

Abstract
The persistence of Mycobacterium tuberculosis is a major cause of concern in tuberculosis (TB) therapy. In the persistent mode the pathogen can resist drug therapy, allowing the possibility of reactivation of the disease. Several protein factors have been identified that contribute to persistence, one of them being the 16-kDa low-molecular-weight mycobacterial heat shock protein Hsp16.3, a homologue of the mammalian eye lens protein alpha-crystallin. It is believed that Hsp16.3 plays a key role in the persistence phase by protecting essential proteins from being irreversibly denatured. Because of the close association of Hsp16.3 with persistence, an attempt has been made to develop inhibitors against it. Random peptide libraries displayed on bacteriophage M13 were screened for Hsp16.3 binding. Two phage clones were identified that bind to the Hsp16.3 protein. The corresponding synthetic peptides, an 11-mer and a 16-mer, were able to bind Hsp16.3 and inhibit its chaperone activity in vitro in a dose-dependent manner. Little or no effect of these peptides was observed on alphaB-crystallin, a homologous protein that is a key component of human eye lens, indicating that there is an element of specificity in the observed inhibition. Two histidine residues appear to be common to the selected peptides. Nuclear magnetic resonance studies performed with the 11-mer peptide indicate that in this case these two histidines may be the crucial binding determinants. The peptide inhibitors of Hsp16.3 thus obtained could serve as the basis for developing potent drugs against persistent TB.
Drug resistance is a major problem in the therapy of tuberculosis (TB). Such resistance arises not only due to mutations but also due to the ability of the TB pathogen Mycobacterium tuberculosis to enter into a dormant phase in which it can persist for prolonged periods of time (9). This happens particularly when it is encapsulated within a granuloma—a structure formed by the activated monocyte-macrophage system of the host. The conditions within the granuloma are far from ideal for mycobacterial growth (24). In particular, M. tuberculosis is an aerobic organism, whereas the conditions within the granuloma are anaerobic. Although under such conditions active growth is halted, the bacteria can persist indefinitely by entering into a dormant phase. Drug therapy further accelerates the shift from the active to the dormant or persistent phase (9, 24). Treatment with the presently available drugs therefore can potentially cause the accumulation of dormant bacilli (17), which can reactivate themselves at a later stage.
Hsp16.3 was initially identified as a 14-kDa immunodominant antigen (26) but subsequently characterized to be a molecular chaperone that functions by preventing the aggregation of proteins under stress conditions (7). Hsp16.3 belongs to the alpha-heat shock protein (HSP) family of proteins represented by the small molecular weight heat shock proteins alpha-crystallin A and B, which play an important role in the maintenance of the transparency of vertebrate eye lenses (25). The expression of the mycobacterial alpha-HSP (Hsp16.3) has been found to be induced during the course of in vitro infection of macrophages (30). When the gene for Hsp16.3 (acr) of M. tuberculosis was deleted, the resulting strain was shown to be equivalent to the wild-type H37Rv in in vitro growth rate and infectivity but was significantly impaired for growth in both mouse bone marrow-derived macrophages and THP-1 cells (31). Deletion of the acr gene also resulted in the virulent strain of M. tuberculosis becoming attenuated. It has been claimed that such attenuated strains, in which Hsp16.3 expression is impaired, could be used as anti-TB vaccines (3a).
Based on the available evidence, it is generally considered that Hsp16.3 is a potentially important component, which facilitates the survival of M. tuberculosis during latent human infection (31). Hence, Hsp16.3 may be regarded as a possible target for anti-TB drug discovery. However, due to the lack of a crystal or nuclear magnetic resonance (NMR) structure, a structure-based drug design is not possible at present. One of the ways to develop specific inhibitors is to look for binding peptides against a target protein by using the phage display technique (22) and then develop peptidomimetics (2) to inhibit its activity. We report here the identification of peptides by phage display that bind to Hsp16.3 protein and inhibit its chaperone activity. The putative binding determinants have also been identified. Although as of now there is little evidence to suggest that the approach would lead to novel therapeutics, it would be interesting to look for anti-Hsp16.3 compounds through peptidomimetics for exploring new methods to combat latent TB.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The protein Hsp16.3 was obtained from the Escherichia coli strain EC-16, which harbors a recombinant plasmid comprising the acr gene (Rv2031c) cloned in the E. coli expression vector pQE-8 (QIAGEN, Inc., Stanford, CA). The strain EC-16 was obtained on the basis of a material transfer agreement with the Medical Research Council of London. The protein is expressed from the recombinant plasmid as a fusion protein with a stretch of His6 residues at the N-terminal end. The recombinant alphaB-crystallin (5) expressed from a similar recombinant construct was obtained as a gift from K. P. Das of Bose Institute, Calcutta, India.
Expression and purification of Hsp16.3.
EC-16 cells were grown at 37°C under shaking conditions in 2× YT medium containing 100 μg of ampicillin/ml and 50 μg of kanamycin/ml and then induced (at an A600 of ∼0.6) with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside; Sigma, St. Louis, MO) for 4 h. Cells were harvested by centrifugation, washed with phosphate-buffered saline, and frozen at −20°C until use. After resuspension in buffer A (50 mM sodium phosphate [pH 7.5], 300 mM NaCl, 10 mM imidazole) containing 1 mM phenylmethylsulfonyl fluoride (Sigma, St. Louis, MO), a protease inhibitor, cells were disrupted by using a Labsonic-L sonicator (B. Braun, Germany), while cooling on ice, by giving 10 pulses (30 s each at 150 W) at intervals of 2 to 3 min. After centrifugation at 14,000 × g, the clear supernatant was loaded onto a 4-ml Ni2+-nitrilotriacetic acid (NTA) agarose column (QIAGEN), preequilibrated with buffer A. The column was washed with about 5 column volumes of buffer B (50 mM sodium phosphate [pH 7.5], 300 mM NaCl, 20 mM imidazole), and the bound protein was eluted with a 50 to 400 mM imidazole gradient in 50 mM sodium phosphate (pH 7.5) buffer containing 300 mM NaCl. Fractions were collected and analyzed by 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The fractions containing pure Hsp16.3 protein were pooled and dialyzed against 50 mM sodium phosphate (pH 7.5) buffer containing 300 mM NaCl. The Hsp16.3 protein obtained by this method was >98% pure, as judged by SDS-PAGE. The concentration of Hsp16.3 protein was determined on the basis of its molar extinction coefficient (3,840 M−1 cm−1) and the Lowry method (16).
Screening of phage display libraries.
Phage display libraries (New England Biolabs, Inc., Beverly, MA) contained rationally designed combinatorial libraries of peptide sequences (7-mer or 12-mer) inserted into the NH2 terminus of the pIII minor coat protein of the M13 bacteriophage. These libraries (termed Ph.D.-7 and Ph.D.-12) contained either 7 amino acids (for the Ph.D.-7 library) or 12 amino acids (for the Ph.D.-12 library). The peptides were followed by a short spacer (Gly-Gly-Gly-Ser) and then the wild-type pIII sequence. The library consists of ∼2.8 × 109 electroporated sequences (compared to 207 = 1.28 × 109 possible 7- or 12-residue sequences). The libraries were screened by biopanning using standard methods with a few modifications. Briefly, a 996-well microtiter plate was coated with 30 μg of Hsp16.3/well in 0.1 M NaHCO3 buffer (pH 8.6) and incubated overnight at 4°C with gentle agitation in a humidified chamber. The coating solution was poured off, and each well was completely filled with blocking buffer (5 mg of bovine serum albumin [BSA]/ml in 0.1 M NaHCO3 buffer [pH 8.6]) and incubated for 1 to 2 h at 4°C. Protein-coated wells were washed with TBST (Tris-buffered saline [TBS] plus 0.1% [vol/vol] Tween 20) at least five to six times before use for biopanning of phage libraries. A Ph.D.-7/Ph.D.-12 phage library containing 1011 PFU was incubated in the wells for 1 h at room temperature on a rocker. Nonbinding phage was poured off, and the wells were washed 10 times with TBST. Phage bound to the Hsp16.3 protein was eluted with 0.2 M glycine-HCl buffer (pH 2.2) containing 1 mg of BSA/ml. After elution the acidic buffer was neutralized with 1 M Tris-HCl (pH 9.1). The eluted phage was then amplified in vivo on an E. coli ER2738 host strain obtained from New England Biolabs, Inc. (Beverly, MA), purified by double precipitation in cold with 1/6 volume of polyethylene glycol (PEG)-NaCl (20% [vol/vol] PEG 8000, 2.5 M NaCl) and used in second round of biopanning. In first, second, and third rounds of biopanning, the Tween 20 concentration was increased progressively from 0.1 to 0.3 to 0.5%, respectively, for better washing off of nonspecifically adhered phage. After three rounds of positive selection, E. coli ER2738 cells were infected with the eluted phage and grown on LB-agar plates coated with X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside)/IPTG. Individual plaques were picked and amplified, and single-stranded phage DNA was isolated for sequencing.
Reverse phage ELISA.
A reverse phage enzyme-linked immunosorbent assay (ELISA) was used to evaluate the ability of individual phage clones to bind to Hsp16.3. Briefly, 30 μg of Hsp16.3/well in 0.1 M NaHCO3 buffer (pH 8.6) was added to a 96-well microtiter plate and blocked with 2% skim milk powder in TBS. BSA (30 μg/well) was used as a negative control. The selected peptide phage clones, amplified and concentrated by the PEG-NaCl precipitation method, were added to each coated well (107 to 108 PFU/well) and incubated for 2 h at room temperature. Unbound phage was removed by washing with TBST (0.5% Tween 20), and bound phage was detected with horseradish peroxidase-conjugated anti-M13 monoclonal antibody (Amersham Pharmacia Biotech, Uppsala, Sweden) at 1:5,000 and with ABTS [2,2′azinobis(3-ethylbenzthiazolinesulfonic acid)] substrate (Roche, Mannheim, Germany). Plates were read at an A405. If the phage clones obtained after screening are true Hsp16.3 binders, then ELISA signals substantially higher than the background are obtained.
Synthesis and labeling of peptides.
Peptides were produced on a 0.12-mmol scale using a Biolynx 4175 peptide synthesizer. They were synthesized by the solid-phase method with the standard Fmoc (9-fluorenylmethoxy carbonyl) chemistry. The peptides were synthesized with their C terminus amidated by using MBHA-Rink amide resin (Novabiochem, San Diego, CA). Amino acids (Novabiochem) except those stated below were used as N-terminally Fmoc protected and C-terminally pentafluorophenyl ester activated with N-hydroxybenzotriazole (Novabiochem, San Diego, CA). Thr, His, Arg, Ile, Ser, Ala and Pro were used in the COOH form using benzotriazole-1-yloxytripyrrolidinophosphoriumhexafluorophosphate (PyBop; Novabiochem, San Diego, CA) as the activating agent with N-hydroxybenzotriazole and N,N-diisopropylethylamine (DIPEA; Fluka Chemie, GmbH) (1:1:2). The Fmoc-protecting group was removed from the N terminus of the peptide resin by 20% piperidine in dimethyl formamide (DMF; Merck, Germany), followed by washes with DMF. The resin was dried under vacuum. The peptides were cleaved by using standard trifluoroacetic acid (TFA; Spectrochem, Mumbai, India) cleavage procedures, followed by multiple ether extractions. Both the peptides were purified by reversed-phase high-pressure liquid chromatography on a C18 column (Vydac, Hesperin, CA) using a 0 to 60% acetonitrile gradient in 0.1% TFA and characterized by NMR. A part of the resin-bound peptides was labeled with fluorescein isothiocyanate (FITC), thus placing the fluorophore on the N terminus of the peptide. One equivalent of dried resin in 2% piperidine in DMF was mixed with 30 eq of FITC (Molecular Probes, Inc., Eugene, OR) and 10 eq of DIPEA in a microcentrifuge tube, and the reaction was allowed to proceed for 2 to 3 h at room temperature with occasional mixing. After several washes with DMF, t-amyl alcohol, glacial acetic acid, and diethyl ether, the resin was dried under vacuum. The dye-conjugated peptides were cleaved from the resin, and the side chain-protecting groups were removed by incubating in reagent K (TFA-phenol-thioanisole-water-ethane dithiol [82.5:5:5:5:2.5]) for 3 h at room temperature. The labeled peptides were purified by reversed-phase high-pressure liquid chromatography as described above. The concentration of peptides was determined by using the Lowry method (16).
Fluorescence measurements: anisotropy and quenching.
All fluorescence studies were performed in a Hitachi F-3010 spectrofluorometer with a facility for spectrum addition and subtraction. The excitation and emission band passes were 5 nm unless stated otherwise. Fluorescence anisotropy measurements (8) were done by using a Hitachi polarizer accessory. The steady-state fluorescence anisotropy (A) was calculated according to the following equation:
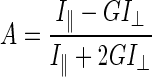 |
(1) |
where I∥ is the intensity when the polarizers were in the same direction, I⊥ is the intensity when the polarizers were crossed, and G is the grating factor that corrects for wavelength-dependent distortion of the polarizing system. For each anisotropy value, three measurements were taken and averaged. FITC-conjugated peptides (800 nM) were titrated separately with increasing concentrations of Hsp16.3 in 50 mM sodium phosphate buffer (pH 7.5) containing 300 mM NaCl at room temperature (25 ± 1°C). The excitation and emission were 495 and 519 nm, respectively.
A single-step binding model was used for the analysis of the binding isotherm of the peptides (10 and 22). The anisotropy (A) data were fitted to the analytical formula (equation 2) by using the Marquardt nonlinear least squares algorithm. The parameters of the fit were the anisotropies of free and bound peptides (Af and Ab, respectively) and the dissociation constant KD.
 |
The total concentrations of peptide 10 or 22, Peptot, and protein Protot were known in these experiments.
For tyrosine fluorescence quenching studies (4, 15) excitation and emission wavelengths were 280 and 311 nm, respectively. The respective band passes were 5 and 10 nm. The observed fluorescence values were corrected for volume change and inner filter effect. The inner filter effect was corrected by using the following formula:
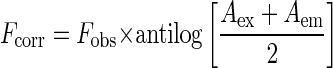 |
(3) |
where Aex is the absorbance at the excitation wavelength and Aem is the absorbance at the emission wavelength. The buffer used for this experiment was the same as mentioned earlier. Binding of peptide 10 (Seq ID 10) was determined from quenching of tyrosine fluorescence of Hsp16.3. Hsp16.3 (5 and 15 μM) was titrated with increasing concentrations of peptide 10. F/F0 was plotted against peptide 10 concentration, where F0 is the initial fluorescence and F is the fluorescence value after quenching at each point. The ratio of F/F0 is assumed to be equal to the degree of binding.
All curve fittings and statistical deductions were done by using Kyplot software (K. Yoshioka, 1997-1999, version 2.0 beta 4). The results have been presented as the mean ± the standard error (SE), calculated from three independent experiments. The significance levels are indicated, wherever relevant, by the P value. P values of <0.05 are considered significant.
In vitro assay for anti-Hsp16.3 activity.
Aggregation of 5 μM ADH at 50°C was measured as the apparent optical density at 360 nm by using a Hitachi spectrophotometer equipped with a thermostated cuvette holder in a total reaction volume of 500 μl. The optical density was monitored continuously until steady-state levels were reached. The absorbance values at 100-s intervals were retrieved and plotted against time. Intra-assay variations of absorbance values with the same protein stock solutions were <1%. The chaperone activity of Hsp16.3 was determined by mixing ADH with Hsp16.3 at a molar ratio of 5:4, and aggregation rates were again determined by continuous spectrophotometric monitoring as described above. The same experiment was then performed in the presence of increasing concentrations of the Hsp16.3 binding peptides. The percent residual chaperone activity was plotted against peptide concentration, and from such a plot the inhibitory concentration at which 50% activity was lost (IC50) was calculated by using a nonlinear regression data analysis in Kyplot (K. Yoshioka, 1997-1999, version 2.0 beta 4).
Formation and analysis of Hsp16.3-substrate complex.
Hsp16.3 was mixed with MDH in a 40:1 molar ratio in 50 mM sodium phosphate buffer (pH 7.5), containing 300 mM NaCl (20 μl total volume) in a 1.5-ml microcentrifuge tube, followed by incubation at 50°C for 30 min. Samples were then centrifuged at room temperature for 15 min at 15,000 × g. Both supernatants and pellet fractions were analyzed by SDS-PAGE on a 15% gel. Band intensities were determined by densitometric scanning using a GS-700 imaging densitometer (Bio-Rad Laboratories) and analyzed by the supplied Molecular Analyst (version 1.5) software.
NMR spectroscopy.
All NMR spectra were obtained with a Bruker DRX-500 NMR spectrometer equipped with a Z-field gradient probe. All measurements were done in high-precision 5-mm NMR tubes in 20 mM sodium phosphate buffer (pH 7.0) containing 250 mM NaCl in 90% H2O and 10% D2O. All NMR experiments were done at 5°C unless stated otherwise. Total correlation spectroscopy (TOCSY) spectra were measured by using standard pulse sequences in the Bruker pulse library using the WATERGATE water suppression method. Standard Bruker software (Xwin-NMR, version 1.3) was used to acquire and process the NMR data. The peptide 10 concentration was 2 mM. When present, the Hsp16.3 concentration was 0.1 mM, thus making a 20-fold peptide excess over the binding sites.
RESULTS
Biological enrichment of phage displaying Hsp16.3 binding peptide sequences through biopanning.
After the third round of panning of the Ph.D.-7 library on Hsp16.3, it was found that out of the eight randomly selected phage clones (Seq IDs 1 to 8), which were sequenced for the peptide-coding region, five were identical (Table 1). Seq ID 9 represents a consensus sequence, Lys-Met-His-Ala-Thr-Asn-His, emerging from this panning experiment. In the case of the Ph.D.-12 library, the results were more striking. Of the 10 clones sequenced after the third panning, 9 clones (Seq IDs 11 to 20) were identical, and the consensus peptide sequence (Seq ID 21) in this case was Tyr-Pro-His-His-Phe-Lys-His-Arg-His-Ile-Pro-Ile. When the two consensus sequences (Seq IDs 9 and 21) were compared, it was found that the third and seventh His residues existed in an aligned position. The binding of phage clones displaying peptides (Seq IDs 9 and 21) to Hsp16.3 was confirmed further by performing a reverse phage ELISA (Fig. 1). For deriving synthetic peptides, the Gly-Gly-Gly-Ser spacer was added to the consensus sequences at the C-terminal ends to obtain the effective binding sequences having IDs 10 and 22.
TABLE 1.
Hsp16.3 binding peptide sequences selected after third-round panning
| Phage display library used | Sequence IDa | Amino acid sequenceb | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ph.D.-7 | 1 | Gly | Val | Glu | Asn | Val | Ser | Trp | |||||||||
| 2 | Lys | Met | His | Ala | Thr | Asn | His | ||||||||||
| 3 | Lys | Met | His | Ala | Thr | Asn | His | ||||||||||
| 4 | Lys | Met | His | Ala | Thr | Asn | His | ||||||||||
| 5 | Leu | Pro | Ala | Lys | Asn | Phe | His | ||||||||||
| 6 | Phe | Pro | Pro | Leu | Lys | Ser | Pro | ||||||||||
| 7 | Lys | Met | His | Ala | Thr | Asn | His | ||||||||||
| 8 | Lys | Met | His | Ala | Thr | Asn | His | ||||||||||
| 9* | Lys | Met | His | Ala | Thr | Asn | His | ||||||||||
| 10† | Lys | Met | His | Ala | Thr | Asn | His | Gly | Gly | Gly | Ser | ||||||
| Ph.D.-12 | 11 | Tyr | Pro | His | His | Phe | Lys | His | Arg | His | Ile | Pro | Ile | ||||
| 12 | Tyr | Pro | His | His | Phe | Lys | His | Arg | His | Ile | Pro | Ile | |||||
| 13 | Tyr | Pro | His | His | Phe | Lys | His | Arg | His | Ile | Pro | Ile | |||||
| 14 | Tyr | Pro | His | His | Phe | Lys | His | Arg | His | Ile | Pro | Ile | |||||
| 15 | Tyr | Pro | His | His | Phe | Lys | His | Arg | His | Ile | Pro | Ile | |||||
| 16 | Ala | Tyr | Lys | Pro | Ile | Ala | His | Phe | Ile | Ser | Pro | Ala | |||||
| 17 | Tyr | Pro | His | His | Phe | Lys | His | Arg | His | Ile | Pro | Ile | |||||
| 18 | Tyr | Pro | His | His | Phe | Lys | His | Arg | His | Ile | Pro | Ile | |||||
| 19 | Tyr | Pro | His | His | Phe | Lys | His | Arg | His | Ile | Pro | Ile | |||||
| 20 | Tyr | Pro | His | His | Phe | Lys | His | Arg | His | Ile | Pro | Ile | |||||
| 21* | Tyr | Pro | His | His | Phe | Lys | His | Arg | His | Ile | Pro | Ile | |||||
| 22† | Tyr | Pro | His | His | Phe | Lys | His | Arg | His | Ile | Pro | Ile | Gly | Gly | Gly | Ser | |
ID, identification number. *, the most frequently occurring sequence in the group: the consensus sequence. †, the spacer sequence (Gly-Gly-Gly-Ser) was added to the C-terminal end of the consensus sequence as suggested by the suppliers of the Ph.D. kits (New England Biolabs) to derive the effective binding sequences.
The His residues that are in aligned positions are in boldface.
FIG. 1.

Reverse phage ELISA to confirm the binding of phage clones displaying peptide sequences, Seq ID 9 and 21, to Hsp16.3. The phage clones were amplified, concentrated by PEG precipitation, and reacted with either Hsp16.3 or nonspecific target BSA coated on the wells of a microtiter plate. Unbound phage was removed by washing with TBST (0.5% Tween 20), and bound phage was detected with horseradish peroxidase-conjugated anti-M13 monoclonal antibody (1:5,000) and ABTS substrate. Color development was monitored spectrophotometrically at 405 nm. Each experiment was done in triplicate. The bar graph shows binding activity of phage clones displaying Seq ID 9 (□) and Seq ID 21 (▪) to Hsp16.3 and BSA (background binding). The error bars represent the SE.
The various Hsp16.3 binding peptide sequences mentioned in the present study, the method of deriving them, and assay of their inhibitory activities are all covered by a patent application (pending) made to the U.S. Patent and Trademark Office.
Binding affinity of the peptide ligands for Hsp16.3.
Seq IDs 10 and 22, referred to as peptides 10 and 22 henceforth, were synthesized chemically and labeled with FITC at the N terminal ends.
The fluoresceinated peptides were used to measure binding with Hsp16.3 by fluorescence anisotropy. Figure 2A and B show the titrations of FITC-conjugated peptides 10 and 22 (800 nM), respectively, with increasing concentrations of Hsp16.3. For each peptide three independent titrations were done, and the mean anisotropy ± the SE was plotted against protein concentration. Anisotropy increased as a function of Hsp16.3 concentration. The data were fitted to the single-site binding equation (equation 2), and the dissociation constant was determined. The KD values thus obtained were found to be 54.2 ± 13.7 μM (P = 0.001) and 45.7 ± 18.0 μM (P = 0.023) for peptides 10 and 22, respectively.
FIG. 2.

Determination of the dissociation constant and stoichiometry of peptide binding to Hsp16.3. Anisotropy increase of FITC-labeled peptide 10 (A) and peptide 22 (B) as a function of Hsp16.3 concentration in 50 mM sodium phosphate buffer (pH 7.5) containing 300 mM NaCl at room temperature. Anisotropy values are means ± the SE (indicated by error bars) of three independent experiments. The excitation and emission wavelengths were at 495 and 519 nm, respectively. In case of peptide 10, stoichiometry was determined (C) from the dose-dependent quenching of the intrinsic tyrosine fluorescence of Hsp16.3 in the same buffer conditions. The lines were drawn to determine binding stoichiometry.
Tyrosine fluorescence quenching of Hsp16.3 protein was also used to study the binding of peptide 10. Since peptide 10 has no tyrosine residues, it can be used as a quencher, but a similar exercise could not be done in the case of peptide 22 since tyrosine forms a part of the sequence. Figure 2C shows the tyrosine fluorescence quenching profile of Hsp16.3 (15 μM) upon binding peptide 10. The abscissa value of the intersection of the asymptotic line and that drawn through the initial F/F0 values represents nPt + KD, where n is the number of peptide binding sites per subunit of Hsp16.3 and Pt is the total protein concentration in terms of monomer. In order to determine n, the quenching experiments were done at two concentrations of Hsp16.3: 15 μM (Fig. 2C) and 5 μM (data not shown). The abscissa values were found to be 20.0 (Fig. 2C) and 10.2 μM, respectively. Hence, we have KD + 15n = 20.0 and KD + 5n = 10.2. By eliminating KD from these two equations, the value of n was determined to be 0.98. It may be concluded that there is one binding site for peptide 10 per subunit of Hsp16.3.
Specific inhibition of Hsp16.3 chaperone activity by the peptides.
Alcohol dehydrogenase (ADH) (5 μM) aggregates when heated to 50°C (Fig. 3A, B, D, and E). This aggregation is inhibited to the extent of ca. 85% when ADH is coincubated with Hsp16.3 (Fig. 3A and D) at a molar ratio of ADH to Hsp16.3 of 5:4. The addition of increasing amounts of peptide 10 resulted in progressive loss of the chaperone activity of Hsp16.3. At the highest concentration of 100 μM, i.e., at an ∼25-fold molar excess, a significant inhibition (>70%) of chaperone activity was observed (Fig. 3A). Similarly, in the case of peptide 22, the extent of inhibition at 100 μM was ca. 60% (Fig. 3D). The addition of peptide alone did not have any effect on aggregation rates of ADH (Fig. 3A and D). Using the same experimental setup, Hsp16.3 was replaced by alphaB-crystallin, and the inhibitory effect, if any, of the peptides on this protein was analyzed. As expected, when ADH (5 μM) was mixed with alphaB-crystallin (2.5 μM), complete protection against aggregation was observed, but this protection could not be reversed by the addition of either peptide 10 or peptide 22 (Fig. 3B and E, respectively) at the concentration of 100 μM, which effectively gives an ∼40-fold molar excess of peptide over the target protein alphaB-crystallin. To obtain a more quantitative interpretation, the percent residual chaperone activity (mean ± the SE of three independent experiments) was plotted against peptide concentration. This yielded a dose-response curve (Fig. 3C and F) from which IC50 values of 53.0 ± 8.47 μM (P = 0.0079) for peptide 10 and of 57.1 ± 4.37 μM (P = 0.0058) for peptide 22 were obtained. The dose-response curves for both peptides also show graphically that in the context of alphaB-crystallin there was almost no effect.
FIG. 3.

Dose-dependent inhibition of chaperone activity of Hsp16.3 by peptides 10 and 22. Aggregation assays were performed by heating 5 μM ADH at 50°C directly in a spectrophotometer using a thermostated cuvette holder in a total reaction volume of 500 μl in 100 mM sodium phosphate buffer (pH 7.0) containing 100 mM NaCl. The absorbance was monitored at 360 nm for 1,200 s, and readings were taken at every 100-s interval. ADH aggregates upon heating (A, B, D, and E; ▴). Incorporation of Hsp16.3 at a molar ratio of 5:4 (A and D) or alphaB-crystallin at a molar ratio of 1:1 (B and E) results in a decline in aggregation activity (•). Then increasing concentrations of Hsp16.3 binding peptide 10 (A and B) and peptide 22 (D and E) were added as indicated at the following concentrations: 25 μM (□), 50 μM (▪), and 100 μM (▵). Stars represent the ADH aggregation in the presence of peptide 10 (A and B) and peptide 22 (D and E). The IC50 for peptide 10 (C) and peptide 22 (F) was calculated from the plot of the percent residual chaperone activity against the peptide concentration. Each datum point in panels C and F is an average of three independently derived results. Error bars indicate the SE.
Peptides affect the stability of Hsp16.3-substrate complexes.
It has been demonstrated earlier that alpha-HSPs form soluble complexes with aggregation-prone proteins when heated together at 50°C (14). In order to investigate the possible mechanism of action of the peptide inhibitors, their effect on Hsp16.3-substrate complex formation was examined by using the model substrate malate dehydrogenase (MDH). MDH was taken at a concentration of 2 μM and heated at 50°C. As expected, it formed aggregates and precipitated out. When the pellet and supernatant were analyzed by SDS-PAGE, the entire protein was found to be present in the pellet fraction (Fig. 4A, lane 2). Hsp16.3 (80 μM) was then added to MDH at a 40-fold molar excess prior to heating to prevent aggregation. The incorporation of this amount gave maximal protection, and most of the MDH was retained in the supernatant (Fig. 4A, lane 3). To investigate the effect of peptide 10 on complex formation, increasing doses of the peptide were added to inactivate Hsp16.3. Figure 4B shows a progressive shift in the MDH band from the supernatant to the pellet fraction (lanes marked by black dots) upon addition of increasing amounts of peptide 10. Hsp16.3, however, remained soluble up to a 3.2 mM concentration of peptide 10 (40-fold molar excess), although at a higher concentration (4.8 mM, i.e., a 60-fold molar excess) it was also precipitated (Fig. 4D). Addition of peptide 22 led to similar consequences (Fig. 4C), but in this case almost complete precipitation of the complex occurred at a 1.2 mM peptide concentration (15-fold molar excess) compared to 4.8 mM (60-fold molar excess) in the case of peptide 10. Another notable difference was that, unlike the previous case, precipitation of Hsp16.3 and MDH occurred in parallel when peptide 22 was used as an inhibitor (Fig. 4E). Since in both cases a detectable loss in the solubility of Hsp16.3 was observed, the effect of these peptides on the solubility of Hsp16.3 itself was examined at the peptide concentrations at which the complexes were precipitated. The results indicate that in the case of peptide 10 (Fig. 4F), ca. 70% of the chaperone protein present became insoluble at a 60-fold molar excess (4.8 mM), whereas in the case of peptide 22 (Fig. 4G), almost 90% of it lost solubility at a 15-fold molar excess (1.2 mM).
FIG. 4.

Effect of peptides on the stability of the Hsp16.3-MDH complex. (A) MDH (2 μM) either alone or after being mixed with Hsp16.3 (80 μM) was heated to 50°C for 30 min, followed by centrifugation at 15,000 × g. The supernatants and pellets were analyzed by SDS-PAGE on a 15% gel. Lanes marked with black dots represent pellet fractions in all cases. The MDH-Hsp16.3 complex was then formed in the presence of inhibitory peptide 10 or peptide 22 (B and C), respectively. The molar excesses of the peptides used relative to Hsp16.3 are indicated below the lanes. The bands were scanned densitometrically, and the fraction of the protein (either Hsp16.3 or MDH) that appeared in the supernatant was plotted as the percent solubility against the molar excess of peptide 10 or peptide 22 (D and E), respectively. (F and G) The effect of peptides 10 and 22 on the solubility of Hsp16.3 was tested using the indicated molar excess. (H) The comparative effect of peptide 22 (lanes marked with black dots) on alphaB-crystallin was determined side by side with the Hsp16.3 control. (I) Similarly, the effect of peptide 22 on the formation of a soluble complex between alphaB-crystallin and MDH was tested alongside an Hsp16.3-MDH control. In the last two experiments, a peptide molar excess of 15 over either Hsp16.3 or alphaB-crystallin was used.
Although the precipitation was observed at relatively high concentrations of peptides, the phenomenon is specific since it does not occur in case of BSA and DnaK (data not shown), nor does it affect the related homologue alphaB-crystallin (Fig. 4H). Thus, when a 15-fold molar excess (1.2 mM) of peptide 22 was added to Hsp16.3 (80 μM), a significant loss of solubility was observed as expected (Fig. 4H, lane 4), but when it was added to alphaB-crystallin, no effect was observed (Fig. 4H, lane 8). The effect of the peptides on the complex between MDH and alphaB-crystallin was also investigated (Fig. 4I). The results presented for peptide 22 show that at the concentration of the peptide at which Hsp16.3-MDH complex precipitates out (Fig. 4I, lane 4), no effect on alphaB-crystallin-MDH complex (Fig. 4I, lane 8) was observed. The assays were repeated several times, and the precipitation pattern was found to be reproducible. These observations further establish that the peptide inhibitors have target specificity.
Precipitation is not an essential feature for inhibition.
In order to investigate whether the loss of chaperone activity of Hsp16.3 observed in the spectrophotometric aggregation assays was due to precipitation of the protein, an assay (Fig. 5A) was first performed essentially as described in the previous sections and then, after reaching steady-state aggregation, the samples were centrifuged, and pellet fractions were analyzed by SDS-15% PAGE (Fig. 5C). The presence of Hsp16.3 (4 μM) prevents aggregation to an extent of 60% (Fig. 5A and B). Addition of peptide 10 or peptide 22 at a 50-fold molar excess (200 μM) restored aggregation to almost the initial level (ADH alone). Hsp16.3 itself does not show any aggregation. SDS-PAGE analysis revealed that in presence of Hsp16.3 there was a 55% drop in the intensity of the ADH band (Fig. 5C and D, lane 3) commensurate with the drop observed in the spectrophotometric assay. In the presence of the peptides, the intensity returned to almost 100%, indicating inhibition of Hsp16.3 activity (Fig. 5C and D, lanes 4 and 5 compared to lane 2). The fluctuations in intensities occurred only in the case of the ADH bands but not for Hsp16.3, the intensity of which remained at the background levels (Fig. 5D). This indicates that under the conditions of the spectrophotometric assay the inactivation by the peptides does not involve aggregation of Hsp16.3. The result suggests that the binding of the peptides alters the conformation of Hsp16.3 so that it becomes less soluble, but the loss of solubility is actually manifested when the concentrations of peptides are high, i.e., in the millimolar range. Precipitation, therefore, is not the phenomenon itself but a reflection of a possible conformational change in Hsp16.3 as a result of peptide binding.
FIG. 5.

Peptide-mediated inactivation of Hsp16.3 under conditions used for spectrophotometric assay of chaperone activity. (A) The chaperone activity was measured spectrophotometrically as described in Fig. 3. Aggregation of ADH (5 μM) without chaperone is represented by the curve marked by white circles, protection by Hsp16.3 (4 μM) by black circles, and inhibition of Hsp16.3 by peptides 10 and 22 (200 μM) by white and black squares, respectively. Aggregation of Hsp16.3 alone is indicated by white triangles. (B) The steady-state aggregations (1,200 s) observed in panel A are graphically represented. (C) The aggregated samples were centrifuged, and the pellet fractions were analyzed by SDS-PAGE on a 15% gel; lanes corresponding to the aggregation curves are indicated by identical symbols. The combinations of proteins and peptides used are indicated on the top. The bar diagram (D) shows the band intensities of the ADH (□) and Hsp16.3 (▪) components in each lane.
Evidence for involvement of His residues in the interaction of peptide with Hsp16.3.
In order to further investigate the interaction between peptides and Hsp16.3, NMR experiments were performed. Figure 6A represents the overlaid TOCSY spectra of the NH-αH region of peptide 10 (2 mM) in the absence of Hsp16.3 (red) and in the presence of Hsp16.3 (blue) at a substoichiometric concentration (0.1 mM). Under these ligand excess conditions, only the peptide peaks are visible. The fast exchange between the receptor bound and free forms allows averaging of the two chemical shifts under two different conditions. Thus, the chemical shift change, compared to the free peptide, represents chemical shifts that are different in the receptor bound state and possible points of interaction between the peptide and the receptor. Figure 6B graphically represents the chemical shift differences of amide protons of peptide 10 residues in buffer solutions in the presence or absence of Hsp16.3. It is clear from the bar diagram that only a few residues shift and that chemical shift of most of the residues remains unchanged. The residue types were assigned from spin connectivity and chemical shift information (data not shown). The peaks for Met were not observed in the TOCSY spectra, probably because of broadening of peaks due to conformational exchange. The peak for Met could, however, be observed when the spectra were derived in water at 27°C (data not shown), confirming that the peptide contained Met. The comparison of TOCSY spectra of peptide 10 obtained in the presence or absence of Hsp16.3 revealed significant chemical shift changes for both His residues of peptide 10 in the presence of Hsp16.3. In the case of Ala, a minor effect was observed, but for the others the effect was negligible. From this result it appears that the His-X-Y-Y-His region of peptide 10 may be necessary for interaction with Hsp16.3, where X stands for Ala or an analogous amino acid and Y represents any other amino acid. A similar analysis with peptide 22 was not possible since the peptide 22-Hsp16.3 complex precipitates out at high concentration, as has been demonstrated earlier.
FIG. 6.

Changes in chemical shift of peptide 10 residues due to interaction with Hsp16.3. (A) Overlay 2D TOCSY spectra in the fingerprint (selected) region of 2 mM peptide 10 in 20 mM sodium phosphate buffer (pH 7.0) containing 250 mM NaCl in 90% H2O and 10% D2O at 5°C in the absence (red) or in the presence (blue) of 0.1 mM Hsp16.3 protein. Amino acids are designated by using the one-letter code. (B) Graphical representation of chemical shift differences (Δ δ Hz) of amide protons of peptide 10 residues in buffer solution in the presence or absence of Hsp16.3, as mentioned in the legend to panel A.
DISCUSSION
Low-molecular-weight HSPs (sHSPs) play a major role in the maintenance of protein stability under stress conditions (6). The most well-known examples of sHSPs are the lens proteins alpha-crystallin A and B, which are implicated in the maintenance of lens transparency. Alpha-crystallin-like proteins (alpha-HSPs) are, however, ubiquitous in nature, being present in animals, plants, and also bacteria. Functionally, these proteins have been classified as chaperones since they can prevent thermal denaturation of proteins. This feature is common to all sHSPs, and hence prevention of aggregation of proteins is considered a standard assay for their activity. Aggregation prevention, however, is not the only function of these proteins, and there is evidence to show that they are implicated in cytomorphological reorganization and signal transduction events (10).
This is possibly the first report where an attempt to develop an inhibitor against an alpha-HSP has been made. The importance of alpha-HSPs in human eye lens physiology demands that all efforts be made to prevent rather than cause inhibition of activity. In the case of bacteria, however, there is a paradigm shift. Since sHSPs play an important role in the survival of bacteria under stress conditions, their inactivation may be an important issue, particularly in the context of intracellular pathogens such as M. tuberculosis, which use sHSPs to survive various challenges (30). The present investigation shows that it should be possible to derive a specific inhibitor against the mycobacterial virulence factor Hsp16.3 without inhibiting functionally similar human counterparts such as alphaB-crystallin (25).
The binding affinity that is represented by the KD is in the range of 50 μM for both entities. The determination of KD was done by performing anisotropic titrations using fluoresceinated peptides. We have used a single site binding equation to obtain an estimate of KD for the peptides. In the case of peptide 10, the assumption that binding stoichiometry is one per subunit of Hsp16.3 seems to work well, as indicated by the low P value (0.001) associated with the determined KD. The quenching experiments also support such a model for peptide 10 since the stoichiometry was determined to be one binding site per subunit of Hsp16.3. In the case of peptide 22, the P value (0.023) of KD determination is relatively higher, which indicates a lower level of significance. It is possible that in the case of peptide 22 the binding may be explained by a more complex model. However, the true binding affinities could be higher than those obtained from anisotropic titrations, as is evident from the quenching experiment performed using peptide 10. Although a precise KD determination from this experiment was not feasible (the protein concentration should be much lower than the KD for its accurate determination), an approximate estimate revealed a value of ∼5 μM, indicating that the unmodified peptide possibly has a higher affinity. The higher KD obtained from anisotropic titrations may be due to steric hindrance contributed by the conjugated fluorophore (i.e., FITC).
Members of the alpha-HSP superfamily include three regions, a characteristic alpha-crystallin domain, which is flanked by a short variable C-terminal extension, and a poorly conserved N-terminal region (18). The alpha-HSPs are generally highly soluble proteins, which is expected since they are required to maintain the solubility of other proteins. However, during chaperone action, the hydrophobic surfaces (29) are exposed, allowing the possibility of loss of solubility. However, this does not happen because of the flexible C-terminal extensions (6), which serve as solubilizers. The C-terminal extensions are rich in polar amino acid residues, and any replacement that gives a hydrophobic character results in severe loss of not only chaperone function but also solubility (23). Solubility and chaperone function of alpha-HSPs are thus interlinked, and any agent that affects one is likely to affect the other. It is therefore not unexpected that the peptides, which cause inhibition of chaperone activity, also cause precipitation.
The loss of chaperone activity occurs at relatively low concentrations, whereas precipitation requires a higher concentration of both peptide and target. The effect of the peptides on the protein appears to be manifested at two levels. At lower concentrations these peptides bind to high-affinity binding sites, with KDs in the micromolar range, resulting in functional inhibition. This inhibition may be due to the peptides competing for the active site in a manner resembling histatins—antimicrobial peptides known to act as inhibitors by serving as pseudosubstrates or by tight binding to the active site, eliminating the accessibility to the natural substrates (11). Alternatively, the mode of action may resemble that of pyrrohocoricin, another antimicrobial peptide that binds not to the active site but to the regulatory region of DnaK, thereby inhibiting its function (20). At higher concentrations it appears that secondary sites are engaged, resulting in the loss of solubility. Since both peptides are highly cationic in nature, it is possible that, subsequent to binding, the peptides disrupt charge-charge interactions that are generally considered necessary for maintaining both the structural integrity and solubility of alpha-HSPs (5). Such a hypothesis accounts for the fact that peptide 22, which is more cationic, could precipitate out Hsp16.3, as well as the Hsp16.3-MDH complex, more efficiently than could peptide 10. However, the destabilization effect seems to have some degree of specificity, since the alphaB-crystallin-MDH complex was not affected by the presence of high concentrations of peptide 22.
The chemical shift changes involving His residues of peptide 10 suggest their involvement in the peptide-protein interactions. It may be noted here that peptide 22, like peptide 10, also contains several histidines, two of these being in aligned positions. When the Protein Information Resource database (3) was scanned for M. tuberculosis proteins carrying a His-X-X-X-His signature (where X stands for any amino acid), a large number of hits were found. Several of these contained signatures that closely resembled either the peptide 10 or peptide 22 sequences. It is speculated that some of these proteins may be the natural substrates of Hsp16.3. Thus, by this approach it may be possible to obtain clues regarding substrate specificities of not only Hsp16.3 but other members of the alpha-HSP family as well. In this context it may be mentioned that a similar approach based on screening phage display libraries had been used earlier to determine substrate preferences of DnaK (12).
The inhibitory effects described have been demonstrated by using in vitro assays. Since no other assay was available, chaperone function was targeted. As has been stressed earlier, Hsp16.3 may have other functions apart from chaperone activity. In that case, the in vitro assays may not reflect or only partly reflect the in vivo situation. A direct demonstration of antimycobacterial activity would thus be necessary. However, it must be emphasized that Hsp16.3 does not have a role to play in the viability of actively growing bacteria. It is only in dormancy that it plays a vital role in survival. Hence, to demonstrate the antimycobacterial activity, these peptides need to be tested with anaerobic models (28). The penetration of the lipid-rich cell envelopes of mycobacteria has always been a major impediment in developing drugs and therefore issues such as penetration of membrane barriers and stability in vivo need to be resolved. Antimicrobial peptides are common in nature (1, 11, 19). Most of these peptides are cationic and amphipathic. Some of these, such as defensins or maganins, function by acting on cell membranes, whereas others, such as histatins or NAP-2 are known to act as pseudo-substrate inhibitors as mentioned earlier. Pyrrhocoricin, the insect-derived antimicrobial peptide, is an interesting case (20). It binds DnaK of gram-negative bacteria without affecting the human counterpart. This is a remarkable example of a naturally occurring antibacterial peptide that targets a well-conserved protein in a species-selective manner. Peptides 10 and 22 thus appear to function in a manner similar to pyrrhocoricin since they, too, inhibit a relatively conserved protein. The absence of reactivity of these peptides toward the lens protein is an indication that although there are elements of conservation, the two proteins are sufficiently diverse to allow inactivation of one (Hsp16.3) without affecting the other (alphaB-crystallin). However, for a peptide to have antimicrobial property it should have some elementary properties (13); one of these is cationic character. It has been suggested that the cationic character helps in membrane disruption. Both of the peptide inhibitors reported here satisfy this criterion. The other important feature is amphipathicity (11), which is necessary for membrane permeability. Apparently, neither of these two peptides has the necessary amphipathicity, as is evident from a search conducted at one website (http://aps.unmc.edu/AP /main.php) (27). An alternative approach would be to look for homologous peptides, which are known to have antimicrobial activity, and to test their activity against Hsp16.3. Two such peptides that could be considered potential candidates are histatin 5 (AP00503) and histatin 8 (AP00523). They are both derived from histatins, which are histidine-rich peptides produced by the salivary glands of humans. It has been suggested that histatins may be used to combat Pseudomonas aeruginosa infections in the airways of cystic fibrosis patients (21). It would be interesting to study the effect of histatins on Hsp16.3 on one hand and on mycobacterial viability on the other.
A point that has been emphasized earlier is that inhibition has been demonstrated only in the context of chaperone activity. However, the protein could have other functions. Whether these other functions would also be inhibited remains to be seen, but it can be argued that the peptides were selected solely on the basis of binding activity; therefore, there is no reason to presume that inhibition would be targeted against one function, leaving aside the others. Even if that happens, it should be possible to expand the repertoire of binding sequences through further screening of phage display libraries. The peptide sequences described here may therefore be considered essentially as “leads.” Their efficacies may be further enhanced through biochemical/biophysical studies and modeling. Even if the peptide inhibitors do not actually evolve into effective drugs, they could be used as tools to understand better the function of Hsp16.3 in particular and alpha-HSPs in general.
Acknowledgments
We thank Juraj Ivanyii of MRC London for the Hsp16.3-expressing clone EC-16. We thank K. P. Das for giving us substantial amounts of recombinant alphaB-crystallin derived from a recombinant plasmid originally developed by W. Boelens and W. W. de Jong. We thank W. Boelens for consent to use the recombinant alphaB-crystallin protein for the comparative studies.
We thank CSIR, Government of India, for financial support under the New Millennium India Technology Leadership Initiative.
REFERENCES
- 1.Andreu, D., and L. Rivas. 1998. Animal antimicrobial peptides: an overview. Biopolymers 47:415-433. [DOI] [PubMed] [Google Scholar]
- 2.Appeldoorn, C. C., T. J. Molenaar, A. Bonnefoy, S. H. van Leeuwen, P. A. Vandervoort, M. F. Hoylaerts, T. J. van Berkel, and E. A. Biessen. 2003. Rational optimization of a short human P-selectin-binding peptide leads to nanomolar affinity antagonists. J. Biol. Chem. 278:10201-10207. [DOI] [PubMed] [Google Scholar]
- 3.Barker, W. C., J. S. Garavelli, H. Huang, P. B. McGarvey, B. C. Orcutt, G. Y. Srinivasarao, C. Xiao, L. L. Yeh, R. S. Ledley, J. F. Janda, F. Pfeiffer, H. Mewes, A. Tsugita, and C. Wu. 2000. The protein information resource (PIR). Nucleic Acids Res. 28:41-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3a.Barry, C., III. November2002. Method of attenuating pathogenic mycobacteria and strains of mycobacteria so attenuated. U.S. patent 6,403,100.
- 4.Bhattacharyya, T., A. Bhattacharyya, and S. Roy. 1991. A fluorescence spectroscopic study of glutaminyl-tRNA synthetase from Escherichia coli and its implications for the enzyme mechanism. Eur. J. Biochem. 200:739-745. [DOI] [PubMed] [Google Scholar]
- 5.Boelens, W. C., Y. Croes, M. de Ruwe, L. de Reu, and W. W. de Jong. 1998. Negative charges in the C-terminal domain stabilize the alphaB-crystallin complex. J. Biol. Chem. 273:28085-28090. [DOI] [PubMed] [Google Scholar]
- 6.Carver, J. A. 1999. Probing the structure and interactions of crystallin proteins by NMR spectroscopy. Prog. Retin. Eye Res. 18:431-462. [DOI] [PubMed] [Google Scholar]
- 7.Chang, Z., T. P. Primm, J. Jakana, I. H. Lee, I. Serysheva, W. Chiu, H. F. Gilbert, and F. A. Quiocho. 1996. Mycobacterium tuberculosis 16-kDa antigen (Hsp16.3) functions as an oligomeric structure in vitro to suppress thermal aggregation. J. Biol. Chem. 271:7218-7223. [PubMed] [Google Scholar]
- 8.Chattopadhyay, R., and S. Roy. 2002. DnaK-sigma 32 interaction is temperature dependent: implication for the mechanism of heat shock response. J. Biol. Chem. 277:33641-33647. [DOI] [PubMed] [Google Scholar]
- 9.Coates, A., Y. Hu, R. Bax, and C. Page. 2002. The future challenges facing the development of new antimicrobial drugs. Nat. Rev. Drug Discov. 1: 895-910. [DOI] [PubMed] [Google Scholar]
- 10.de Jong, W. W., J. A. Leunissen, and C. E. Voorter. 1993. Evolution of the alpha-crystallin/small heat-shock protein family. Mol. Biol. Evol. 10:103-126. [DOI] [PubMed] [Google Scholar]
- 11.Epand, R. M., and H. J. Vogel. 1999. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta 1462:11-28. [DOI] [PubMed] [Google Scholar]
- 12.Gragerov, A., L. Zeng, X. Zhao, W. Burkholder, and M. E. Gottesman. 1994. Specificity of DnaK-peptide binding. J. Mol. Biol. 235:848-854. [DOI] [PubMed] [Google Scholar]
- 13.Groisman, E. A. 1996. Bacterial responses to host-defense peptides. Trends Microbiol. 4:127-129. [DOI] [PubMed] [Google Scholar]
- 14.Lee, G. J., A. M. Roseman, H. R. Saibil, and E. Vierling. 1997. A small heat shock protein stably binds heat-denatured model substrates and can maintain a substrate in a folding-competent state. EMBO J. 16:659-671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin, S. X., Q. Wang, and Y. L. Wang. 1988. Interactions between Escherichia coli arginyl-tRNA synthetase and its substrates. Biochemistry 27:6348-6353. [DOI] [PubMed] [Google Scholar]
- 16.Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275. [PubMed] [Google Scholar]
- 17.Michele, T. M., C. Ko, and W. R. Bishai. 1999. Exposure to antibiotics induces expression of the Mycobacterium tuberculosis sigF gene: implications for chemotherapy against mycobacterial persistors. Antimicrob. Agents Chemother. 43:218-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narberhaus, F. 2002. Alpha-crystallin-type heat shock proteins: socializing minichaperones in the context of a multichaperone network. Microbiol. Mol. Biol. Rev. 66:64-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otvos, L., Jr. 2000. Antibacterial peptides isolated from insects. J. Pept. Sci. 6:497-511. [DOI] [PubMed] [Google Scholar]
- 20.Otvos, L., Jr., I. O, M. E. Rogers, P. J. Consolvo, B. A. Condie, S. Lovas, P. Bulet, and M. Blaszczyk-Thurin. 2000. Interaction between heat shock proteins and antimicrobial peptides. Biochemistry 39:14150-14159. [DOI] [PubMed] [Google Scholar]
- 21.Sajjan, U. S., L. T. Tran, N. Sole, C. Rovaldi, A. Akiyama, P. M. Friden, J. F. Forstner, and D. M. Rothstein. 2001. P-113D, an antimicrobial peptide active against Pseudomonas aeruginosa, retains activity in the presence of sputum from cystic fibrosis patients. Antimicrob. Agents Chemother. 45:3437-3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott, J. K., and G. P. Smith. 1990. Searching for peptide ligands with an epitope library. Science 249:386-390. [DOI] [PubMed] [Google Scholar]
- 23.Smulders, R. H. P. H., J. A. Carver, R. A. Lindner, M. A. van Boekel, H. Bloemendal, and W. W. de Jong. 1996. Immobilization of the C-terminal extension of bovine alphaA-crystallin reduces chaperone-like activity. J. Biol. Chem. 271:29060-29066. [DOI] [PubMed] [Google Scholar]
- 24.Stewart, G. R., B. D. Robertson, and D. B. Young. 2003. Tuberculosis: a problem with persistence. Nat. Rev. Microbiol. 1:97-105. [DOI] [PubMed] [Google Scholar]
- 25.Valdez, M. M., J. I. Clark, G. J. Wu, and P. J. Muchowski. 2002. Functional similarities between the small heat shock proteins Mycobacterium tuberculosis HSP 16.3 and human alphaB-crystallin. Eur. J. Biochem. 269:1806-1813. [DOI] [PubMed] [Google Scholar]
- 26.Verbon, A., R. A. Hartskeerl, A. Schuitema, A. H. Kolk, D. B. Young, and R. Lathigra. 1992. The 14,000-molecular-weight antigen of Mycobacterium tuberculosis is related to the alpha-crystallin family of low-molecular-weight heat shock proteins. J. Bacteriol. 174:1352-1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang, Z., and G. Wang. 2004. APD: the antimicrobial peptide database. Nucleic Acids Res. 32(database issue):D590-D592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wayne, L. G., and L. G. Hayes. 1996. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun. 64:2062-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang, H., S. Huang, H. Dai, Y. Gong, C. Zheng, and Z. Chang. 1999. The Mycobacterium tuberculosis small heat shock protein Hsp16.3 exposes hydrophobic surfaces at mild conditions: conformational flexibility and molecular chaperone activity. Protein Sci. 8:174-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan, Y., D. D. Crane, and C. E. Barry III. 1996. Stationary phase-associated protein expression in Mycobacterium tuberculosis: function of the mycobacterial alpha-crystallin homolog. J. Bacteriol. 178:4484-4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuan, Y., D. D. Crane, R. M. Simpson, Y. Q. Zhu, M. J. Hickey, D. R. Sherman, and C. E. Barry III. 1998. The 16-kDa alpha-crystallin (Acr) protein of Mycobacterium tuberculosis is required for growth in macrophages. Proc. Natl. Acad. Sci. USA 95:9578-9583. [DOI] [PMC free article] [PubMed] [Google Scholar]