayton DA (1995) Molecular genetic aspects of human mitochondrial disorders. Annu Rev Genet 29:151–178 [DOI] [PubMed] [Google Scholar]
- Macaulay V, Richards M, Hickey E, Vega E, Cruciani F, Guida V, Scozzari R, Bonne-Tamir B, Sykes B, Torroni A (1999) The emerging tree of West Eurasian mtDNAs: a synthesis of control- region sequences and RFLPs. Am J Hum Genet 64:232–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majamaa K, Finnila S, Turkka J, Hassinen IE (1998) Mitochondrial DNA haplogroup U as a risk factor for occipital stroke in migraine. Lancet 352:455–456 [DOI] [PubMed] [Google Scholar]
- Marzuki S, Noer AS, Lertrit P, Thyagarajan D, Kapsa R, Utthanaphol P, Byrne E (1991) Normal variants of human mitochondrial DNA and translation products: the building of a reference data base. Hum Genet 88:139–145 [DOI] [PubMed] [Google Scholar]
- Mayr-Wohlfart U, Paulus C, Henneberg A, Rodel G (1996) Mitochondrial DNA mutations in multiple sclerosis patients with severe optic involvement. Acta Neurol Scand 94:167–171 [DOI] [PubMed] [Google Scholar]
- Ohlenbusch A, Wilichowski E, Hanefeld F (1998) Characterization of the mitochondrial genome in childhood multiple sclerosis I. Optic neuritis and LHON mutations. Neuropediatrics 29:175–179 [DOI] [PubMed] [Google Scholar]
- Pascual ML, Cebrian-Perez JA, Lopez-Perez MJ, Muino-Blanco T (1996) Short-term inhibition of the energy metabolism affects motility but not surface properties of sperm cells. Biosci Rep 16:35–40 [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Swift RG, Swift M (1994) Linkage of the gene for Wolfram syndrome to markers on the short arm of chromosome 4. Nat Genet 8:95–97 [DOI] [PubMed] [Google Scholar]
- Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Roteer JI, Shohat M, Fischel-Ghodsian N (1993) Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet 4:289–294 [DOI] [PubMed] [Google Scholar]
- Reid FM, Rovio A, Holt IJ, Jacobs HT (1997) Molecular phenotype of a human lymphoblastoid cell-line homoplasmic for the np 7445 deafness-associated mitochondrial mutation. Hum Mol Genet 6:443–449 [DOI] [PubMed] [Google Scholar]
- Reid FM, Vernham GA, Jacobs HT (1994) A novel mitochondrial point mutation in a maternal pedigree with sensorineural deafness. Hum Mutat 3:243–247 [DOI] [PubMed] [Google Scholar]
- Richards M, Corte-Real H, Forster P, Macaulay V, Wilkinson-Herbots H, Demaine A, Papiha S, Hedges R, Bandelt HJ, Sykes B (1996) Paleolithic and neolithic lineages in the European mitochondrial gene pool. Am J Hum Genet 59:185–203 [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Segade S, de la Pena CA, Paz JM, Del Rio R (1985) Determination of L-carnitine in serum, and implementation on the ABA-100 and CentrifiChem 600. Clin Chem 31:754–757 [PubMed] [Google Scholar]
- Rötig A, Cormier V, Chatelain P, Francois R, Saudubray JM, Rustin P, Munnich A (1993) Deletion of Mitochondrial DNA in a case of early-onset diabetes-mellitus, optic atrophy, and deafness (Wolfram syndrome, MIM 222300). J Clin Invest 91:1095–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Pesini E, Diez C, Lapena AC, Perez-Martos A, Montoya J, Alvarez E, Arenas J, López-Pérez M (1998) Correlation of sperm motility with mitochondrial enzymatic activities. Clin Chem 44:1616–1620 [PubMed] [Google Scholar]
- Schon EA, Bonilla E, DiMauro S (1997) Mitochondrial DNA mutations and pathogenesis. J Bioenerg Biomembrane 29:131–149 [DOI] [PubMed] [Google Scholar]
- St. John JC, Cooke ID, Barratt CL (1997) Mitochondrial mutations and male infertility. Nat Med 3:124–125 [DOI] [PubMed] [Google Scholar]
- Torroni A, Huoponen K, Francalacci P, Petrozzi M, Morelli L, Scozzari R, Obinu D, Savontaus ML, Wallace DC (1996) Classification of European mtDNAs from an analysis of three European populations. Genetics 144:1835–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroni A, Petrozzi M, D'Urbano L, Sellitto D, Zeviani M, Carrara F, Carducci C, Leuzzi V, Carelli V, Barboni P, De Negri A, Scozzari R (1997) Haplotype and phylogenetic analyses suggest that one European-specific mtDNA background plays a role in the expression of Leber hereditary optic neuropathy by increasing the penetrance of the primary mutations 11778 and 14484. Am J Hum Genet 60:1107–1121 [PMC free article] [PubMed] [Google Scholar]
- Villani G, Attardi G (1997) In vivo control of respiration by cytochrome c oxidase in wild-type and mitochondrial DNA mutation-carrying human cells. Proc Natl Acad Sci USA 94:1166–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (1995) Mitochondrial DNA variation in human evolution, degenerative disease, and aging. Am J Hum Genet 57:201–223 [PMC free article] [PubMed] [Google Scholar]
- WHO (1992) World Health Organization laboratory manual for the examination of human semen and sperm-cervical mucus interactions. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- Zeviani M, Antozzi C (1997) Mitochondrial disorders. Mol Hum Reprod 3:133–148 [DOI] [PubMed] [Google Scholar]