

Abstract
Pseudohyphal and invasive growth in the yeast Saccharomyces cerevisiae is regulated by the kelch repeat-containing proteins Gpb1p and Gpb2p, which act downstream of the G protein α-subunit Gpa2p. Here we show that deletion of GPB1 and GPB2 causes increased haploid invasive growth in cells containing any one of the three protein kinase A (PKA) catalytic subunits, suggesting that Gpb1p and Gpb2p are able to inhibit each of these kinases. Cells containing gpb1Δ gpb2Δ mutations also display increased phosphorylation of the PKA substrates Sfl1p and Msn2p, indicating that Gpb1p and Gpb2p are negative regulators of PKA substrate phosphorylation. Stimulation of PKA-dependent signaling by gpb1Δ gpb2Δ mutations occurs in cells that lack both adenylyl cyclase and the high-affinity cyclic AMP (cAMP) phosphodiesterase. This effect is also seen in cells that lack the low-affinity cAMP phosphodiesterase. Given that these three enzymes control the synthesis and degradation of cAMP, these results indicate that the effect of Gpb1p and Gpb2p on PKA substrate phosphorylation does not occur by regulating the intracellular cAMP concentration. These findings suggest that Gpb1p and Gpb2p mediate their effects on the cAMP/PKA signaling pathway either by inhibiting the activity of PKA in a cAMP-independent manner or by activating phosphatases that act on PKA substrates.
Activation of protein kinase A (PKA) by cyclic AMP (cAMP) is a conserved feature of eukaryotic signaling systems. In the yeast Saccharomyces cerevisiae, PKA regulates cell growth and morphology in response to nutrient and stress signals (31). Yeast PKA functions downstream of the monomeric G proteins Ras1p and Ras2p, which are activated by an unknown mechanism (3). Ras1p and Ras2p stimulate adenylyl cyclase to produce cAMP (8, 9, 35), which activates PKA through the well-established mechanism of binding to the regulatory subunit of PKA and releasing active catalytic subunits (33). There are three isoforms of the catalytic subunit, called Tpk1p, Tpk2p, and Tpk3p, which are encoded by different genes (34). Deletion of the three genes encoding the catalytic subunits is lethal, but any one of the three genes can provide the essential function of PKA. There is one regulatory subunit, called Bcy1p, which is thought to bind to each of the three catalytic subunits (33). Activation of PKA results in phosphorylation of substrates that are involved in intermediary metabolism, stress responses, and filamentous growth (10, 31).
Yeast PKA also appears to function downstream of the G protein α-subunit Gpa2p (5, 17, 21). Gpa2p is coupled to a cell surface receptor, called Gpr1p, that contains seven membrane-spanning domains, a structure that is found in other G protein-coupled receptors (16, 36, 37). The effects conferred by a deletion of GPR1 are suppressed by constitutive activation of Gpa2p, as would be expected for a G protein that acts downstream of its coupled receptor (22, 30). In contrast to the situation with Ras proteins, little is known about the way in which the Gpa2p pathway regulates cAMP/PKA signaling. The effect of Gpa2p on PKA responses has recently been shown to involve the kelch repeat-containing proteins Gpb1p and Gpb2p (also called Krh2p and Krh1p, respectively) (1, 14). Gpb1p and Gpb2p physically interact with Gpa2p, suggesting that they function in the signaling pathway. Deletion of GPB1 and GPB2 results in phenotypes that are characteristic of increased PKA signaling, indicating that Gpb1p and Gpb2p inhibit a component of the cAMP/PKA pathway. Gpb1p and Gpb2p appear to transmit the signal generated by Gpa2p to downstream components, because gpb1Δ gpb2Δ mutations substantially suppress the defects in pseudohyphal and invasive growth conferred by a gpa2Δ mutation. Gpb1p and Gpb2p appear to act upstream of PKA, because the increase in signaling conferred by gpb1Δ gpb2Δ mutations is substantially blocked by deletion of TPK2, which encodes a PKA catalytic isoform. These results are consistent with a model in which activation of Gpa2p relieves the inhibition of PKA that is either directly or indirectly mediated by Gpb1p and Gpb2p, resulting in increased PKA activity.
Here we show that Gpb1p and Gpb2p affect signaling by altering the level of phosphorylation of PKA substrates. However, the function of Gpb1p and Gpb2p does not require changes in the intracellular concentration of cAMP. These results imply that the signaling process initiated by the Gpa2p α-subunit does not constitute a linear pathway that acts solely by stimulating the production of cAMP but rather has at least one component that acts downstream of adenylyl cyclase.
MATERIALS AND METHODS
Plasmid construction.
To construct a HIS3 disruption of TPK1, a 1.7-kb XbaI fragment from pHIS3-Bs.1 was cloned into the XbaI sites of plasmid pTPK1-50.1, which consists of vector YCp50 with a 1.7-kb insert containing the TPK1 gene, to produce plasmid ptpk1-1::HIS3. To construct a HIS3 disruption of TPK2, a 1.7-kb EcoRI-NotI fragment from pHIS3-Bs.2 was cloned into the EcoRI-NotI sites of plasmid pTPK2N-Bs.1 to produce plasmid ptpk2-2::HIS3. Plasmid pTPK2N-Bs.1 consists of a 2.0-kb BglII fragment containing the TPK2 gene cloned into the BamHI site of pBluescript, in which a NotI site was inserted before the stop codon by site-directed mutagenesis. To construct a HIS3 disruption of TPK3, a 1.7-kb EcoRI-XhoI fragment from pHIS3-Bs.1 was cloned into the EcoRI-XhoI sites of a plasmid with a 2.7-kb HindIII fragment containing the TPK3 gene to produce plasmid ptpk3-3::HIS3. Plasmid pCYR1.Bs contains a 4.5-kb KpnI-XbaI fragment from pCYR1 cloned into the KpnI-XbaI sites of pBluescript. To construct a URA3 disruption of CYR1, a 1.0-kb HindIII-NsiI fragment from pURA3.Bs (1) was cloned into the HindIII-NsiI sites of plasmid pCYR1.Bs to produce pcyr1::URA3.Bs.
Strain construction and media.
Strains used in the present study are listed in Table 1. Construction of strains containing the pde2::HIS3 allele (36) and the gpb1::URA3 and gpb2::HIS3 alleles (1) was described previously. The tpk1-1::HIS3 allele was made by transformation of cells with the 2.4-kb EcoRV-AatII fragment from plasmid ptpk1-1::HIS3. The tpk2-2::HIS3 allele was made by transformation of cells with the 2.6-kb SacI-SalI fragment from plasmid ptpk2-2::HIS3. The tpk3-3::HIS3 allele was made by transformation of cells with the 2.7-kb NcoI-EcoRV fragment from plasmid ptpk3-3::HIS3. The gpb2::TRP1 allele was made by transformation of a gpb2::HIS3 strain with a 3.8-kb SmaI/XhoI fragment from marker swap plasmid pHT6 (7). The cyr1::URA3 allele was made by transformation of cells with the 2.9-kb KpnI-XbaI fragment from plasmid pcyr1::URA3.Bs. The cyr1::TRP1 allele was made by transformation of cells with the 3.7-kb SmaI fragment from marker swap plasmid pUT11 (7). The pde1::kanMX allele was made by transformation of cells with a pde1::kanMX cassette amplified from genomic DNA of a strain containing this allele (Open Biosystems). Strain constructions involving transformations were confirmed by Southern blot or PCR.
TABLE 1.
Strains used in this study
| Straina | Genotype | Source |
|---|---|---|
| SKY762 | MATaura3-52 trp1::hisG leu2::hisG his3::hisG | S. Palecek and S. Kron |
| SKY762.p1k* | MATapde1::kanMX | This study |
| HS182-3B* | MATagpb1::URA3 gpb2::HIS3 | This study |
| HS182-3B.p1k* | MATagpb1::URA3 gpb2::HIS3 pde1::kanMX | This study |
| HS192-2C* | MATatpk2-2::HIS3 | This study |
| HS196-5A* | MATagpb1::URA3 gpb2::HIS3 tpk2-2::HIS3 | This study |
| HS222-3A* | MATα cyr1::URA3 pde2::HIS3 | This study |
| HS222-3A.T* | MATα cyr1::TRP1 pde2::HIS3 | This study |
| HS224-3D* | MATα gpb1::URA3 gpb2::HIS3 cyr1::URA3 pde2::HIS3 | This study |
| HS229-11D* | MATagpb1::URA3 gpb2::HIS3 tpk1-1::TRP1 tpk3-3::HIS3 | This study |
| HS230-15B* | MATatpk1-1::TRP1 tpk3-3::HIS3 | This study |
| HS231-29A* | MATagpb1::URA3 gpb2::HIS3 tpk2-2::TRP1 tpk3-3::HIS3 | This study |
| HS232-2B* | MATagpb1::URA3 gpb2::HIS3 tpk1-1::HIS3 tpk2-2::TRP1 | This study |
| HS233-8B* | MATatpk2-2::TRP1 tpk3-3::HIS3 | This study |
| HS234-1C* | MATatpk1-1::HIS3 tpk2-2::TRP1 | This study |
Strains with a SKY762 background are indicated with an asterisk. SKY762 is derived from Σ1278b (19).
Strains were grown on YEPD (yeast extract-peptone-dextrose) with 2% glucose, and strains under selection were grown on synthetic dropout media, as described previously (13).
Yeast methods and protein assays.
Invasive growth assays were performed according to the method of Kuchin et al. (18). Yeast transformations were performed by the lithium acetate method according to standard procedures (13).
Cell lysates for immunoblots were prepared from cells growing in log phase (optical density at 600 nm = 0.5 to 0.85). Cells were washed once with cold water and resuspended in lysis buffer, made essentially as described previously (27), with the exception that 25 mM β-glycerophosphate was substituted by 15 mM p-nitrophenyl phosphate. Cells were disrupted by shaking in the presence of glass beads for 5 min at 4°C and cleared by centrifugation for 3 min. For immunoblots, proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose. Blots were probed with the following antibodies: anti-myc 9E10 at a dilution of 1/1,000, anti-phospho-CREB (Ser133) (Cell Signaling) at a dilution of 1/1,000, and anti-green fluorescent protein (GFP) serum (Molecular Probes) at a dilution of 1/1,000. Secondary antibodies were donkey anti-rabbit immunoglobulin conjugated to horseradish peroxidase (Amersham) at a dilution of 1/10,000 and goat anti-mouse immunoglobulin conjugated to horseradish peroxidase (Amersham) at a dilution of 1/10,000. Immune complexes were detected with an ECL or an ECL Plus kit (Amersham).
Heat shock assays and yeast RNA extraction were performed as described previously (36) except that cells were incubated for 15 min at 50°C. Northern blots were performed as described previously (1).
RESULTS
Effect of gpb1Δ gpb2Δ mutations is not specific to Tpk2p.
Deletion of GPB1 and GPB2 causes an increase in diploid pseudohyphal growth and haploid invasive growth, phenotypes that are characteristic of increased activation of the cAMP/PKA signaling pathway (1, 14). It was therefore of interest to determine which pathway component is the target of inhibition by Gpb1p and Gpb2p. In yeast, total PKA activity results from the combined activities of the three catalytic subunits of cAMP-dependent kinase, Tpk1p, Tpk2p, and Tpk3p (34). To test the effects of gpb1Δ gpb2Δ mutations on the individual PKA catalytic isoforms, phenotypes associated with filamentous growth were investigated in strains in which two of the three TPK genes were deleted. Previous results have shown that TPK2 has a positive effect on filamentous growth and that TPK1 and TPK3 have negative effects on filamentous growth when TPK2 is present (26, 28). However, in cells containing either TPK1 only or TPK3 only, a small but reproducible increase in invasive growth was observed in gpb1Δ gpb2Δ cells compared to wild-type cells (Fig. 1A). The most straightforward interpretation of these results is that TPK1 and TPK3 have a modest positive effect on invasive growth in the absence of TPK2 and that GPB1 and GPB2 inhibit that effect. Expression of FLO11, which encodes a cell surface flocculin that is required for both pseudohyphal and invasive growth (20), was undetectable in strains containing either TPK1 only or TPK3 only (Fig. 1B, lanes 3, 4, 7, and 8), indicating that the signal generated in cells lacking TPK2 is quite low. Cells containing TPK2 only display very high levels of invasive growth and FLO11 RNA that were not affected by gpb1Δ gpb2Δ mutations (Fig. 1A and B, lanes 5 and 6). In this strain, the high level of Tpk2p activity is due to lack of inhibition by Tpk1p and Tpk3p. However, detectable FLO11 RNA abundance in wild-type cells depends on TPK2, and gpb1Δ gpb2Δ mutations increase the level of FLO11 RNA in wild-type cells (Fig. 1B, lanes 1 and 2). These results imply that the presence of GPB1 and GPB2 causes a decrease in Tpk2p activity. In summary, these findings are consistent with the idea that GPB1 and GPB2 are able to inhibit each of the PKA catalytic isoforms.
FIG. 1.

PKA activity is increased by gpb1Δ gpb2Δ mutations. (A) The following strains were patched onto YEPD-2.5% agar medium, incubated for 4 days at 25°C, and photographed before (Total growth) and after (Invasive growth) rubbing the surface of the plate with a glass rod under a stream of water: wild-type strain SKY762, gpb1Δ gpb2Δ strain HS182-3B, tpk1Δ tpk2Δ strain HS234-1C, gpb1Δ gpb2Δ tpk1Δ tpk2Δ strain HS232-2B, tpk1Δ tpk3Δ strain HS230-15B, gpb1Δ gpb2Δ tpk1Δ tpk3Δ strain HS229-11D, tpk2Δ tpk3Δ strain HS233-8B, and gpb1Δ gpb2Δ tpk2Δ tpk3Δ strain HS231-29A. (B) RNA isolated from the log-phase cultures of the strains described in panel A was transferred to nylon membrane, hybridized with a FLO11 probe, and rehybridized with an ACT1 probe.
PKA activity is increased in gpb1Δ gpb2Δ cells.
The experiments described above indicate that strains containing gpb1Δ gpb2Δ mutations display phenotypes that are characteristic of increased activation of PKA. However, these experiments do not demonstrate whether gpb1Δ gpb2Δ mutations affect the activity of PKA itself. The results are also consistent with the possibility that gpb1Δ gpb2Δ mutations affect the activity of a related kinase that regulates the same physiological processes as PKA, such as Sch9p (32). To test directly whether Tpk2p activity is affected by the presence of Gpb1p and Gpb2p, the phosphorylation state of a Tpk2p substrate was examined in different mutant backgrounds.
Expression of FLO11 is regulated by the transcriptional repressor Sfl1p, which is thought to be an in vivo substrate of Tpk2p based on the following observations. Sfl1p acts downstream of Tpk2p because deletion of SFL1 restores FLO11 expression in tpk2Δ mutant cells (27, 28). Sfl1p binds specifically to Tpk2p, and in vitro phosphorylation of Sfl1p by Tpk2p inhibits its binding to the FLO11 promoter (6, 27, 28). Finally, phosphorylation of Sfl1p in vivo requires the presence of Tpk2p (27). Phosphorylation of Sfl1p by Tpk2p can be detected as a change in mobility on a gel (6, 27, 28). In wild-type cells, deletion of GPB1 and GPB2 results in a significant increase in the phosphorylated form of Sfl1p (Fig. 2A, lanes 1 and 2). There was no detectable phosphorylated Sfl1p in cells containing a tpk2Δ mutation, and deletion of GPB1 and GPB2 had no effect in this mutant background (Fig. 2A, lanes 3 and 4).
FIG. 2.
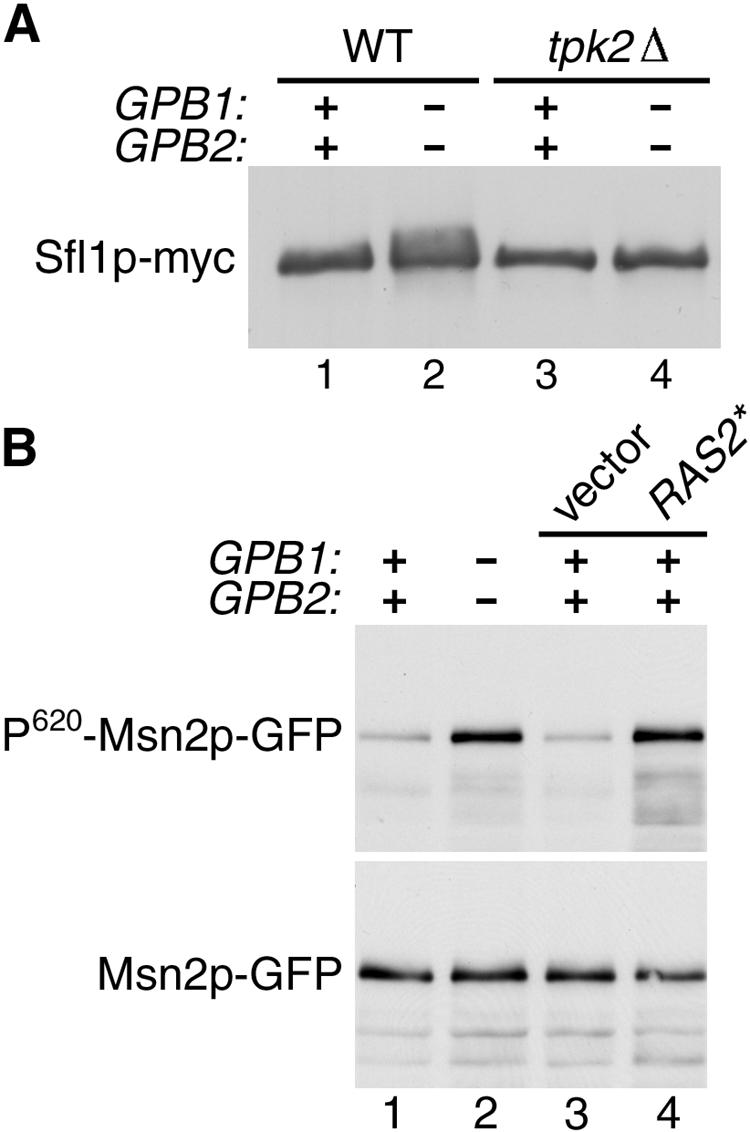
Phosphorylation of in vivo PKA substrates is increased by gpb1Δ gpb2Δ mutations. (A) Extracts made from the following strains expressing Sfl1p-myc from plasmid pXP181 (27) (provided by X. Pan and J. Heitman) were analyzed by SDS-PAGE and immunoblotting with anti-myc antibody 9E10: wild-type strain SKY762 (lane 1), gpb1Δ gpb2Δ strain HS182-3B (lane 2), tpk2Δ strain HS192-2C (lane 3), and gpb1Δ gpb2Δ tpk2Δ strain HS196-5A (lane 4). (B) Extracts made from the following strains expressing Msn2p-GFP fusion protein from plasmid pAdh1-Msn2-GFP (12) (provided by C. Schüller and W. Reiter) were analyzed by SDS-PAGE and immunoblotting with antibodies to P-CREB (S133) and GFP: wild-type strain SKY762 (lane 1), gpb1Δ gpb2Δ strain HS182-3B (lane 2), SKY762 carrying vector YCplac33 (lane 3), and SKY762 carrying plasmid YCp50-RAS2ala18val19 (lane 4).
One phenotype that is affected by all three forms of PKA is the response to stress, which is mediated, in part, by the PKA substrate Msn2p. Msn2p is a stress-activated transcription factor that is negatively regulated by PKA phosphorylation (11, 29). Msn2p is phosphorylated on serine 620 by PKA, and the phosphorylated epitope is recognized by an antibody specific for the phosphorylated form of mammalian CREB (12). Strains transformed with a construct expressing an Msn2p-GFP fusion protein were used to assay the level of Msn2p phosphorylation in different mutant backgrounds by comparing the signal obtained with the anti-phospho-CREB antibody to that obtained with an anti-GFP antibody. A substantial increase in the level of Msn2p phosphorylation at serine 620 was observed in cells containing gpb1Δ gpb2Δ mutations compared to wild-type cells (Fig. 2B, lanes 1 and 2). A pronounced effect on Msn2p phosphorylation was also seen in cells containing an activated allele of RAS2, which is known to increase PKA activity by stimulation of adenylyl cyclase activity (Fig. 2B, lanes 3 and 4). These results establish that gpb1Δ gpb2Δ mutations cause an increase in phosphorylation of PKA substrates in vivo at sites that are specifically phosphorylated by PKA.
Effect of gpb1Δ gpb2Δ mutations is independent of adenylyl cyclase and the high- and low-affinity cAMP phosphodiesterases.
The observation that gpb1Δ gpb2Δ mutations affect the phosphorylation level of PKA substrates raises the question of whether Gpb1p and Gpb2p function by inhibiting the activity of adenylyl cyclase. Adenylyl cyclase activates PKA by producing cAMP, which binds to the PKA regulatory subunit, resulting in the release of active PKA catalytic subunits. The PKA regulatory subunit, Bcy1p, is common to all three PKA catalytic isoforms (33). Therefore, if Gpb1p and Gpb2p were to function by inhibiting production of cAMP, all three PKA catalytic isoforms would be expected to be affected by deletion of GPB1 and GPB2. To test this idea directly, the effect of deleting GPB1 and GPB2 was determined in a strain containing a deletion of the CYR1 gene, which encodes adenylyl cyclase. The viability of cells containing a cyr1Δ mutation can be maintained by deletion of PDE2, a gene encoding a high-affinity cAMP phosphodiesterase (2). In these cells, viability is maintained by supplementing the medium with exogenous cAMP. Cells containing cyr1Δ pde2Δ mutations were viable when supplied with 1.5 mM cAMP but did not grow on 0.75 or 0.5 mM cAMP (Fig. 3, top panels). In contrast, cells lacking adenylyl cyclase and carrying gpb1Δ gpb2Δ mutations were viable on 0.75 mM cAMP. However, cyr1Δ pde2Δ gpb1Δ gpb2Δ cells were inviable on 0.5 mM cAMP. Wild-type and pde2Δ cells were viable at all concentrations of cAMP (data not shown). Therefore, gpb1Δ gpb2Δ mutations do not confer viability to cells containing very low levels of cAMP but are able to remediate the loss of viability at an intermediate concentration of cAMP. cyr1Δ pde2Δ cells containing either an activated allele of RAS2 or empty vector were viable when supplied with 1.5 mM cAMP but did not grow on 0.75 mM or 0.5 mM cAMP (Fig. 3, bottom panels). Therefore, activation of Ras has no effect in cells lacking adenylyl cyclase, a finding consistent with previous results showing that Ras2p directly activates adenylyl cyclase (35). The finding that deletion of GPB1 and GPB2 has an effect on cells that cannot synthesize cAMP eliminates the possibility that the inhibitory function of Gpb1p and Gpb2p acts solely on adenylyl cyclase.
FIG. 3.

Effect of gpb1Δ gpb2Δ mutations is independent of adenylyl cyclase and the high-affinity cAMP phosphodiesterase. Tenfold dilutions of the following strains were spotted onto medium containing the indicated concentrations of cAMP: HS222-3A (cyr1Δ pde2Δ) and HS224-3D (gpb1Δ gpb2Δ cyr1Δ pde2Δ) grown in synthetic complete medium containing 2 mM cAMP (upper panel), and HS222-3A.T (cyr1Δ pde2Δ) carrying vector YEp352 or plasmid YCp50-RAS2ala18val19 grown in medium lacking uracil and containing 2 mM cAMP (lower panel). Plates were photographed after 2 days of growth.
The level of cAMP in cells results from the combined effect of adenylyl cyclase-mediated synthesis of cAMP and phosphodiesterase-mediated degradation of cAMP. The results shown above demonstrate that gpb1Δ gpb2Δ mutations have an effect on cells that lack adenylyl cyclase and the high-affinity cAMP phosphodiesterase Pde2p. In addition to these enzymes, the level of cellular cAMP is regulated by the low-affinity cAMP phosphodiesterase Pde1p (23). To test the involvement of Pde1p in mediating the effect of the kelch repeat proteins, strains containing a pde1Δ mutation in combination with other mutations that affect cAMP levels were constructed. However, strains containing cyr1Δ pde1Δ pde2Δ mutations grew extremely slowly in several different cAMP concentrations, and they rapidly generated faster growing variants that were likely to contain suppressor mutations. In addition, strains containing cyr1Δ pde1Δ mutations were inviable at all concentrations of cAMP tested (data not shown). Therefore, the effect of gpb1Δ gpb2Δ mutations was tested in a strain containing only a pde1Δ mutation.
In strains containing a pde1Δ mutation, Msn2p phosphorylation at serine 620 was substantially increased in gpb1Δ gpb2Δ cells compared to GPB1 GPB2 cells (Fig. 4A, lanes 3 and 4). This effect is similar to that conferred by gpb1Δ gpb2Δ mutations in a wild-type strain (Fig. 4A, lanes 1 and 2). Therefore, Pde1p is not required for the decrease in Msn2p phosphorylation caused by the presence of Gpb1p and Gpb2p. To determine whether PDE1 is involved in the effect of gpb1Δ gpb2Δ mutations on a physiological phenotype that is controlled by PKA, a heat shock experiment was performed. Deletion of GPB1 and GPB2 results in a very large increase in heat shock sensitivity in a wild-type background (1). In strains containing a pde1Δ mutation, the effect of gpb1Δ gpb2Δ mutations on heat shock sensitivity was the same as that seen in wild-type strains (Fig. 4B). These results demonstrate that the inhibitory function of Gpb1p and Gpb2p does not act solely on the low-affinity cAMP phosphodiesterase Pde1p.
FIG. 4.

Effect of gpb1Δ gpb2Δ mutations is independent of the low-affinity cAMP phosphodiesterase. (A) Extracts made from the following strains expressing Msn2p-GFP fusion protein from plasmid pAdh1-Msn2-GFP were analyzed by SDS-PAGE and immunoblotting with antibodies to P-CREB (S133) and GFP: wild-type strain SKY762 (lane 1), gpb1Δ gpb2Δ strain HS182-3B (lane 2), pde1Δ strain SKY.p1k (lane 3), gpb1Δ gpb2Δ pde1Δ strain HS182-3B.p1k (lane 4). (B) The strains described in panel A were grown to saturation for 2 days in synthetic complete medium, incubated at 50°C for 15 min, and diluted and plated to determine the percent survival. Values for GPB1 GPB2 strains are represented by the open bars; values for gpb1Δ gpb2Δ strains are represented by the filled bars. Values shown are the mean and standard deviation from three independent experiments.
DISCUSSION
The kelch repeat-containing proteins Gpb1p and Gpb2p are negative regulators of the cAMP/PKA pathway that were identified on the basis of their interaction with the G protein α-subunit Gpa2p (1, 14). Here we show that deletion of GPB1 and GPB2 causes an increase in the phosphorylation level of physiologically relevant PKA substrates. These effects correlate with changes in PKA-dependent characteristics such as invasive growth and heat shock sensitivity. However, the increase in signaling conferred by gpb1Δ gpb2Δ mutations does not require any of the three enzymes known to be involved in cAMP metabolism, which include adenylyl cyclase and the high- and low-affinity cAMP phosphodiesterases. The most straightforward interpretation of these results is that Gpb1p and Gpb2p either directly or indirectly inhibit a component of the cAMP/PKA pathway that acts downstream of cAMP. In fact, signaling through Gpb1p and Gpb2p could account for the observation that there is a cAMP-independent mechanism for regulating PKA-dependent phenotypes (4). Potential targets of Gpb1p and Gpb2p include the PKA regulatory subunit, PKA catalytic subunits, and phosphatases that act on PKA substrates.
These results appear to contradict an earlier report showing that mutation of GPB1 and GPB2 causes changes in the intracellular concentration of cAMP, which would suggest that Gpb1p and Gpb2p modulate the activity of enzymes involved in cAMP metabolism (14). In the previous report, it was shown that wild-type cells and cells containing either a gpb1Δ or gpb2Δ mutation display a twofold increase in cAMP levels upon readdition of glucose to starved cells. However, whereas in wild-type cells the concentration of cAMP returns to its basal level after one minute, in mutant cells it remains at the elevated level for several minutes. In contrast, cells containing double gpb1Δ gpb2Δ mutations do not display a twofold increase in cAMP levels upon readdition of glucose to starved cells. Therefore, deletion of either GPB1 or GPB2 results in slightly higher cAMP levels, and deletion of both genes results in slightly lower cAMP levels. These observations are somewhat unexpected given that the defects conferred by individual gpb1Δ and gpb2Δ mutations are similar to, though less severe than, the defects conferred by double gpb1Δ gpb2Δ mutations for all other phenotypes tested, including pseudohyphal growth, invasive growth, FLO11 expression, heat shock sensitivity, sporulation efficiency, and glycogen accumulation (1, 14). Moreover, all of the observed phenotypes of gpb1Δ gpb2Δ cells are consistent with an increase in PKA activity, rather than a decrease. Given that the glucose-induced spike in cAMP levels is extremely sensitive to the nutritional status of the cells (5), it is possible that the reported differences between wild-type, gpb1Δ, gpb2Δ, and gpb1Δ gpb2Δ cells were caused by differences in the nutritional state of the cells in the culture sample used for the assay. Alternatively, the effects of GPB1 and GPB2 mutant alleles on cAMP levels could be due to a feedback mechanism that regulates the concentration of cAMP in response to PKA activity (25). In any case, the finding that loss of Gpb1p and Gpb2p causes significant effects in cyr1Δ pde2Δ and pde1Δ strains indicates that the kelch repeat proteins can function independently of cAMP metabolism.
The G protein α-subunit Gpa2p has been thought to mediate signaling through direct activation of adenylyl cyclase, like mammalian Gαs, based on the following observations. First, the glucose-induced spike in cAMP concentration is increased in cells that overexpress GPA2, and it is eliminated in cells that contain a gpa2Δ mutation when the cells are grown under certain nutritional conditions (5, 22, 24). Second, the addition of cAMP compensates for the defect in pseudohyphal growth conferred by a gpa2Δ mutation (17, 21). However, here we show that the Gpa2p-interacting proteins Gpb1p and Gpb2p affect signaling through the cAMP/PKA pathway by a process that is independent of cAMP metabolism. These observations can be reconciled if Gpa2p and the kelch repeat proteins impinge on the cAMP/PKA pathway at different points. Such a situation might be expected based on the complex relationship between the functions of Gpb1p, Gpb2p, and Gpa2p. For example, although it has been proposed that Gpb1p and Gpb2p bind to Gpa2p in a manner that mimics G protein β-subunits (14), deletion of GPB1 and GPB2 in either wild-type cells or in gpa2Δ cells causes a substantial increase in PKA-dependent phenotypes (1, 14). These results suggest that Gpb1p and Gpb2p inhibit PKA signaling in both the presence and the absence of Gpa2p. Therefore, one possibility is that the kelch repeat proteins act as effectors of the G protein. However, deletion of GPA2 in gpb1Δ gpb2Δ cells causes a two- to threefold decrease in FLO11 RNA abundance compared to that seen in gpb1Δ gpb2Δ cells. Similarly, gpa2Δ gpb1Δ gpb2Δ cells display a decreased level of pseudohyphal growth compared to gpb1Δ gpb2Δ cells. Therefore, part of the signal present in gpb1Δ gpb2Δ cells is dependent on Gpa2p. One possible model to explain these results is that Gpb1p and Gpb2p negatively regulate both Gpa2p and another component of the PKA signaling pathway that is downstream of adenylyl cyclase. Deletion of GPB1 and GPB2 activates both Gpa2p and the other target, resulting in very high signal generation. In a gpb1Δ gpb2Δ mutant background, deletion of GPA2 eliminates the part of the signal that originates with Gpa2p but leaves intact the part of the signal resulting from the other target of Gpb1p and Gpb2p. Direct activation of adenylyl cyclase by Gpa2p would be consistent with this model, if the other target of Gpb1p and Gpb2p acts downstream of adenylyl cyclase.
The relationship between Gpa2p and the kelch repeat proteins has also been investigated by using an assay that detects changes in the subcellular location of GFP-Gpb2p. A recent study showed that overexpression of GPA2 causes GFP-Gpb2p to accumulate at the cell periphery and that overexpression of the nonactivatable GPA2G299A allele has a similar effect on GFP-Gpb2p localization (15). These results are not consistent with the idea that Gpb1p and Gpb2p act as effectors of Gpa2p, because an effector would be expected to bind the GTP-bound form of a G protein α-subunit and the G299A version of Gpa2p is expected to be present predominantly in the GDP-bound form. However, that study did not investigate whether a constitutively active allele of GPA2 has any effect on GFP-Gpb2p localization. A comparison between the effects of nonactivatable and constitutively active versions of Gpa2p on Gpb2p localization would provide information about whether the function of Gpb1p and Gpb2p is more similar to that of Gpa2p effectors or G protein β-subunits.
Signaling through the Gpa2p pathway displays several unusual features. In contrast to essentially all known G protein-mediated systems, the Gpa2p α-subunit does not appear to form a heterotrimer with classical βγ-subunits. Moreover, here we show that the Gpa2p-interacting proteins Gpb1p and Gpb2p affect cAMP/PKA signaling at a step downstream of adenylyl cyclase. Gpb1p and Gpb2p may also have a function that negatively regulates Gpa2p activity. Given that kelch repeat-containing proteins of unknown function are present in all eukaryotes, it will be of interest to determine whether the alternative signaling mechanisms used in the Gpa2p pathway are also seen in other G protein-mediated pathways.
Acknowledgments
This study was supported by Grant-In-Aid 9951029T from the American Heart Association, Heritage Affiliate, and by grant GM60332 from the National Institutes of Health.
We thank S. Palecek, S. Kron, X. Pan, J. Heitman, C. Schüller, W.Reiter, and S. Garrett for plasmids and strains used in this study. We also thank S. Piñol-Roma for critical comments on the manuscript.
REFERENCES
- 1.Batlle, M., A. Lu, D. A. Green, Y. Xue, and J. P. Hirsch. 2003. Krh1p and Krh2p act downstream of the Gpa2p Gα subunit to negatively regulate haploid invasive growth. J. Cell Sci. 116:701-710. [DOI] [PubMed] [Google Scholar]
- 2.Boy-Marcotte, E., H. Garreau, and M. Jacquet. 1987. Cyclic AMP controls the switch between division cycle and resting state programs in response to ammonium availability in Saccharomyces cerevisiae. Yeast 3:85-93. [DOI] [PubMed] [Google Scholar]
- 3.Broach, J. R. 1991. RAS genes in Saccharomyces cerevisiae: signal transduction in search of a pathway. Trends Genet. 7:28-33. [DOI] [PubMed] [Google Scholar]
- 4.Cameron, S., L. Levin, M. Zoller, and M. Wigler. 1988. cAMP-independent control of sporulation, glycogen metabolism, and heat shock resistance in Saccharomyces cerevisiae. Cell 53:555-566. [DOI] [PubMed] [Google Scholar]
- 5.Colombo, S., P. Ma, L. Cauwenberg, J. Winderickx, M. Crauwels, A. Teunissen, D. Nauwelaers, J. H. de Winde, M.-F. Gorwa, D. Colavizza, and J. M. Thevelein. 1998. Involvement of distinct G-proteins, Gpa2 and Ras, in glucose- and intracellular acidification-induced cAMP signalling in the yeast Saccharomyces cerevisiae. EMBO J. 17:3326-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conlan, R. S., and D. Tzamarias. 2001. Sfl1 functions via the co-repressor Ssn6-Tup1 and the cAMP-dependent protein kinase Tpk2. J. Mol. Biol. 309:1007-1015. [DOI] [PubMed] [Google Scholar]
- 7.Cross, F. R. 1997. ‘Marker swap’ plasmids: convenient tools for budding yeast molecular genetics. Yeast 13:647-653. [DOI] [PubMed] [Google Scholar]
- 8.De Vendittis, E., A. Vitelli, R. Zahn, and O. Fasano. 1986. Suppression of defective RAS1 and RAS2 functions in yeast by an adenylate cyclase activated by a single amino acid change. EMBO J. 5:3657-3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Field, J., J. Nikawa, D. Broek, B. MacDonald, L. Rodgers, I. A. Wilson, R. A. Lerner, and M. Wigler. 1988. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol. Cell. Biol. 8:2159-2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gancedo, J. M. 2001. Control of pseudohyphae formation in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 25:107-123. [DOI] [PubMed] [Google Scholar]
- 11.Görner, W., E. Durchschlag, M. T. Martinez-Pastor, F. Estruch, G. Ammerer, B. Hamilton, H. Ruis, and C. Schüller. 1998. Nuclear localization of the C2H2 zinc finger protein Msn2p is regulated by stress and protein kinase A activity. Genes Dev. 12:586-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Görner, W., E. Durchschlag, J. Wolf, E. L. Brown, G. Ammerer, H. Ruis, and C. Schüller. 2002. Acute glucose starvation activates the nuclear localization signal of a stress-specific yeast transcription factor. EMBO J. 21:135-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guthrie, C., and G. R. Fink. 1991. Guide to yeast genetics and molecular biology. Academic Press, Inc., San Diego, Calif.
- 14.Harashima, T., and J. Heitman. 2002. The Gα protein gpa2 controls yeast differentiation by interacting with kelch repeat proteins that mimic Gβ subunits. Mol. Cell 10:163-173. [DOI] [PubMed] [Google Scholar]
- 15.Harashima, T., and J. Heitman. 2005. Gα subunit Gpa2 recruits kelch repeat subunits that inhibit receptor-G protein coupling during cAMP-induced dimorphic transitions in Saccharomyces cerevisiae. Mol. Biol. Cell 16:4557-4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kraakman, L., K. Lemaire, P. Ma, A. W. R. H. Teunissen, M. C. V. Donaton, P. Van Dijck, J. Winderickx, J. H. de Winde, and J. M. Thevelein. 1999. A Saccharomyces cerevisiae G-protein coupled receptor, Gpr1, is specifically required for glucose activation of the cAMP pathway during the transition to growth on glucose. Mol. Microbiol. 32:1002-1012. [DOI] [PubMed] [Google Scholar]
- 17.Kübler, E., H.-U. Mösch, S. Rupp, and M. P. Lisanti. 1997. Gpa2p, a G-protein α-subunit, regulates growth and pseudohyphal development in Saccharomyces cerevisiae via a cAMP-dependent mechanism. J. Biol. Chem. 272:20321-20323. [DOI] [PubMed] [Google Scholar]
- 18.Kuchin, S., V. K. Vyas, and M. Carlson. 2002. Snf1 protein kinase and the repressors Nrg1 and Nrg2 regulate FLO11, haploid invasive growth, and diploid pseudohyphal differentiation. Mol. Cell. Biol. 22:3994-4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu, H., C. A. Styles, and G. R. Fink. 1993. Elements of the yeast pheromone response pathway required for filamentous growth of diploids. Science 262:1741-1744. [DOI] [PubMed] [Google Scholar]
- 20.Lo, W. S., and A. M. Dranginis. 1998. The cell surface flocculin Flo11 is required for pseudohyphae formation and invasion by Saccharomyces cerevisiae. Mol. Biol. Cell 9:161-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lorenz, M. C., and J. Heitman. 1997. Yeast pseudohyphal growth is regulated by GPA2, a G protein α homolog. EMBO J. 16:7008-7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lorenz, M. C., X. Pan, T. Harashima, M. E. Cardenas, Y. Xue, J. P. Hirsch, and J. Heitman. 2000. The G protein-coupled receptor Gpr1 is a nutrient sensor that regulates pseudohyphal differentiation in Saccharomyces cerevisiae. Genetics 154:609-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma, P., S. Wera, P. Van Dijck, and J. M. Thevelein. 1999. The PDE1-encoded low-affinity phosphodiesterase in the yeast Saccharomyces cerevisiae has a specific function in controlling agonist-induced cAMP signaling. Mol. Biol. Cell 10:91-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakafuku, M., T. Obara, K. Kaibuchi, I. Miyajima, A. Miyajima, H. Itoh, S. Nakamura, K. Arai, K. Matsumoto, and Y. Kaziro. 1988. Isolation of a second yeast Saccharomyces cerevisiae gene (GPA2) coding for guanine nucleotide-binding regulatory protein: studies on its structure and possible functions. Proc. Natl. Acad. Sci. USA 85:1374-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikawa, J., S. Cameron, T. Toda, K. M. Ferguson, and M. Wigler. 1987. Rigorous feedback control of cAMP levels in Saccharomyces cerevisiae. Genes Dev. 1:931-937. [DOI] [PubMed] [Google Scholar]
- 26.Pan, X., and J. Heitman. 1999. Cyclic AMP-dependent protein kinase regulates pseudohyphal differentiation in Saccharomyces cerevisiae. Mol. Cell. Biol. 19:4874-4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan, X., and J. Heitman. 2002. Protein kinase A operates a molecular switch that governs yeast pseudohyphal differentiation. Mol. Cell. Biol. 22:3981-3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robertson, L. S., and G. R. Fink. 1998. The three yeast A kinases have specific signaling functions in pseudohyphal growth. Proc. Natl. Acad. Sci. USA 95:13783-13787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith, A., M. P. Ward, and S. Garrett. 1998. Yeast PKA represses Msn2p/Msn4p-dependent gene expression to regulate growth, stress response and glycogen accumulation. EMBO J. 17:3556-3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tamaki, H., T. Miwa, M. Shinozaki, M. Saito, C.-W. Yun, K. Yamamoto, and H. Kumagai. 2000. GPR1 regulates filamentous growth through FLO11 in yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 267:164-168. [DOI] [PubMed] [Google Scholar]
- 31.Thevelein, J. M., and J. H. de Winde. 1999. Novel sensing mechanisms and targets for the cAMP-protein kinase A pathway in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 33:904-918. [DOI] [PubMed] [Google Scholar]
- 32.Toda, T., S. Cameron, P. Sass, and M. Wigler. 1988. SCH9, a gene of Saccharomyces cerevisiae that encodes a protein distinct from, but functionally and structurally related to, cAMP-dependent protein kinase catalytic subunits. Genes Dev. 2:517-527. [DOI] [PubMed] [Google Scholar]
- 33.Toda, T., S. Cameron, P. Sass, M. Zoller, J. D. Scott, B. McMullen, M. Hurwitz, E. G. Krebs, and M. Wigler. 1987. Cloning and characterization of BCY1, a locus encoding a regulatory subunit of the cAMP-dependent protein kinase in yeast. Mol. Cell. Biol. 7:1371-1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toda, T., S. Cameron, P. Sass, M. Zoller, and M. Wigler. 1987. Three different genes in S. cerevisiae encode the catalytic subunits of the cAMP-dependent protein kinase. Cell 50:277-287. [DOI] [PubMed] [Google Scholar]
- 35.Toda, T., I. Uno, T. Ishikawa, S. Powers, T. Kataoka, D. Broek, S. Cameron, J. Broach, K. Matsumoto, and M. Wigler. 1985. In yeast, RAS proteins are controlling elements of adenylate cyclase. Cell 40:27-36. [DOI] [PubMed] [Google Scholar]
- 36.Xue, Y., M. Batlle, and J. P. Hirsch. 1998. GPR1 encodes a putative G protein-coupled receptor that associates with the Gpa2p Gα subunit and functions in a Ras-independent pathway. EMBO J. 17:1996-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yun, C.-W., H. Tamaki, R. Nakayama, K. Yamamoto, and H. Kumagai. 1997. G-protein coupled receptor from yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 240:287-292. [DOI] [PubMed] [Google Scholar]