

Abstract
The ispA gene encoding farnesyl pyrophosphate (FPP) synthase from Escherichia coli and the crtM gene encoding 4,4′-diapophytoene (DAP) synthase from Staphylococcus aureus were overexpressed and purified for use in vitro. Steady-state kinetics for FPP synthase and DAP synthase, individually and in sequence, were determined under optimized reaction conditions. For the two-step reaction, the DAP product was unstable in aqueous buffer; however, in situ extraction using an aqueous-organic two-phase system resulted in a 100% conversion of isopentenyl pyrophosphate and dimethylallyl pyrophosphate into DAP. This aqueous-organic two-phase system is the first demonstration of an in vitro carotenoid synthesis pathway performed with in situ extraction, which enables quantitative conversions. This approach, if extended to a wide range of isoprenoid-based pathways, could lead to the synthesis of novel carotenoids and their derivatives.
Carotenoids are naturally occurring pigments found in a wide variety of plants and microorganisms (22, 23). Recent studies indicate that these isoprenoid-based natural products possess biologically active properties (21, 24), thereby making them of interest to the medicinal chemist. Most carotenoids belong to the C30 and C40 classes, being distinguished by the number of iterative steps of isopentenyl pyrophosphate (IPP) condensation to add C5 isoprene units. The initial synthetic step is the condensation of IPP with γ,γ-dimethylallyl pyrophosphate (DMAPP) catalyzed by farnesyl pyrophosphate (FPP) synthase (ispA) to give geranyl pyrophosphate (GPP) and, sequentially, FPP (9). This C15 compound serves as a central node for the synthesis of sterols (29), farnesylated proteins (4), hemes (20), sesquiterpenes (6), and dolichols (5), as well as the crucial precursor for carotenoids.
C30 carotenoids are present in the nonphotosynthetic bacteria, such as Streptococcus faecium, Staphylococcus aureus, and Methylobacterium rhodinum, and in the photosynthetic Heliobacterium species (16, 25, 27, 28). Only the genes encoding 4,4′-diapophytoene (DAP) synthase (crtM) and 4,4′-diapophytoene desaturase (crtN) from S. aureus have been cloned and functionally expressed in Escherichia coli, resulting in the yellow 4,4′-diaponeurosporene from FPP (30) (Fig. 1). The first committed step in C30 carotenoid biosynthesis is the condensation of two molecules of FPP catalyzed by DAP synthase to form the colorless carotenoid 4,4′-diapophytoene.
FIG. 1.

Pathways of C30 carotenoid biosynthesis. The first committed step in C30 carotenoid biosynthesis is the extension of the isoprenoid pathway by FPP synthase (ispA) and diapophytoene synthase (crtM). OPP, O-linked pyrophosphate.
Until now, only a few reports have discussed the formation of DAP through a reconstituted C30 carotenoid metabolic pathway in vitro (13, 17), and none have provided a full kinetic analysis or reaction optimization of the in vitro pathway. Nevertheless, such a study would be valuable in order to design novel carotenoids and carotenoid-based hybrid natural products. Therefore, in the current work, we set out to optimize the C30 carotenoid pathway reconstructed in vitro for the synthesis of DAP from IPP and DMAPP. Furthermore, we elucidated the kinetic behavior of both the FPP synthase and DAP synthase and developed a unique optimization technique for carotenoid synthesis involving two-phase biocatalysis in the presence of a hydrophobic organic solvent (1, 2). This technique allowed us to overcome product instability and resulted in a nearly 100% conversion of IPP and DMAPP into DAP, thereby providing a new route to the high-yield synthesis of isoprenoids.
MATERIALS AND METHODS
Materials.
ispA (farnesyl pyrophosphate synthase) was cloned from E. coli strain JM109 (Stratagene, La Jolla, CA), and crtM (4,4′-diapophytoene synthase) was cloned from S. aureus genomic DNA (ATCC). E. coli type III alkaline phosphatase, IPP, DMAPP, GPP, FPP, LB broth, ampicillin, and arabinose were purchased from Sigma (St. Louis, MO). All other chemicals and reagents were obtained commercially at the highest purity available and used without further purification.
Generation, overexpression, and purification of recombinant His-tagged proteins
The vector pThioHis-TOPO (Invitrogen, Carlsbad, CA) was modified by using TOPO cloning of a linker insert introducing a 5′ KpnI site and a 3′ EcoRI site downstream from the His patch-thioredoxin open reading frame. The ispA and crtM genes were amplified with forward and reverse primers that introduce corresponding KpnI and EcoRI sites so that the His patch-thioredoxin leader peptide remained in frame. The PCR products were digested with Acc651 and EcoRI, gel purified, and ligated into similarly treated vector. These ligations were transformed into chemically competent E. coli strain JM109, and the insert containing the plasmids was isolated and sequenced to confirm that no PCR errors were present.
Cells were grown at 37°C in LB medium containing 100 μg/ml ampicillin with shaking at 200 rpm. Overnight LB-ampicillin starter cultures (2 ml) were added to 50 ml LB-ampicillin and grown for 2 h (until reaching an optical density at 600 nm of 0.6 to 0.8). The expression of recombinant proteins was induced with 2 mg/ml sterilized arabinose for 4 h. The cells were then harvested by centrifugation. All subsequent procedures were performed at 4°C. The cell pellets were resuspended in 8 ml native binding buffer supplied with the Probond purification kit (Invitrogen, CA) with 8 mg lysozyme and incubated on ice for 30 min. Resuspended cells were lysed by four rapid freeze-thaw cycles using liquid nitrogen and a 25°C water bath. By use of the native purification conditions according to the manufacturer's instructions, expressed cell lysates and purified proteins were obtained and confirmed by sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis (Invitrogen, CA). Protein concentrations in the cell lysates and purified preparations were determined by the microbicinchoninic acid protein assay reagent (Pierce, Rockford, IL) with bovine serum albumin as a standard.
Enzyme activity assays and product analysis
FPP synthase activity was determined using an assay developed by Davisson et al. (7) with a slight modification. The standard reaction mixture for FPP synthase catalysis at 37°C contained 20 mM Tris-HCl buffer (pH 7.7), 3 mM MgCl2, 10 μg/ml phosphatidylcholine, 40 μM IPP, 20 μM DMAPP, and 9.2 μg purified FPP synthase in a 500-μl reaction volume contained within 3-ml vials. Reactions for FPP synthase starting with GPP were performed in identical reaction mixtures, except that the substrates consisted of 50 μM IPP and 50 μM GPP. Kinetic parameters for FPP synthase were determined in reaction mixtures containing 10 to 50 μM IPP and 5 to 30 μM DMAPP. FPP synthase-catalyzed condensation of GPP with IPP was performed with 2.5 to 50 μM GPP and 10 to 50 μM IPP. Reaction mixtures were incubated for 30 min at 37°C, after which 0.2 M lysine hydrochloride buffer (10%, vol/vol; pH 10.5) was added to terminate the enzymatic reaction. The reaction mixture was then incubated with 50 units E. coli type III alkaline phosphatase for 1 h before the extraction of the reaction products with hexane. The hydrolyzed products were analyzed by reverse-phase high-pressure liquid chromatography (HPLC) using an Altima C18 column (4.6 by 150 mm, 3-μm particle diameter; Alltech, Deerfield, IL) with a total flow rate of 0.8 ml/min. Solvent A was water, and solvent B was acetonitrile-methanol-isopropanol (85:10:5). The mobile-phase concentration of solvent B was initially maintained at 70% for 2 min and then was increased linearly to 100% in 8 min, held at 100% for 25 min, decreased to 70% in 30 min, and then held at 70% for 40 min. Detection was performed with a UV/visible-spectrum photodiode array detector at 254 nm. Unless otherwise indicated, all experiments were conducted in triplicate.
For DAP synthase catalysis, reaction mixtures contained 2 mM NADPH, 50 μM FPP as the substrate, and 18.2 μg purified DAP synthase in 500 μl of aqueous buffer (the same as for FPP synthase). The reaction mixtures were incubated at 37°C for 60 min in 3-ml vials, which were brown to prevent potential light-induced product degradation. The DAP product was extracted with hexane, analyzed by HPLC using the aforementioned Altima C18 column, and eluted with acetonitrile-methanol-isopropanol (85:10:5) at a flow rate of 1.0 ml/min. UV and visible spectra were obtained with a photodiode array detector with 200- to 700-nm wavelength scanning, and DAP was detected at 286 nm (ɛ = 1,050 M−1 cm−1). Mass spectrometry (HPLC/atmospheric pressure chemical ionization) analysis was performed on an Agilent 100 series HPLC equipped with an MS 1100 series LC/MSD (Palo Alto, CA). Kinetic analysis of DAP synthase involved 2.5 to 50 μM FPP as the substrate in the standard reaction mixture.
Effects of water-immiscible solvents on FPP synthase and DAP synthase
Biphasic reactions were performed with hexane, toluene, cyclohexane, or ethyl acetate in 1:1 volumes with aqueous buffer. The reaction mixtures contained 9.2 μg FPP synthase and either 18.2 μg DAP synthase with 40 μM IPP and 20 μM DMAPP or 18.2 μg DAP synthase with 20 μM FPP. Assay mixtures were incubated for up to 48 h at 37°C, and then the organic phases were analyzed by HPLC as detailed above.
RESULTS AND DISCUSSION
Cloning, expression, and purification of FPP synthase and DAP synthase.
For expression in E. coli and subsequent purification, the ispA gene (0.9 kb) encoding FPP synthase from E. coli and the crtM gene (0.9 kb) encoding DAP synthase from S. aureus were cloned into the arabinose-inducible expression vector pThioHis. The expression of these genes as six-His-thioredoxin fusion proteins enabled rapid purification to give relatively pure enzymes according to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (data not shown), with ca. 22- and 18-fold enrichments in specific activities for FPP synthase and DAP synthase, respectively (Table 1).
TABLE 1.
Purification of the FPP synthase (ispA) from E. coli and the DAP synthase (crtM) from S. aureus
| Step | Protein
|
Enzyme
|
|||
|---|---|---|---|---|---|
| Vol of fraction (ml) | Concn (mg/ml) | Total amt (mg) | Sp act (nmol/min/mg) | Purification (fold) | |
| IspA | |||||
| Crude lysate | 8.0 | 4.64 | 37.12 | 0.961a | 1.00 |
| Purified product | 1.5 | 0.184 | 0.276 | 20.85a | 21.70 |
| CrtM | |||||
| Crude lysate | 8.0 | 2.25 | 18.0 | 0.284b | 1.00 |
| Purified product | 1.5 | 0.242 | 0.363 | 4.98b | 17.55 |
FPP synthase activity was determined from the rate of FPP synthesis from GPP and IPP, as represented in the amount of farnesol produced upon hydrolysis of the FPP with 50 units of alkaline phosphatase.
DAP synthase activity was determined from the rate of DAP formation when FPP was used as the substrate.
Enzyme kinetics for FPP synthase and DAP synthase.
The kinetics of FPP synthase has been reported previously (8, 19) specifically for the condensation of IPP and DMAPP in a 2:1 molar ratio. However, a complete kinetic analysis of FPP formation through the discrete GPP intermediate has not been done. Because of the importance of FPP synthase as the first step in isoprenoid synthesis, we sought a more detailed kinetic understanding of FPP synthase, which included the kinetics of both GPP and FPP formation. The kinetic parameters of the formation of GPP from IPP and DMAPP and the subsequent kinetics of FPP synthesis from the condensation of GPP with IPP were determined from Lineweaver-Burk plots (Fig. 2). Parallel plots were obtained for GPP formation, indicating a classical ping-pong-bi-bi kinetic mechanism (Fig. 2A). Secondary replots yielded a Vmax of 10.7 μM/h and Km values for IPP and DMAPP of 1.3 and 29.3 μM, respectively (Fig. 2A, inset, and Table 2). Because the FPP yield was <5% during the initial rate analysis, the condensation of IPP with GPP was minor and did not influence the observed reaction kinetics for GPP synthesis. These results appear to be the first report to date that describes the kinetics of the FPP synthase-catalyzed formation of GPP.
FIG. 2.

Lineweaver-Burk plots for GPP and FPP syntheses catalyzed by FPP synthase (conditions described in Materials and Methods). (A) Lineweaver-Burk plot, secondary replot (box), and proposed kinetic mechanism for DMAPP as the allylic substrate. (B) GPP as the allylic substrate with FPP synthase. Symbols: •, 10 μM IPP; ▿, 20 μM IPP; ▪, 30 μM IPP; and ⋄, 50 μM IPP.
TABLE 2.
Kinetic parameters of FPP synthase and DAP synthase reactions
| Product | Vmax (μM/h) |
Km (μM)
|
Vmax/Km,allylic (h−1) | |||
|---|---|---|---|---|---|---|
| IPP | DMAPP | GPP | FPP | |||
| GPPa | 10.7 | 1.3 | 29.3 | 0.36 | ||
| FPPb | 23.8 | 10.3 | 5.5 | 4.3 | ||
| DAPc | 3.2 | 1.2 | ||||
GPP from IPP and DMAPP.
FPP from IPP and GPP.
DAP from FPP.
FPP formation from IPP and GPP (Fig. 2B) revealed an ordered bi-bi reaction kinetic mechanism, wherein GPP binds first to the enzyme's active site, followed by the binding of IPP and the subsequent release of pyrophosphate. These results are consistent with the literature for the formation of FPP from GPP for FPP synthases from several organisms (14, 15, 18, 26). The maximal reactivity for the second step of FPP synthase catalysis (formation of FPP) was more than twice that of the first step (formation of GPP). Moreover, evaluation of the Vmax/Km values for the allylic substrates (10-fold higher for GPP than for DMAPP [Table 2]) suggests that the enzyme has greater binding-energy interaction with the larger substrate (12). Since it is likely that the same binding pocket is used for DMAPP and GPP, the former is too small to take advantage of the substrate-protein interactions at the binding site and, therefore, shows lower binding affinity, as reflected in the higher Km value for DMAPP than for GPP. The parameters for DAP synthase from FPP showed typical Michaelis-Menten kinetics for a single-substrate system (data not shown).
In vitro C30 pathway catalysis.
The combined action of FPP synthase and DAP synthase provides the simplest in vitro C30 carotenoid pathway. The theoretical time course formations of GPP, FPP, and DAP can be solved using a Runge-Kutta method. Concentrations of DMAPP and IPP at any time, tt, are determined using simple reaction stoichiometry (equations 1 and 2, respectively). Rate expressions for the formation of GPP, FPP, and DAP are shown in equations 3 through 5, respectively, and these expressions can be integrated to provide time course predictions for each compound synthesized, based solely on initial rate kinetics.
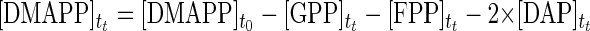 |
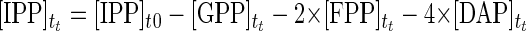 |
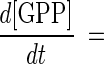 |
 |
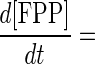 |
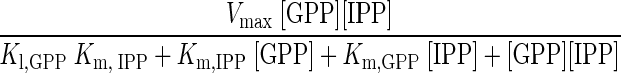 |
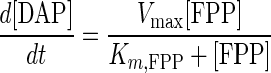 |
Here, Vmax is the maximum reaction velocity from Table 2, and KI,GPP, 71.4 μM, is the dissociation constant of GPP obtained from the secondary plot (Fig. 2B, inset). Figure 3A and B show a numerical simulation and the experimental data, respectively, for the combined FPP synthase and DAP synthase reaction. At early times (<20 min) in both cases, GPP is the predominant product, which, as expected, is further consumed to give FPP and DAP. Experimentally, DAP formation maximizes at ca. 60 min, along with that of the total measurable products, even though there remain substantial concentrations of unreacted DMAPP and IPP. This indicates a general instability of the reaction products in aqueous solution. Extending the reaction time to 48 h (data not shown) resulted in the complete consumption of the DMAPP, yet, essentially, no observable product. Analysis of the reaction mixture by mass spectrometry did not reveal end products other than DAP nor any intermediates other than GPP and FPP. These results were quite distinct from the predicted values generated through use of the initial rate kinetics (Table 2). As shown in Fig. 3B, the series reaction from DMAPP and IPP to DAP does seem to semiquantitatively match the theoretical values only at early reaction times (<30 min). However, past this point, there is a clear loss of product. The lower yields of the intermediates and product of the bienzymatic reaction may be due to product instability. A similar result was obtained by Kanasawud and Crouzet for lycopene and β-carotene synthesis (10, 11), where it was observed that these C40 products were degraded in aqueous buffer into volatile compounds at 30°C. Therefore, it is not surprising that DAP, which is structurally similar to the C40 products, would also be unstable.
FIG. 3.

Time course of GPP, FPP, and DAP synthesis under the conditions described in Materials and Methods. (A) Predictions of kinetic modeling from equations 1 through 5. The dash, dash-dot, and solid lines represent GPP, FPP, and DAP, respectively. (B) Experimental time course using 40 μM IPP and 20 μM DMAPP. Symbols: ○, GPP (C10); ▾, FPP (C15); and •, DAP (C30).
To confirm similar behavior for DAP, we examined the loss of a known concentration of DAP under DAP synthase reaction conditions. Figure 4A shows the loss of DAP from aqueous buffer in the presence of DAP synthase at 37°C, which clearly demonstrates the instability of DAP. From this result, an expression for the degradation rate of DAP in the presence of enzyme was obtained (equation 6). The combined rate expression of DAP, taking into account the degradation of DAP, is shown in equation 7. Based on the degradation rate of DAP, we compared our experimental data for DAP production from 20 μM FPP with equation 7. As shown in Fig. 4B, the values for the reaction from FPP to DAP do seem to quantitatively match the observed values.
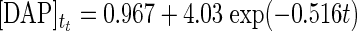 |
(6) |
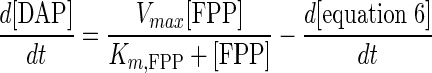 |
FIG. 4.

Time course of DAP (5 μM) degradation in aqueous buffer in the presence of DAP synthase at 37°C (A) and kinetic modeling (B) taking into account the rate of degradation of DAP (equation 7). For comparison of modeling results, 20 μM FPP was used as the substrate for experimental data. The symbols indicate the experimental data (•) and modeling equations (—) for equation 6 (A) and equation 7 (B).
In vitro C30 pathway under biphasic conditions.
The instability of DAP from the enzymatic reaction mixture limits the in vitro approach to generating C30-based products. To overcome this limitation, we reasoned that removal of the DAP from the aqueous reaction mixture, once it is generated, could result in high DAP yields. Therefore, we examined two-phase enzymatic catalysis, using a water-immiscible organic solvent as the second phase. Four solvents were examined, including hexane, cyclohexane, toluene, and ethyl acetate.
As shown in Fig. 5, when hexane and cyclohexane were used as the biphasic solvents, the reaction yields were nearly 100% for DAP synthase (Fig. 5A) and 100% for the FPP synthase-DAP synthase bienzyme system (Fig. 5B), respectively. This contrasts greatly with the results of the aqueous reactions (Fig. 3B), as well as the findings of Raisig and Sandmann, who observed a <20% yield for DAP synthesis (17). Importantly, the formation of DAP was fast in both solvent systems. Therefore, the removal of the DAP product once it is generated does enable high conversion rates to be achieved. Reactions were more efficient in hydrophobic solvents; reactions in toluene were less efficient than in hexane or cyclohexane, and essentially no activity was observed with ethyl acetate as the cosolvent. This is likely due to the relatively high solubility of ethyl acetate in the aqueous phase, which is known to deactivate enzymes (3).
FIG. 5.

Effect of biphasic solvent systems on C30 carotenoid enzyme reactions. The symbols represent results for aqueous buffer solution (•) and 1:1 (vol/vol) (with aqueous buffer) hexane (▿), cyclohexane (▪), ethyl acetate (⋄), and toluene (▴). (A) DAP synthase reaction with 20 μM FPP; (B) FPP synthase and DAP synthase with 40 μM IPP and 20 μM DMAPP.
In conclusion, we have reported the kinetic characterization of FPP synthase (ispA) from E. coli and DAP synthase (crtM) from S. aureus. The results suggest that FPP synthase was more active on GPP than on DMAPP and showed the different mechanisms for GPP and FPP formation. This result provides further information on the nature of the in vivo metabolic pathway. In particular, one significant finding is that the binding of the allylic substrate increases as the pathway proceeds from DMAPP to GPP to FPP. Such increased binding energy may provide a thermodynamic driving force to complete the C30 pathway once the initial condensation of IPP and DMAPP occurs. On a more applied level, while the in vitro pathway is challenged by the instability of DAP, the use of a biphasic reaction system, including hexane or cyclohexane as the product recovery organic phase, results in essentially 100% conversion from the IPP and DMAPP precursors. This approach, if generalized, may prove useful in performing carotenoid and other isoprenoid syntheses upon the isolation and further characterization of key pathway enzymes, particularly from C40 and related pathways.
Acknowledgments
This work was supported by the Defense Advanced Research Projects Agency (N66001-02-1-8926).
REFERENCES
- 1.Barberis, S., E. Quiroga, M. C. Arribere, and N. Priolo. 2002. Peptide synthesis in aqueous-organic biphasic systems catalyzed by a protease isolated from Morrenia brachystephana (Asclepiadaceae). J. Mol. Catal. B 17:39-47. [Google Scholar]
- 2.Barros, M. T., M. G. V. Carvalho, F. A. P. Garcia, and E. M. V. Pires. 1992. Stability performance of Cynara cardunculus L. acid protease in aqueous-organic biphasic systems. Biotechnol. Lett. 14:179-184. [Google Scholar]
- 3.Cantarella, M., L. Cantarella, and F. Alfani. 1991. Hydrolytic reactions in two-phase systems. Effect of water-immiscible organic solvents on stability and activity of acid phosphatase, beta-glucosidase, and beta-fructofuranosidase. Enzyme Microb. Technol. 13:547-553. [DOI] [PubMed] [Google Scholar]
- 4.Chehade, K. A. H., K. Kiegiel, R. J. Isaacs, J. S. Pickett, K. E. Bowers, C. A. Fierke, D. A. Andres, and H. P. Spielmann. 2002. Photoaffinity analogues of farnesyl pyrophosphate transferable by protein farnesyl transferase. J. Am. Chem. Soc. 124:8206-8219. [DOI] [PubMed] [Google Scholar]
- 5.Crick, D. C., J. R. Scocca, J. S. Rush, D. W. Frank, S. S. Krag, and C. J. Waechter. 1994. Induction of dolichyl-saccharide intermediate biosynthesis corresponds to increased long chain cis-isoprenyltransferase activity during the mitogenic response in mouse B cells. J. Biol. Chem. 269:10559-10565. [PubMed] [Google Scholar]
- 6.Crick, D. C., and C. J. Waechter. 1994. Long-chain cis-isoprenyltransferase activity is induced early in the developmental program for protein N-glycosylation in embryonic rat brain cells. J. Neurochem. 62:247-256. [DOI] [PubMed] [Google Scholar]
- 7.Davisson, V. J., T. R. Neal, and C. D. Poulter. 1993. Farnesyl-diphosphate synthase. Catalysis of an intramolecular prenyl transfer with bisubstrate analogs. J. Am. Chem. Soc. 115:1235-1245. [Google Scholar]
- 8.Ding, V. D. H., B. T. Sheares, J. D. Bergstrom, M. M. Ponpipom, L. B. Perez, and C. D. Poulter. 1991. Purification and characterization of recombinant human farnesyl diphosphate synthase expressed in Escherichia coli. Biochem. J. 275:61-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujisaki, S., T. Nishino, and H. Katsuki. 1986. Isoprenoid synthesis in Escherichia coli. Separation and partial purification of four enzymes involved in the synthesis. J. Biochem. 99:1327-1337. [DOI] [PubMed] [Google Scholar]
- 10.Kanasawud, P., and J. C. Crouzet. 1990. Mechanism of formation of volatile compounds by thermal degradation of carotenoids in aqueous medium. 1. β-Carotene degradation. J. Agric. Food Chem. 38:237-243. [Google Scholar]
- 11.Kanasawud, P., and J. C. Crouzet. 1990. Mechanism of formation of volatile compounds by thermal degradation of carotenoids in aqueous medium. 2. Lycopene degradation. J. Agric. Food Chem. 38:1238-1242. [Google Scholar]
- 12.Kawaguchi, S., Y. Nobe, J. Yasuoka, T. Wakamiya, S. Kusumoto, and S. Kuramitsu. 1997. Enzyme flexibility: a new concept in recognition of hydrophobic substrates. J. Biochem. 122:55-63. [DOI] [PubMed] [Google Scholar]
- 13.Lee, P. C., A. Z. R. Momen, B. N. Mijts, and C. Schmidt-Dannert. 2003. Biosynthesis of structurally novel carotenoids in Escherichia coli. Chem. Biol. 10:453-462. [DOI] [PubMed] [Google Scholar]
- 14.Popjak, G., P. W. Holloway, R. P. Bond, and M. Roberts. 1969. Analogues of geranyl pyrophosphate as inhibitors of prenyltransferase. Biochem. J. 111:333-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poulter, C. D., P. L. Wiggins, and A. T. Le. 1981. Farnesylpyrophosphate synthetase. A stepwise mechanism for the 1′-4 condensation reaction. J. Am. Chem. Soc. 103:3926-3927. [Google Scholar]
- 16.Raisig, A., and G. Sandmann. 1999. 4,4′-Diapophytoene desaturase: catalytic properties of an enzyme from the C30 carotenoid pathway of Staphylococcus aureus. J. Bacteriol. 181:6184-6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raisig, A., and G. Sandmann. 2001. Functional properties of diapophytoene and related desaturases of C30 and C40 carotenoid biosynthetic pathways. Biochim. Biophys. Acta 1533:164-170. [DOI] [PubMed] [Google Scholar]
- 18.Reed, B. C., and H. C. Rilling. 1975. Crystallization and partial characterization of prenyltransferase from avian liver. Biochemistry 14:50-54. [DOI] [PubMed] [Google Scholar]
- 19.Reed, B. C., and H. C. Rilling. 1976. Substrate binding of avian liver prenyltransferase. Biochemistry 15:3739-3745. [DOI] [PubMed] [Google Scholar]
- 20.Saiki, K., T. Mogi, K. Ogura, and Y. Anraku. 1993. In vitro heme O synthesis by the cyoE gene product from Escherichia coli. J. Biol. Chem. 268:26041-26044. [PubMed] [Google Scholar]
- 21.Sandmann, G. 2002. Combinatorial biosynthesis of carotenoids in a heterologous host: a powerful approach for the biosynthesis of novel structures. ChemBioChem 3:629-635. [DOI] [PubMed] [Google Scholar]
- 22.Sandmann, G. 2001. Genetic manipulation of carotenoid biosynthesis: strategies, problems and achievements. Trends Plant Sci. 6:14-17. [DOI] [PubMed] [Google Scholar]
- 23.Sandmann, G., M. Albrecht, G. Schnurr, O. Knorzer, and P. Boger. 1999. The biotechnological potential and design of novel carotenoids by gene combination in Escherichia coli. Trends Biotechnol. 17:233-237. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt-Dannert, C., D. Umeno, and F. H. Arnold. 2000. Molecular breeding of carotenoid biosynthetic pathways. Nat. Biotechnol. 18:750-753. [DOI] [PubMed] [Google Scholar]
- 25.Takaichi, S., K. Inoue, M. Akaike, M. Kobayashi, H. Oh-Oka, and M. T. Madigan. 1997. The major carotenoid in all known species of heliobacteria is the C30 carotenoid 4,4′-diaponeurosporene, not neurosporene. Arch. Microbiol. 168:277-281. [DOI] [PubMed] [Google Scholar]
- 26.Tarshis, L. C., P. J. Proteau, B. A. Kellogg, J. C. Sacchettini, and C. D. Poulter. 1996. Regulation of product chain length by isoprenyl diphosphate synthases. Proc. Natl. Acad. Sci. USA 93:15018-15023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor, R. F. 1984. Bacterial triterpenoids. Microbiol. Rev. 48:181-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor, R. F., and B. H. Davies. 1976. Triterpenoid carotenoids and related lipids. Triterpenoid carotenoid aldehydes from Streptococcus faecium UNH 564P. Biochem. J. 153:233-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogeli, U., and J. Chappell. 1988. Induction of sesquiterpene cyclase and suppression of squalene synthetase activities in plant cell cultures treated with fungal elicitor. Plant Physiol. 88:1291-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wieland, B., C. Feil, E. Gloria-Maercker, G. Thumm, M. Lechner, J.-M. Bravo, K. Poralla, and F. Götz. 1994. Genetic and biochemical analyses of the biosynthesis of the yellow carotenoid 4,4′-diaponeurosporene of Staphylococcus aureus. J. Bacteriol. 176:7719-7726. [DOI] [PMC free article] [PubMed] [Google Scholar]