

Abstract
To tailor leucine aminopeptidase from Streptomyces septatus TH-2 (SSAP) to become a convenient biocatalyst, we are interested in Phe221 of SSAP, which is thought to interact with the side chain of the N-terminal residue of the substrate. By using saturation mutagenesis, the feasibility of altering the performance of SSAP was evaluated. The hydrolytic activities of 19 mutants were investigated using aminoacyl p-nitroanilide (pNA) derivatives as substrates. Replacement of Phe221 resulted in changes in the activities of all the mutants. Three of these mutants, F221G, F221A, and F221S, specifically hydrolyzed l-Phe-pNA, and F221I SSAP exhibited hydrolytic activity with l-Leu-pNA exceeding that of the wild type. Although the hydrolytic activities with peptide substrates decreased, the hydrolytic activities with amide and methyl ester substrates were proportional to the changes in the hydrolytic activities with pNA derivatives. Furthermore, based on a comparative kinetic study, the mechanism underlying the alteration in the preference of SSAP from leucine to phenylalanine is discussed.
Leucine aminopeptidase (LAP) is produced by various microbial species. One of the roles of bacterial leucine aminopeptidases (bLAPs) is the provision of readily available nutrients required for growth (13). In general, enzymes belonging to the LAP family have strict enantioselectivity. Furthermore, some bLAPs exhibit high thermal stability and function as amidases and esterases (3, 26). Consequently, bLAPs are considered enzymes that are available for the synthesis of pharmaceutical compounds and as versatile chiral building blocks for the synthesis of pharmaceuticals, agrochemicals, and feed additives. For instance, some bLAPs have been shown to convert l-homophenylalanyl amide to l-homophenylalanine, the intermediate for a class of angiotension 1-converting enzyme inhibitors, such as enalapril and benzapril (10). Therefore, regulation of bLAP performance is crucial for the synthesis of pharmaceuticals or chiral building blocks.
Recently, we identified a thermostable LAP secreted by Streptomyces septatus TH-2 (SSAP) and succeeded in overproducing it using recombinant Escherichia coli (2). In addition, we showed that SSAP can function as an amidase (Arima, Uesugi, Iwabuchi, and Hatanaka, submitted for publication). The primary structure of SSAP exhibits 71% identity with the primary structure of LAP from Streptomyces griseus (SGAP), which is a calcium-activated enzyme (22, 23, 24, 25). The three-dimensional structure of SGAP has been determined by X-ray crystallography (8), and the double-zinc-coordinated active center has been extensively studied by inhibition analysis, structural analysis, and site-direct mutagenesis (5, 6, 7, 14, 18, 20).
Because of the sequence similarity, we predict that the overall structure of SSAP is similar to that of SGAP. Although SSAP is not activated by calcium, its substrate specificity is almost the same as that of SGAP (2). In the predicted structure of SSAP, there are several residues around the side chain of the substrate (Fig. 1). Among these residues, we focused on one bulky residue, Phe221, which likely interacts with the side chain of the substrate. The distance between the side chain of Phe221 and the side chain of the substrate is estimated to be about 3.3 to 3.9 Å. Based on a previous study of the crystal of SGAP binding with Trp or p-iodo-phenylalanine, the phenyl ring of the residue corresponding to Phe221 of SSAP is thought to be involved in stacking interactions with the aromatic ring of the bound affector (20). Thus, we predicted that replacement of this residue would result in an alteration in the specificity for the N-terminal residue of a substrate.
FIG. 1.

Predicted local three-dimensional structure of SSAP around the side chain of the substrate. The partial enzyme structure is indicated by a ribbon diagram. The residues surrounding the side chain of a substrate (free l-Phe, l-Leu, or l-Met) are indicated by a stick model. The bound substrate is indicated by a dark gray stick model. Distances between the side chain of the substrate and surrounding residues were calculated using Swiss-PdbViewer 3.7.
In this study, we first demonstrated that SSAP also functions as an esterase. Next, we prepared F221X SSAP mutants by saturation mutagenesis to alter SSAP properties. The substrate specificity was changed by this mutagenesis, and we obtained two mutants that exhibited high performance; one of these mutants specifically hydrolyzed phenylalanyl derivatives, and the other exhibited hydrolytic activity with leucyl derivatives that exceeded the activity of the wild type. Furthermore, based on a comparative kinetic study, the mechanism underlying the alteration in SSAP preference from leucine to phenylalanine is discussed.
MATERIALS AND METHODS
Materials, bacterial strains, and plasmids.
l-Leu-p-nitroanilide (l-Leu-pNA), l-Phe-pNA, l-Leu-methyl ester (l-Leu-O-Me), l-Phe-O-Me, and snake venom l-amino acid oxidase were purchased from Sigma Chemicals. Other aminoacyl-pNAs and peptides were purchased from Bachem AG. Aminoacyl amides were purchased from the Peptide Institute. Horseradish peroxidase was purchased from Wako Chemicals.
Plasmid pGEM-T Easy (Promega) was used for cloning PCR products. Plasmid pET-SSAP:His (2) (with the SSAP gene inserted into the NcoI-BamHI gap of pET-KmS2 [17]) was used for expression of the wild-type enzyme and as a template of mutagenesis. Escherichia coli JM109 (27) was used as the host strain for general cloning procedures. E. coli BL21(DE3) was used as the host strain for gene expression.
Structural model of wild-type and mutant SSAPs.
The sequence of the primary structure of SSAP (DDBJ accession no. AB125216) was aligned with the sequences of SGAP (Protein Data Bank accession no. 1F2oA.pdb, 1F2pA.pdb, and 1qq9A.pdb) and aminopeptidase from Aeromonas proteolytica (Protein Data Bank accession no. 1txrA.pdb). The alignment data were submitted to SWISS-MODEL (9, 21) for generation of a homology model of SSAP based on the template of the structures of SGAP and A. proteolytica aminopeptidase using Swiss-PdbViewer 3.7 as the interface. The structural model obtained was analyzed using Swiss-PdbViewer.
Saturation mutagenesis.
The fragments of a partial SSAP gene (about 240 bp of the 3′-terminal domain and 300 bp of the internal domain) were amplified using the following primers: 5′-CGATCGGACCACGCGCCTTTCCAGAACGTCGGCATACCCGTCGGCGGGCTCNN(GC)AGCGGC-3′ and 5′-GGATCCGACGTGGCGCTGCCGCTCAGCGACCACACCGCACCCGCGATGGCATC-3′ for amplification of the 3′-terminal domain of the SSAP gene and 5′-TCTCCGCGGGCCCCGGCATCAACGACAAC-3′ and 5′-CGATCGGCCGTCGCCCTCGTGGTCGACCTC-3′ for amplification of the internal domain of the SSAP gene (the underlined regions are PvuI, BamHI, and SacII sites, respectively; the boldface type indicates silent mutations; and the italics indicate the saturation mutagenesis sites). The PCR products were cloned into pGEM-T Easy and confirmed by sequencing. The insert fragment of each plasmid was separated by restriction digestion and gel extraction. The two fragments of the partial SSAP gene were ligated into the SacII-BamHI gap of pET-SSAP:His.
Preparation of wild-type and F221X SSAPs.
E. coli strain BL21(DE3) harboring the constructed plasmid was cultivated at 30°C for 12 h in 3 ml of LB medium containing 50 μg/ml kanamycin. Cells were then inoculated into 100 ml synthetic medium (17) and cultivated at 22°C for 12 h. Isopropyl-β-thiogalactopyranoside was then added at a final concentration of 0.5 mM, and cultivation was continued for another 24 h under the same conditions. The culture was centrifuged to remove cells and brought to 80% ammonium sulfate saturation. The precipitate obtained by centrifugation was dissolved in 10 mM Tris-HCl containing 1 mM CaCl2 (pH 8.0). Then it was heated at 60°C for 30 min with occasional stirring. After centrifugation to remove the precipitate, the solution was dialyzed against 20 mM potassium phosphate buffer (pH 7.0). The dialysate was passed through a hydroxyapatite column (Bio-Rad) equilibrated with the same buffer. The fractions were pooled and dialyzed against 10 mM Tris-HCl (pH 8.0). The dialysate was then loaded onto a Vivapure-Q spin column (Millipore) equilibrated with the same buffer. The bound protein was eluted with 0.2 M NaCl in 10 mM Tris-HCl (pH 8.0). The eluates were pooled and dialyzed against 10 mM Tris-HCl (pH 8.0). The dialysate was used as the purified enzyme preparation. The chromatographic behavior, solubility, and stability of mutants in storage were similar to those of the wild type. The purified enzymes were confirmed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) under denaturing conditions (11). The purities of the different enzyme preparations were estimated from SDS-PAGE results using the Scion Image software. Almost all of the enzyme preparations were >94% pure; F221P SSAP was 68% pure.
Hydrolytic activities with aminoacyl-pNA derivatives.
Specific activities with various aminoacyl-pNAs were determined under the following conditions. An enzyme solution (0.1 ml, 1 to 100 μg/ml) and a substrate solution (0.1 ml, 3.2 or 1.0 mM) were added to 0.8 ml of 100 mM Tris-HCl (pH 8.0), and the mixture was incubated at 37°C. The increase in absorbance at 405 nm per minute due to the release of p-nitroaniline was monitored continuously using a U2800 spectrophotometer (Hitachi). The initial rate of activity was determined from the linear portion of the optical density profile (ɛ405 = 10,600 M−1 cm−1 [19]). Kinetic parameters of aminoacyl-pNA hydrolysis were determined using a reaction mixture that contained 0.1 ml of a 2-μg/ml purified enzyme solution, 0.1 ml of a 1 to 32 mM (l-Leu-pNA) or 0.5 to 12 mM (l-Phe-pNA) substrate solution, and 0.8 ml of 100 mM Tris-HCl buffer (pH 8.0) under the conditions described above.
Hydrolytic activities with methyl esters.
Specific activities with l-Leu-O-Me and l-Phe-O-Me were determined under the following conditions. An enzyme solution (0.1 ml, 100 μg/ml) and a substrate solution (0.1 ml, 100 mM) were added to 0.8 ml of 100 mM Tris-HCl (pH 8.0), and the mixture was incubated at 37°C. The decrease in absorbance at 230 nm per minute due to the cleavage of methyl esters was monitored continuously using a U2800 spectrophotometer (Hitachi). The initial rate of activity was determined from the linear portion of the optical density profile (ɛ230 = 47.8 M−1 cm−1 and ɛ230 = 147.7 M−1 cm−1 for l-Leu-O-Me and l-Phe-O-Me, respectively). Kinetic parameters of ester hydrolysis were determined using a reaction mixture that contained 0.1 ml of a purified enzyme solution (100 to 1,000 μg/ml), 0.1 ml of a 40 to 320 mM (l-Leu-O-Me) or 10 to 80 mM (l-Phe-O-Me) substrate solution, and 0.8 ml of 100 mM Tris-HCl (pH 8.0) under the conditions described above.
Hydrolytic activities with aminoacyl amides and peptides.
Activities with amide and peptide substrates were determined under the conditions described by Arima et al. (submitted). An enzyme solution (0.1 ml, 2 μg/ml) and a substrate solution (0.1 ml, 100 mM) were added to 0.8 ml of 100 mM Tris-HCl (pH 8.0). The reaction mixture was incubated at 37°C for an appropriate time, and then the reaction was stopped by heat treatment (95°C, 15 min). The liberated free amino acids were detected by the 4-aminoantipyrine phenol method (1) coupled with the reaction of l-amino acid oxidase. After centrifugation for 5 min, 100 μl of the supernatant was added to 0.9 ml of a mixture containing 100 mM Tris-HCl (pH 8.0), 0.5 mM 4-aminoantipyrine, 1.7 mM phenol, 5 U/ml horseradish peroxidase, and 0.1 U/ml snake venom l-amino acid oxidase, which can oxidize a wide range of hydrophobic amino acids (15). After incubation for 30 min at 37°C, the absorbance at 505 nm of the solution was determined. Kinetic parameters of amide hydrolysis were determined in a reaction mixture that contained 0.1 ml of a purified enzyme solution (5 to 50 μg/ml), 0.1 ml of a 10 to 160 mM substrate solution, and 0.8 ml of 100 mM Tris-HCl (pH 8.0) under the conditions described above.
Other analytical procedures.
Thermal stability analysis was performed by incubating 200 μl of an enzyme sample (1 μg/ml protein) in 10 mM Tris-HCl (pH 8.0) at temperatures between 30 and 90°C for 30 min. Residual activity was measured under the conditions described above for hydrolytic activity with aminoacyl-pNA derivatives using l-Leu-pNA as the substrate. Fluorescence emission spectra were recorded using an F4500 fluorescence spectrophotometer (Hitachi). The excitation wavelength was 284 nm, and emission spectra were recorded between 300 and 400 nm in cuvettes with a path length of 1 cm. Circular dichroism (CD) spectroscopy was performed using a Jasco J-720WI spectrometer. The absorbance at 280 nm was determined for all samples and used for fine adjustment of the protein concentrations in the samples used for CD spectroscopy. The far-UV spectra of the proteins were determined from 200 to 260 nm in 10 mM Tris-HCl at 20°C and pH 8.0 with the following instrument settings: response, 1 s; sensitivity, 100 millidegrees; and speed, 50 nm/min. The average of 20 scans was determined. Δɛ (M−1 · cm−1) was calculated from the observed instrument output (φ) (in millidegrees), the protein concentration (c) (molar), and the number of peptide bonds (n) using the formula Δɛ= φ/3300cn (4).
RESULTS
Amidase and esterase activities of SSAP.
To confirm that SSAP can hydrolyze ester substrates, we assayed SSAP activity using l-Leu-O-Me as the substrate and compared this activity with the activity when l-Leu-pNA and l-Leu-NH2 were used as substrates. As shown in Fig. 2, SSAP could catalyze the hydrolysis of methyl ester substrates with a specific activity of 48.4 ± 2.4 μmol/min/mg. Although the activity with l-Leu-O-Me was lower than the activities with l-Leu-pNA and l-Leu-NH2, the results indicated that SSAP could function as an esterase. The activity with l-Phe-O-Me was also investigated, and it was almost the same as that with l-Phe-pNA (data not shown).
FIG. 2.

Time dependence of l-Leu-pNA hydrolysis (A), l-Leu-NH2 hydrolysis (B), and l-Leu-O-Me hydrolysis (C) by wild-type SSAP. (A) Continuous spectrophotometric assay for l-Leu-pNA hydrolysis. The y axis shows the amount of released p-nitroaniline determined from the increase in absorbance at 405 nm. (B) Time dependence of l-Leu-NH2 hydrolysis. (C) Continuous spectrophotometric assay of l-Leu-O-Me hydrolysis. The y axis shows the absorbance at 230 nm. The values are averages of three independent experiments. In all cases, the standard deviation was less than 5% of the mean.
Saturation mutagenesis.
On the basis of the predicted structure of SSAP, we focused on residue Phe221, which likely interacts with the side chain of the substrate (Fig. 1). To investigate the feasibility of altering the performance, we replaced the Phe at position 221 of SSAP with 19 other amino acids. When the mutant enzymes were expressed in E. coli, they were secreted at concentrations of 30 to 100 mg/liter (data not shown). Although their quantities were different, as judged by SDS-PAGE, all the mutants could be purified as described in Materials and Methods (Fig. 3A).
FIG. 3.
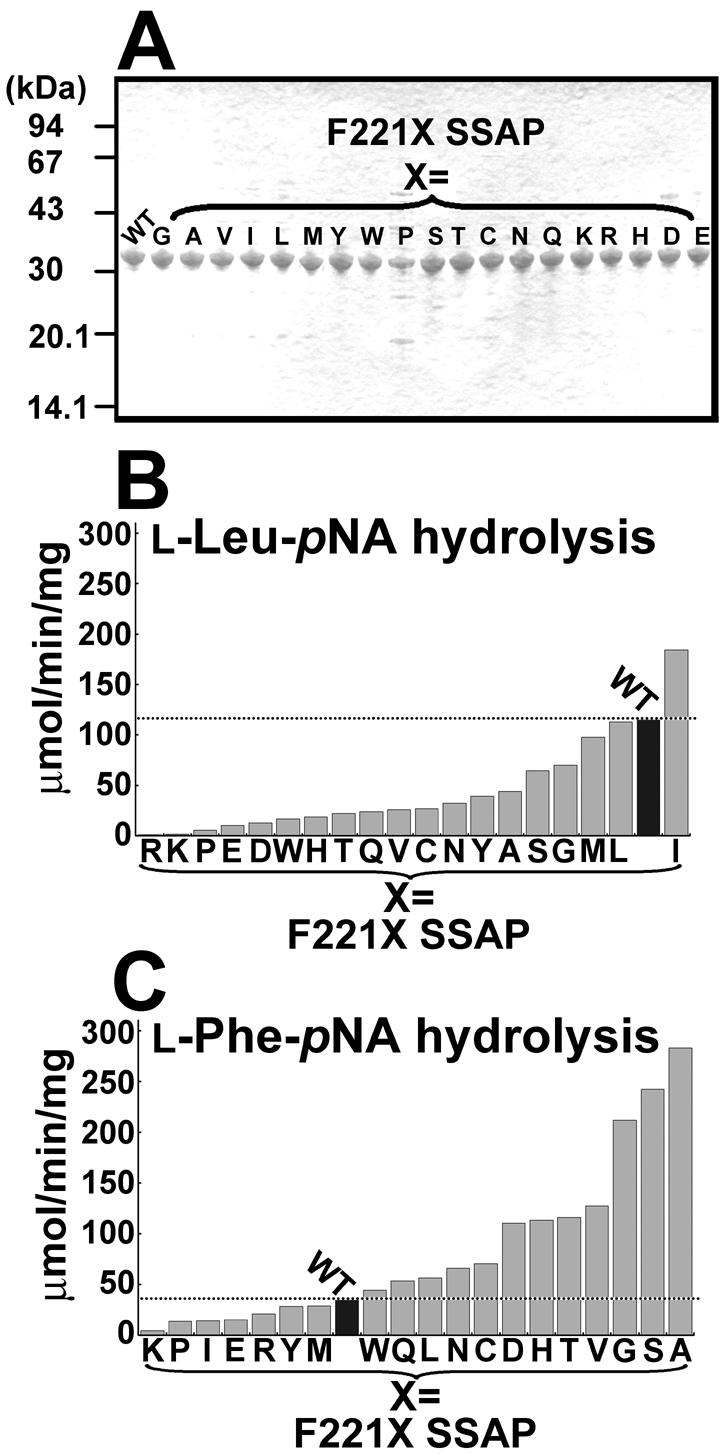
SDS-PAGE and hydrolytic activities of wild-type and F221X SSAPs. (A) SDS-PAGE of purified F221X SSAPs. Samples (1.5 μg of enzyme) were loaded on a 12% gel. (B) Hydrolytic activities of mutants with l-Leu-pNA. (C) Hydrolytic activities of mutants with l-Phe-pNA. In panels B and C, the values are representative of three independent experiments. In all cases, the standard deviation was less than 5% of the mean. WT, wild type.
Effects of mutations on hydrolytic activities with aminoacyl-pNA derivatives.
The activities of all mutants with aminoacyl-pNAs were investigated. As shown in Fig. 3B, for almost all mutants, mutations resulted in a decrease in l-Leu-pNA hydrolytic activity. Only the activity of F221I SSAP was higher than that of the parental enzyme (1.6-fold higher). In contrast, the l-Phe-pNA hydrolytic activities were enhanced in 12 SSAP variants; in particular, the Gly, Ser, and Ala mutants exhibited activities that were about twofold higher than the activity of the wild type with l-Leu-pNA (Fig. 3C).
Figure 4A shows the activities of the wild-type, F221A, and F221I SSAPs with several aminoacyl-pNAs. Wild-type SSAP exhibited the highest specific activity with l-Leu-pNA (117 ± 2.2 μmol/min/mg), and the specific activity with l-Phe-pNA was about fourfold lower than that with l-Leu-pNA (35.2 ± 1.8 μmol/min/mg). Compared with the wild type, F221I SSAP exhibited strict specificity with l-Leu-pNA; the maximum activity was observed with l-Leu-pNA (184 ± 5.7 μmol/min/mg). In contrast, F221A SSAP exhibited the highest activity of all enzymes with l-Phe-pNA (283 ± 11 μmol/min/mg), indicating that a single mutation could convert the leucine aminopeptidase into a phenylalanine aminopeptidase. The Gly and Ser derivatives exhibited properties similar to those of the F221A mutant (data not shown).
FIG. 4.

Substrate specificities of wild-type, F221A, and F221I SSAPs. (A) Hydrolytic activities with aminoacyl-pNA derivatives. The values are representative of three independent experiments. (B) Hydrolytic activities with leucyl and phenylalanyl derivatives. Hydrolytic activities with pNA derivatives are indicated by gray bars, and hydrolytic activities with other derivatives and peptides are indicated by solid bars. The values are representative of three independent experiments. In all cases, the standard deviation was less than 5% of the mean.
Amidase, esterase, and peptidase activities of mutant enzymes.
To confirm that the effect of mutations on the activities with amide and methyl ester substrates was similar to the effect of mutations on the activities with the pNA derivatives, we investigated the activities of wild-type, F221A, and F221I SSAPs with l-Leu-NH2, l-Leu-O-Me, l-Phe-NH2, and l-Phe-O-Me. As shown in Fig. 4B, the activity of F221I SSAP with each leucyl derivative was two- to threefold higher than that of the wild type, indicating that its activities with amide and methyl ester substrates are proportional to its activities with pNA derivatives. Although the activities of F221A SSAP with l-Phe-NH2 and l-Phe-O-Me were also threefold higher than those of the wild type, these activities were twofold lower than the l-Phe-pNA hydrolytic activity. Furthermore, F221A SSAP exhibited four- to sixfold-lower activities with l-Leu-NH2 and l-Leu-O-Me than with l-Leu-pNA.
We also compared the peptidase activities of these mutant enzymes with those of wild-type SSAP using Leu-Phe and Phe-Phe as the substrates. As shown in Fig. 4B, the wild type exhibited higher activities with peptides than with the pNA derivatives. Furthermore, its activity with Phe-Phe was twofold higher than its activity with Leu-Phe. These results agree with those of our previous study on peptide hydrolysis (Arima et al., submitted). Although replacement of Phe221 could alter the activities with aminoacyl derivatives, as in the case of F221I and F221A SSAPs, both of these enzymes exhibited 3.3- to 1.5-fold-lower activities with the peptides than the wild type (Fig. 4B). These results indicate that recognition of the penultimate residue of the peptide substrate was affected negatively by the mutations. We speculate that the lower activities of F221A SSAP with amide and ester substrates than with pNA derivatives are also caused by the negative effect of the mutations on the specificity for the flanking moieties.
Kinetic analysis.
We also characterized the kinetics of the mutants, and the results are summarized in Table 1. Because the affinity of F221I SSAP for l-Phe-O-Me and the affinity of F221A SSAP for l-Leu-O-Me were significantly low, we could not determine the kinetic parameters of their activities.
TABLE 1.
Kinetic parameters for wild-type, F221A, and F221I SSAPsa
| Substrate | SSAP variant | kcat (s−1) | kcat relative to wild type | Km (mM) | Km relative to wild type | kcat/Km (mM−1 s−1) | kcat/Km relative to wild type |
|---|---|---|---|---|---|---|---|
| l-Leu-pNA | Wild type | 65.9 ± 0.6 | 1 | 0.36 ± 0.01 | 1 | 184 | 1 |
| F221A | 34.0 ± 1.0 | 0.52 | 0.68 ± 0.03 | 1.89 | 49.9 | 0.27 | |
| F221I | 108 ± 2.9 | 1.64 | 0.25 ± 0.01 | 0.69 | 428 | 2.32 | |
| l-Phe-pNA | Wild type | 17.6 ± 1.3 | 1 | 0.34 ± 0.04 | 1 | 51.4 | 1 |
| F221A | 229 ± 26 | 13.0 | 0.44 ± 0.06 | 1.29 | 525 | 10.2 | |
| F221I | 19.3 ± 0.57 | 1.10 | 1.85 ± 0.06 | 5.44 | 10.4 | 0.20 | |
| l-Leu-NH2 | Wild type | 104 ± 9.2 | 1 | 15.6 ± 1.8 | 1 | 6.67 | 1 |
| F221A | 31.7 ± 7.3 | 0.30 | 42.7 ± 10.6 | 2.74 | 0.74 | 0.11 | |
| F221I | 155 ± 16.2 | 1.49 | 8.31 ± 1.29 | 0.53 | 18.7 | 2.80 | |
| l-Phe-NH2 | Wild type | 43.8 ± 2.5 | 1 | 6.98 ± 0.6 | 1 | 6.28 | 1 |
| F221A | 115 ± 5.7 | 2.63 | 7.68 ± 0.53 | 1.09 | 15.0 | 2.39 | |
| F221I | 10.7 ± 1.15 | 0.24 | 24.2 ± 3.1 | 3.47 | 0.44 | 0.07 | |
| l-Leu-O-Me | Wild type | 64.2 ± 6.7 | 1 | 15.8 ± 1.4 | 1 | 4.06 | 1 |
| F221A | NDb | ND | ND | ||||
| F221I | 169 ± 2.6 | 2.63 | 16.1 ± 0.33 | 1.02 | 10.5 | 2.59 | |
| l-Phe-O-Me | Wild type | 54.3 ± 7.5 | 1 | 6.90 ± 0.76 | 1 | 7.87 | 1 |
| F221A | 224 ± 7.9 | 4.12 | 12.8 ± 0.60 | 1.86 | 17.5 | 2.22 | |
| F221I | ND | ND | ND |
Km and kcat were calculated from a nonlinear regression fit to the Michaelis-Menten equation, using initial estimates from double-reciprocal plots. The values are means ± standard deviations for three independent experiments.
ND, not detectable.
In the case of F221I SSAP, the activities with leucyl derivatives were higher than those of the wild type. Specifically, the Km values of F221I SSAP were lower than (for l-Leu-pNA and l-Leu-NH2 hydrolysis) or almost the same as (for l-Leu-O-Me hydrolysis) those of the wild type. Moreover, this mutant exhibited higher kcat values with leucyl derivatives than the wild type, resulting in 2.3- to 2.8-fold increases in catalytic efficiency for leucyl derivatives caused by the replacement of Phe221 by Ile (Table 1). In the case of F221A SSAP, the enzyme preferred phenylalanyl derivatives rather than leucyl derivatives. In particular, the kcat values with phenylalanyl derivatives were 2.6- to 13-fold higher than those of the wild type, while the affinities of F221A SSAP for phenylalanyl derivatives were lower than those of the wild type.
F221A SSAP showed the maximum catalytic efficiency (kcat/Km, 525 mM−1 s−1) with l-Phe-pNA, and this efficiency was approximately 10-fold higher than that of the wild type. The catalytic efficiencies of this mutant with other phenylalanyl derivatives were twofold higher than those of the wild type. In contrast to this result, when leucyl derivatives were used, this mutant exhibited 4- to 10-fold-lower catalytic efficiencies than the wild type; this resulted from F221A SSAP being altered from a leucine aminopeptidase to a phenylalanine aminopeptidase by replacement of Phe221 by Ala.
Thermal stability and structural analysis of mutant enzymes.
To evaluate the thermal stabilities of mutant enzymes, the residual activities of wild-type, F221A, and F221I SSAPs after heat treatment for 30 min were measured. As shown in Fig. 5A, the thermal stabilities of these mutants were lower than that of the wild type. The half-inactivation temperature of the wild type was approximately 77°C, and those of F221A and F221I SSAPs were 69 and 65°C, respectively. However, several mutants (for example, F221W and F221Y SSAPs) exhibited almost the same thermal stability as wild-type SSAP (data not shown). Consequently, the aromatic ring of Phe221 of SSAP is thought to contribute to the thermal stability of the enzyme.
FIG. 5.

Thermal stabilities and structural analysis of wild-type, F221A, and F221I SSAPs. (A) Effect of temperature on stability. The values are averages ± standard deviations of three independent experiments. (B) CD spectra of wild-type, F221A, and F221I SSAPs. (C) Fluorescence spectra of wild-type, F221A, and F221I SSAPs. a.u., arbitrary uints.
Figures 5B and 5C show CD and fluorescence spectra for wild-type, F221A, and F221I SSAPs. The spectra for the wild-type and mutant enzymes are similar, suggesting that the secondary and overall structures were not significantly changed by replacement of Phe221.
DISCUSSION
There has been only one report of a change in the substrate specificity of an aminopeptidase which has activity similar to that of SSAP by site-direct mutagenesis (16). However, this enzyme is not closely related to SSAP, and hydrolysis of esters and amides was not described in the previous study. Because SSAP could hydrolyze ester and amide substrates (Fig. 2), we focused on this enzyme as a tool for the synthesis of various compounds, and we attempted to alter its substrate preference to obtain a convenient biocatalyst.
By saturation mutagenesis of SSAP position 221, we obtained several mutant enzymes that exhibit high activities with aminoacyl-pNA derivatives. Because the phenyl ring of Phe221 is close to the side chain of the substrate (Fig. 1), one possible reason for the change in activity following mutation of Phe221 is alteration of the environment around the side chain of a substrate; that is, a change in the electrostatic environment or steric hindrance caused by the mutation allows the change in the interaction between the enzyme and the side chain of a substrate.
Because the Ala and Ile mutants exhibited the highest activities when the wild type and mutants were examined, we chose these two enzymes to investigate differences in activity and substrate specificity compared with the wild type. These differences are thought to be related to structures around the substrate-binding region of the enzymes. Although the activities of F221I and F221A SSAPs are higher than those of the wild type, a decrease in thermal stability was observed for these mutants (Fig. 5A). CD and fluorescence spectral analysis revealed that these mutant enzymes have the same secondary and overall structures as the wild type (Fig. 5B and 5C). Because the Trp and Tyr mutants exhibited almost the same stability as the wild type (data not shown), the replacement of Phe221 with Ala or Ile led to a decrease in structural stability, probably due to disruption of the hydrophobic core by loss of the aromatic ring.
As shown in Fig. 6, there is a cavity around the side chain of the bound substrate in both mutants. This cavity emerges following replacement of Phe221 by other residues. The bound substrate seems to be highly flexible because of the space formed by the substitution. We speculate that a subtle orientation shift of the bound substrate occurs in the mutants, and this shift leads to a change in the distance between the substrate and the catalytic residues (Glu133 and Tyr248). In the case of F221A SSAP, it is thought that activity with phenylalanyl derivatives is enhanced by suitable flexibility. On the other hand, the F221A mutant exhibited lower affinities for the substrates than the wild type (Table 1), indicating that a cavity that is too large reduces the interaction between the enzyme and the substrates. The Gly and Ser mutants had properties similar to those of the F221A mutant (data not shown), suggesting that replacement of Phe221 by a small residue changes leucine aminopeptidase to phenylalanine aminopeptidase. In contrast, F221I SSAP exhibited higher activities with leucyl derivatives than the wild type. The cavity of F221I around the side chain of the bound substrate is smaller than that of F221A SSAP, and the side chain of bound leucine seems to fit in this small cavity (Fig. 6C).
FIG. 6.

Predicted local three-dimensional structures of wild-type (A), F221A (B), and F221I (C) SSAPs around bound substrate. The bound substrate is indicated by a purple stick model. The side chains of Phe221 of the wild type and the residues at position 221 of mutants are indicated by green. The proposed catalytic residues are indicated by sticks that are colored according to the atom type. The two bound zinc atoms are indicated by yellow balls. The cavity formed by the mutation is indicated by a circle shadow in panels B and C.
F221A SSAP exhibited twofold-lower activities with amide and methyl ester derivatives than with pNA derivatives. In addition, the activities with leucyl and phenylalanyl peptides were decreased by mutation of Phe221 (Fig. 4B). In another study we demonstrated that specificity for the N-terminal residue of bLAPs is affected by the penultimate residue, flanking moiety, and length of the peptide substrate (Arima et al., submitted). Furthermore, bLAPs, including SSAP, cannot hydrolyze peptides whose penultimate residue is proline or a d-amino acid (12, 24, 26). Thus, we believe that there is a region that is associated with recognition of the penultimate residue and the flanking moiety of a substrate in bLAPs. The replacement of Phe221 by other residues is thought to change the flexibility of the bound substrate. This change in flexibility may also affect the interaction between the penultimate residue recognition region in an enzyme and the flanking moiety of a substrate.
In the present study, we demonstrated that a single mutation at position 221 can alter the activities of SSAP, particularly the activities with amide and ester substrates. To develop Streptomyces aminopeptidase as an excellent biocatalyst, further studies of other amino acid residues related to enzymatic performance are needed.
REFERENCES
- 1.Allain, C. C., L. S. Poon, C. S. Chan, W. Richmond, and P. C. Fu. 1974. Enzymatic determination of total serum cholesterol. Clin. Chem. 20:470-475. [PubMed] [Google Scholar]
- 2.Arima, J., M. Iwabuchi, and T. Hatanaka. 2004. Gene cloning and overproduction of an aminopeptidase from Streptomyces septatus TH-2, and comparison with a calcium-activated enzyme from Streptomyces griseus. Biochem. Biophys. Res. Commun. 317:531-538. [DOI] [PubMed] [Google Scholar]
- 3.Bienvenue, D. L., R. S. Mathew, D. Ringe, and R. C. Holz. 2002. The aminopeptidase from Aeromonas proteolytica can function as an esterase. J. Biol. Inorg. Chem. 7:129-135. [DOI] [PubMed] [Google Scholar]
- 4.Compton, L. A., and W. C. Johnson. 1986. Analysis of protein circular dichroism spectra for secondary structure using a simple matrix multiplication. Anal. Biochem. 155:155-167. [DOI] [PubMed] [Google Scholar]
- 5.Fundoiano-Hershcovitz, Y., L. Rabinovitch, Y. Langut, V. Reiland, G. Shoham, and Y. Shoham. 2004. Identification of the catalytic residues in the double-zinc aminopeptidase from Streptomyces griseus. FEBS Lett. 571:192-196. [DOI] [PubMed] [Google Scholar]
- 6.Gilboa, R., H. M. Greenblatt, M. Perach, A. Spungin-Bialik, U. Lessel, G. Wohlfahrt, D. Schomburg, S. Blumberg, and G. Shoham. 2000. Interactions of Streptomyces griseus aminopeptidase with a methionine product analogue: a structural study at 1.53 Å resolution. Acta Crystallogr. Sect. D 56:551-558. [DOI] [PubMed] [Google Scholar]
- 7.Gilboa, R., A. Spungin-Bialik, G. Wohlfahrt, D. Schomburg, S. Blumberg, and G. Shoham. 2001. Interactions of Streptomyces griseus aminopeptidase with amino acid reaction products and their implications toward a catalytic mechanism. Proteins 44:490-504. [DOI] [PubMed] [Google Scholar]
- 8.Greenblatt, H. M., O. Almog, B. Maras, A. Spungin-Bialik, D. Barra, S. Blumberg, and G. Shoham. 1997. Streptomyces griseus aminopeptidase: X-ray crystallographic structure at 1.75 Å resolution. J. Mol. Biol. 265:620-636. [DOI] [PubMed] [Google Scholar]
- 9.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18:2714-2723. [DOI] [PubMed] [Google Scholar]
- 10.Kamphuis, J., E. M. Meijer, W. H. J. Boesten, Q. B. Broxterman, B. Kaptein, H. F. M. Hermes, and H. E. Schoemaker. 1992. Production of natural and synthetic l- and d-amino acids by aminopeptidases and aminoamidases, p. 178-206. In J. D. Rozzell and F. Wagner (ed.), Biocatalytic production of amino acids and derivatives. Wiley, New York, N.Y.
- 11.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 12.Lowther, W. T., and B. W. Matthews. 2002. Metalloaminopeptidases: common functional themes in disparate structural surroundings. Chem. Rev. 102:4581-4607. [DOI] [PubMed] [Google Scholar]
- 13.Maeda, H., and T. Yamamoto. 1996. Pathogenic mechanisms induced by microbial proteases in microbial infections. Biol. Chem. Hoppe-Seyler 377:217-226. [DOI] [PubMed] [Google Scholar]
- 14.Maras, B., H. M. Greenblatt, G. Shoham, A. Spungin-Bialik, S. Blumberg, and D. Barra. 1996. Aminopeptidase from Streptomyces griseus: primary structure and comparison with other zinc-containing aminopeptidases. Eur. J. Biochem. 236:843-846. [DOI] [PubMed] [Google Scholar]
- 15.Massay, V., and B. Curti. 1967. On the reaction mechanism of Crotalus adamanteus l-amino acid oxidase. J. Biol. Chem. 242:1259-1264. [PubMed] [Google Scholar]
- 16.Mathew, Z., T. M. Knox, and C. G. Miller. 2000. Salmonella enterica serovar Typhimurium peptidase B is a leucyl aminopeptidase with specificity for acidic amino acids. J. Bacteriol. 182:3383-3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mishima, N., K. Mizumoto, Y. Iwasaki, H. Nakano, and T. Yamane. 1997. Insertion of stabilizing loci in vectors of T7 RNA polymerase-mediated Escherichia coli expression systems: a case study on the plasmids involving foreign phospholipase D gene. Biotechnol. Prog. 13:864-868. [DOI] [PubMed] [Google Scholar]
- 18.Papir, G., A. Spungin-Bialik, D. Ben-Meir, E. Fudim, R. Gilboa, H. M. Greenblatt, G. Shoham, U. Lessel, D. Schomburg, R. Ashkenazi, and S. Blumberg. 1998. Inhibition of Streptomyces griseus aminopeptidase and effects of calcium ions on catalysis and binding comparisons with the homologous enzyme Aeromonas proteolytica aminopeptidase. Eur. J. Biochem. 258:313-319. [DOI] [PubMed] [Google Scholar]
- 19.Prescott, J. M., and S. H. Wilkes. 1976. Aeromonas aminopeptidase. Methods Enzymol. 45:530-543. [DOI] [PubMed] [Google Scholar]
- 20.Reiland, V., R. Gilboa, A. Spungin-Bialik, D. Schomburg, Y. Shoham, S. Blumberg, and G. Shoham. 2004. Binding of inhibitory aromatic amino acids to Streptomyces griseus aminopeptidase. Acta Crystallogr. Sect. D 60:1738-1746. [DOI] [PubMed] [Google Scholar]
- 21.Schwede, T., J. Kopp, N. Guex, and M. C. Peitsch. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spungin, A., and S. Blumberg. 1989. Streptomyces griseus aminopeptidase is a calcium-activated zinc metalloprotein. Purification and properties of the enzyme. Eur. J. Biochem. 183:471-477. [DOI] [PubMed] [Google Scholar]
- 23.Vosbeck, K. D., K. F. Chow, and W. M. Awad, Jr. 1973. The proteolytic enzymes of the K-1 strain of Streptomyces griseus obtained from a commercial preparation (Pronase). Purification and characterization of the aminopeptidases. J. Biol. Chem. 248:6029-6034. [PubMed] [Google Scholar]
- 24.Vosbeck, K. D., B. D. Greenberg, and W. M. Awad, Jr. 1975. The proteolytic enzymes of the K-1 strain of Streptomyces griseus obtained from a commercial preparation (Pronase). Specificity and immobilization of aminopeptidase. J. Biol. Chem. 250:3981-3987. [PubMed] [Google Scholar]
- 25.Vosbeck, K. D., B. D. Greenberg, M. S. Ochoa, P. L. Whitney, and W. M. Awad, Jr. 1978. Proteolytic enzymes of the K-1 strain of Streptomyces griseus obtained from a commercial preparation (Pronase). Effect of pH, metal ions, and amino acids on aminopeptidase activity. J. Biol. Chem. 253:257-260. [PubMed] [Google Scholar]
- 26.Wagner, F. W., S. H. Wilkes, and J. M. Prescott. 1972. Specificity of Aeromonas aminopeptidase toward amino acid amides and dipeptides. J. Biol. Chem. 247:1208-1210. [PubMed] [Google Scholar]
- 27.Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103-119. [DOI] [PubMed] [Google Scholar]