

Abstract
Transmission/disequilibrium tests have attracted much attention in genetic studies of complex traits because (a) their power to detect genes having small to moderate effects may be greater than that of other linkage methods and (b) they are robust against population stratification. Highly polymorphic markers have become available throughout the human genome, and many such markers can be studied within short physical distances. Studies using multiple tightly linked markers are more informative than those using single markers. However, such information has not been fully utilized by existing statistical methods, resulting in possibly substantial loss of information in the identification of genes underlying complex traits. In this article, we propose novel statistical methods to analyze multiple tightly linked markers. Simulation studies comparing our methods versus existing methods suggest that our methods are more powerful. Finally, we apply the proposed methods to study genetic linkage between the dopamine D2 receptor locus and alcoholism.
Introduction
The lack of success, by either model-dependent parametric methods or model-independent allele-sharing methods, in the identification of genes for complex traits has led researchers to question whether such studies have enough power to detect genes with small to moderate effects (Risch and Merikangas 1996). Although case-control association studies commonly have been used to study the association between diseases and candidate genes, there is always the possibility of population stratification as a cause of the observed association. This is especially a concern for studies in heterogeneous populations, such as the population in the United States.
To reduce the effects of population stratification, many family-based association methods have been proposed (Rubinstein et al. 1981; Falk and Rubinstein 1987; Ott 1989; Terwilliger and Ott 1992; Spielman et al. 1993; Thomson 1995). Although some of these methods are not robust to population stratification, the transmission/disequilibrium test (TDT), introduced by Spielman et al. (1993), is a valid test for linkage in structured populations, irrespective of whether the families are simplex, multiplex, or multigenerational (Spielman and Ewens 1996). Power studies have shown that, for the detection of linkage of complex traits, the TDT may have greater power than do allele-sharing methods (Risch and Merikangas 1996).
With the rapid progress in the Human Genome Project, many genetic markers can now be identified and genotyped within a very short physical distance, and the study of multiple markers will be likely to yield more genetic information than the study of single markers. However, as we will illustrate in the next section, available statistical methods either are not able to analyze multiple markers simultaneously or have been developed under assumptions that are not met by real data. To take full advantage of multiple tightly linked markers, we propose novel statistical methods to analyze multisite parental transmission data. We first review available methods that can be used to analyze multiple tightly linked markers, and we point out their deficiencies in the handling of real data. We then describe our approach for analysis of such data. The new methods are compared with the existing methods through simulation studies, and they are then applied to the study of genetic linkage between the dopamine D2 receptor locus (DRD2) and alcoholism.
Methods
In this section, we first will describe existing methods that can be used to analyze multiple markers, point out their limitations, and then propose new methods with which to simultaneously analyze tightly linked markers.
Method of Lazzeroni and Lange (1998)
When multiple markers within a candidate region are studied, one strategy would be to analyze each marker separately and then, by the Bonferroni correction, adjust for multiple comparisons, to obtain an overall statistical significance level for linkage. Lazzeroni and Lange (1998) suggested the following method, which is less conservative than the standard Bonferroni correction for multiple tests. Suppose that the TDT is conducted at m markers 𝒜1,𝒜2,…,𝒜m. Denote the test statistic at marker 𝒜i as Ti, and denote the corresponding P value as pi. The adjusted P value defined by Lazzeroni and Lange (1998) is 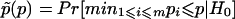 , where H0 is the combined null hypothesis that there is no linkage at any one of the markers. In the following discussion, we denote this single-marker–based testing procedure as Ts.
, where H0 is the combined null hypothesis that there is no linkage at any one of the markers. In the following discussion, we denote this single-marker–based testing procedure as Ts.
This approach ignores possible dependence among the markers, and such dependence may provide valuable information for linkage. Consider a hypothetical two-marker system with alleles A and a at marker 𝒜 and with alleles B and b at marker ℬ. Suppose that each of the four haplotypes (AB, ab, Ab, and aB) has an equal frequency in the population. If having haplotype Ab or aB increases the disease risk, and if having haplotype AB or ab reduces the disease risk, then the TDT applied to each marker separately would reveal no evidence for linkage, although strong evidence would be likely to emerge from a joint analysis.
Ambiguities in Haplotypes for Multilocus Data
Sethuraman (1997), Wilson (1997), and Clayton and Jones (1999) proposed TDTs that use multiple markers jointly. Their methods assume that the haplotypes are known in the parents and are not applicable to haplotype-unknown data. However, for data collected on nuclear families, haplotypes in the parents may not be uniquely resolved. In our genetic studies of alcoholism, three RFLPs spanning 30 kb within the DRD2 locus were genotyped: TaqIB, TaqID, and TaqIA. It is known that linkage disequilibrium exists across this locus (Kidd et al. 1998). We denote the alleles at each marker by integers. Consider the following family:
 |
Under the reasonable assumption of no recombinations among these markers in this family, the following two haplotype scenarios, (A) and (B), are both compatible with the observed set of individual site genotypes:
 |
or
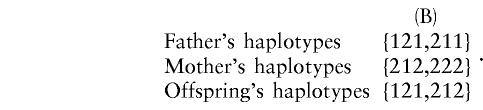 |
The probabilities of scenarios (A) and (B) in the example given above depend on many parameters related to the population structure under study, as well as on parameters related to the disease model. In general, the two scenarios do not have the same probability. As has been pointed out by Dudbridge et al. (2000), a necessary condition for haplotype ambiguity is that there is a locus for which both parents and offspring have the same heterozygous genotype and that there is another locus for which both parents and offspring do not have the same homozygous genotype. Unless there is complete disequilibrium among the markers, such that the testing of additional markers does not increase the number of ambiguous families, the proportion of ambiguous families increases with the number of markers studied.
Method of Clayton (1999)
Clayton (1999) has proposed to estimate haplotype frequencies and to construct a likelihood that considers all possible solutions. However, his method is not robust to population stratification, which is not in keeping with the basic principle for family-based association studies.
Method of Dudbridge et al. (2000)
In a recent report, Dudbridge et al. (2000) have proposed an unbiased test for individual haplotypes, by calculation of the correct variance for the transmission count within a family, using information from multiple siblings if the latter are available. However, families with ambiguous haplotypes have to be discarded from the analysis, resulting in loss of information.
Proposed Methods
Let
 |
When there is no linkage between the marker and the disease genes and there is no segregation distortion, Pik,jl=Pjl,ik. If the transmission patterns are not gender specific; that is, if there is no difference between maternal transmission and paternal transmission, then Pik,jl=Pki,lj. If the haplotypes in each parent could be identified, TDTs could be carried out, on the basis of the following h×h transmission/nontransmission table T:
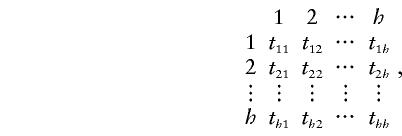 |
where tγδ is the number of parents with haplotypes HγHδ who transmit Hγ to the affected offspring and where h is the total number of possible haplotypes. We use different subscripts here to make it clear that the transmission/nontransmission table is constructed by pooling, in the same table, the contributions from both parents. One test that can be derived from the data in this table is
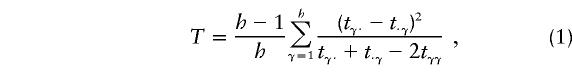 |
where 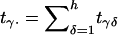 and
and 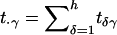 (Spielman and Ewens 1996). This statistic is a test for marginal homogeneity; that is, the γth-row sum in the table is the same as the γth-column sum in this table, for every γ=1,…,h. As noted by Schaid (1996), Sham (1997), and Lazzeroni and Lange (1998), this test statistic may not have a χ2 distribution with k-1 df. However, simulation methods can be used to assess the statistical significance of the observed test statistic.
(Spielman and Ewens 1996). This statistic is a test for marginal homogeneity; that is, the γth-row sum in the table is the same as the γth-column sum in this table, for every γ=1,…,h. As noted by Schaid (1996), Sham (1997), and Lazzeroni and Lange (1998), this test statistic may not have a χ2 distribution with k-1 df. However, simulation methods can be used to assess the statistical significance of the observed test statistic.
Because of the ambiguities in the parental haplotypes, the tγδ values are not directly observable for all families, and the desired table shown above cannot be derived. Instead, we observe only sets of genotypes g=1,…,G, where G is the number of distinct sets of genotypes across all markers. Here each set of genotypes g refers to the observed genotypes of the individual markers of the two parents and the affected offspring. Let {ik,jl} denote the event that the transmitted haplotype in the father is Hi and the nontransmitted haplotype is Hj and that the transmitted haplotype in the mother is Hk and the nontransmitted haplotype is Hl. In the discussion that follows, we designate {ik,jl} as one haplotype group. Suppose that the haplotype groups {isks,jsls} all correspond to the same set of genotypes g. Then the probability for this set of genotypes g is 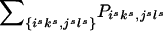 . For an arbitrary set of haplotype frequencies {hi}, we can construct a transmission/nontransmission table
. For an arbitrary set of haplotype frequencies {hi}, we can construct a transmission/nontransmission table 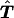 whose expectation is symmetrical under the null hypothesis of no linkage, as follows:
whose expectation is symmetrical under the null hypothesis of no linkage, as follows:
-
1. Suppose that haplotype group {ik,jl} is compatible with the set of genotypes g and that the number of families with the set of genotypes g is ng; then, define
where {isks,jsls}∈g denotes that haplotype group {isks,jsls} is compatible with the set of genotypes g. The value of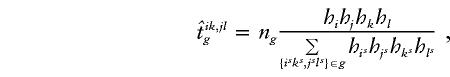
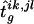 is the estimated number of families in which the father has haplotypes {Hi,Hj} and transmits Hi and in which the mother has haplotypes {Hk,Hl} and transmits Hk, for the set of haplotype frequencies {hi}.
is the estimated number of families in which the father has haplotypes {Hi,Hj} and transmits Hi and in which the mother has haplotypes {Hk,Hl} and transmits Hk, for the set of haplotype frequencies {hi}. -
2. The reconstructed table
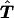 is
is
The value of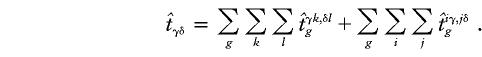
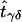 is the estimated number of parents who have haplotypes {Hγ,Hδ} and who transmit Hγ to the affected offspring. Under the null hypothesis of no linkage, the expected unobservable “true” T is symmetrical; that is, Pγ,δ=Pδ,γ, where Pγ,δ=E(tγδ) and Pδ,γ=E(tδγ). In Appendix A, we prove that, for an arbitrary set of haplotype frequencies, the expected transmission/nontransmission table
is the estimated number of parents who have haplotypes {Hγ,Hδ} and who transmit Hγ to the affected offspring. Under the null hypothesis of no linkage, the expected unobservable “true” T is symmetrical; that is, Pγ,δ=Pδ,γ, where Pγ,δ=E(tγδ) and Pδ,γ=E(tδγ). In Appendix A, we prove that, for an arbitrary set of haplotype frequencies, the expected transmission/nontransmission table 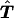 constructed by use of the approach discussed above is also symmetrical; that is,
constructed by use of the approach discussed above is also symmetrical; that is, 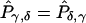 , where
, where 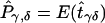 and
and 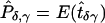 .
.
Therefore, to test linkage, we can test symmetry for the reconstructed transmission/nontransmission table 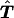 . The symmetry of the table
. The symmetry of the table 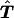 will be tested in the following discussion, by use of the marginal-homogeneity test statistic (1). Because the matrix
will be tested in the following discussion, by use of the marginal-homogeneity test statistic (1). Because the matrix 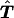 is symmetrical under the null hypothesis of no linkage, regardless of the choice of the hi, particular choices of hi affect only the power—and not the validity—of our proposed TDT. We consider three counting schemes to estimate haplotype frequencies. Let YdHiHj=1 if haplotypes HiHj in the father of the dth nuclear family are compatible with the observed set of genotypes g and if Hi is the transmitted haplotype; that is, haplotype group {ik,jl} is compatible with g for some k and l. Let YdHiHj=0 otherwise. Let XdHiHj be similarly defined for the mother. Also, let cd denote the number of haplotype groups compatible with the observed set of genotypes for the dth family. The three different counting schemes for assignment of haplotype frequencies are as follows:
is symmetrical under the null hypothesis of no linkage, regardless of the choice of the hi, particular choices of hi affect only the power—and not the validity—of our proposed TDT. We consider three counting schemes to estimate haplotype frequencies. Let YdHiHj=1 if haplotypes HiHj in the father of the dth nuclear family are compatible with the observed set of genotypes g and if Hi is the transmitted haplotype; that is, haplotype group {ik,jl} is compatible with g for some k and l. Let YdHiHj=0 otherwise. Let XdHiHj be similarly defined for the mother. Also, let cd denote the number of haplotype groups compatible with the observed set of genotypes for the dth family. The three different counting schemes for assignment of haplotype frequencies are as follows:
-
1. Haplotype frequencies are estimated by use of families with unambiguous haplotypes; that is,
where ncd=1 is the number of unambiguous families. The test statistic derived from this counting scheme is denoted as Tu.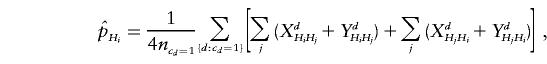
-
2. Haplotype frequencies are estimated by use of both unambiguous families and ambiguous families, where the haplotype groups compatible with the observed set of genotypes in each ambiguous family are assigned equal weight; that is,
where n is the total number of families. The test statistic derived from this counting scheme is denoted as Tc.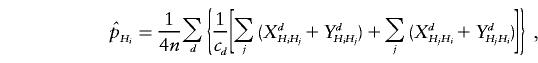
-
3.
Haplotype frequencies are estimated by treating all parents as a random sample of unrelated individuals from a population with Hardy-Weinberg equilibrium. Under this assumption, maximum-likelihood estimates of haplotype frequencies can be obtained by the expectation-maximization algorithm (Hawley and Kidd 1995). The test statistic derived from this counting scheme is denoted as Tml.
Other Approaches to Resolution of Ambiguities
Given the uncertainty with regard to parental haplotypes, one approach is to analyze only those families in which unambiguous haplotypes can be inferred in the parents. In Appendix B we show that, when we construct the transmission/nontransmission table, the discarding of ambiguous families will result in a symmetrical table. Therefore, we can test the symmetry of the reconstructed table for genetic linkage, and the resulting test is unbiased if the statistical significance level is controlled by use of the simulation procedure described below. We denote this multilocus test statistic as Td. However, as the number of markers increases, a substantial number of families may have to be discarded from the analysis, resulting in a potential loss of information.
An alternative method is to assign to each ambiguous family its most likely haplotype group under the homogenous-population assumption. This procedure works as follows. For any set of haplotype frequencies {hi}, suppose that the haplotype groups {isks,jsls} are all compatible with the observed set of genotypes g. The probability of each possible haplotype group under Hardy-Weinberg equilibrium and random mating is proportional to hishjshkshls. We may choose the haplotype group that has the largest probability, and we reconstruct the table by assigning to this haplotype group all families with the observed set of genotypes g; that is, 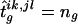 . In Appendix C, we show that this procedure also results in a symmetrical transmission/nontransmission table. Therefore, statistical tests based on this table are unbiased if the statistical significance level is appropriately controlled by use of the randomization procedure described below.
. In Appendix C, we show that this procedure also results in a symmetrical transmission/nontransmission table. Therefore, statistical tests based on this table are unbiased if the statistical significance level is appropriately controlled by use of the randomization procedure described below.
Simulation Results
In this section, we compare our methods versus existing methods, through simulations. Because the entries in table  are calculated on the basis of the observed genotype data and are based on a set of haplotype frequencies, the cell counts in the table are not independent. Therefore, standard asymptotic distributions will not be valid. To avoid possible bias, we estimate the significance level of the test statistics, using the following randomization procedure, by generating many sets of simulated samples. Each simulated sample is obtained by randomly assigning to each affected offspring, with equal chance, either the observed genotypes at all sites or the nontransmitted genotypes at all sites. The test statistics are calculated for each simulated sample. The statistical significance level of the observed test statistics can be estimated by comparison of the observed values versus the test statistic values evaluated on the basis of the simulated samples. For example, for the example discussed before, in which there are two compatible haplotype groups, the randomization procedure will generate, with equal probability, the following two types of family trios:
are calculated on the basis of the observed genotype data and are based on a set of haplotype frequencies, the cell counts in the table are not independent. Therefore, standard asymptotic distributions will not be valid. To avoid possible bias, we estimate the significance level of the test statistics, using the following randomization procedure, by generating many sets of simulated samples. Each simulated sample is obtained by randomly assigning to each affected offspring, with equal chance, either the observed genotypes at all sites or the nontransmitted genotypes at all sites. The test statistics are calculated for each simulated sample. The statistical significance level of the observed test statistics can be estimated by comparison of the observed values versus the test statistic values evaluated on the basis of the simulated samples. For example, for the example discussed before, in which there are two compatible haplotype groups, the randomization procedure will generate, with equal probability, the following two types of family trios:
 |
and
 |
The test statistic is evaluated for each randomized sample. The empirical distribution of the test statistics from these randomized samples is then used to estimate the significance level of the observed test statistic.
Statistical Tests
In our simulation studies, we compare the five test statistics discussed in the Methods section: Ts, Td, Tu, Tc, and Tml. In addition, we also consider the multilocus test statistic, Th, which is calculated under the assumption that haplotypes in the parents could be identified for all the families. The power of Th represents the best power achievable with the collected families. These test statistics are summarized in table 1.
Table 1.
Summary of Test Statistics Compared in This Article
| Test Statistic | Description |
| Ts | Studies each marker separately |
| Td | Discards ambiguous families |
| Th | Assumes that haplotype information is known |
| Tu | Estimates haplotype frequencies only by use of unambiguous families |
| Tc | Estimates haplotype frequencies by use of both unambiguous families and ambiguous families, by assigning each compatible haplotype group equal probability for each ambiguous family |
| Tml | Estimates haplotype frequencies by assuming that parents are a random sample of individuals from a population with Hardy-Weinberg equilibrium |
Simulation Models
In our simulations, we consider a variety of genetic models. The parameters include the number of populations (NP=1 or 2), the attributable risk of the genetic system in each population (AR=0%, 10%, 15%, or 20%), the relative risk for the high-risk genotypes (r=2, 4, or 10), and the genetic model (dominant or recessive). Schaid (1996) studied similar simulation models and described how to calculate haplotype frequencies on the basis of the model parameters. For each population, we assume Hardy-Weinberg equilibrium and random mating and that the families are ascertained through one affected offspring. For each simulation model, 2,000 independent samples are generated in our study of type I errors and power. For each sample, the six test statistics are calculated. In our study, the statistical significance levels are estimated by the randomization procedure, on the basis of 2,000 randomly generated samples for type I error rates and on the basis of 20,000 randomly generated samples for power comparisons.
Type I Errors
We first verify that all the statistical tests have the correct nominal false-positive rates. In our simulations, we consider a three-marker system, with each marker having two alleles. There are eight haplotypes for this system: 111, 112, 121, 122, 211, 212, 221, and 222, with 111 and 222 considered as group I and with the other six haplotypes considered as group II. The haplotypes within each group are assumed to have the same haplotype frequency. We assume that the families are ascertained from two populations, with equal probability. In the first population, the frequency of each haplotype in group I is .10 (1/10), the frequency of each haplotype in group II is .13 (2/15), and all genotypes have the same risk for the disease. For the second population, we vary the frequency of each haplotype in group I (q=.1, .2, .3, and .4). We assume that all genotypes also have the same risk in the second population, but this common risk relative to the common disease risk in the first population is varied: r=2, 3, or 4. We also vary the number of families ascertained from these two populations. In table 2, we summarize the estimated type I error rates for all six statistical tests, for each model and sample size. The statistical significance level is set at .005. This level of significance is appropriate if a candidate gene is studied. However, a more stringent criterion is needed if a genomewide search is performed (e.g., see Risch and Merikangas 1996). We choose this significance level here because our main purpose is to demonstrate the validity of the testing procedures and because a more stringent level would require much more extensive simulation efforts. For 2,000 replicated samples, the standard error for the type I error rate estimate is 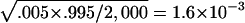 when the true error rate is at the nominal level (.005). We can see from this table that the estimated type I error rates are not statistically significantly different from the nominal level.
when the true error rate is at the nominal level (.005). We can see from this table that the estimated type I error rates are not statistically significantly different from the nominal level.
Table 2.
Observed Type I Error Rates for Different Sample Sizes and Different Population Structures
|
Type I Error Rate, for(%) |
||||||||||||
|
N=100 |
N=200 |
|||||||||||
| r | Ts | Td | Th | Tu | Tc | Tml | Ts | Td | Th | Tu | Tc | Tml |
| q=.1: | ||||||||||||
| 2 | .5 | .35 | .35 | .35 | .3 | .35 | .35 | .35 | .3 | .3 | .3 | .35 |
| 3 | .65 | .6 | .65 | .65 | .55 | .55 | .35 | .45 | .5 | .45 | .45 | .45 |
| 4 | .4 | .4 | .5 | .5 | .4 | .45 | .45 | .6 | .4 | .4 | .4 | .4 |
| q=.2: | ||||||||||||
| 2 | .6 | .6 | .4 | .6 | .55 | .55 | .6 | .55 | .55 | .3 | .3 | .35 |
| 3 | .6 | .65 | .5 | .65 | .65 | .6 | .7 | .4 | .55 | .7 | .65 | .65 |
| 4 | .65 | .45 | .4 | .35 | .35 | .35 | .6 | .4 | .5 | .6 | .65 | .6 |
| q=.3: | ||||||||||||
| 2 | .55 | .55 | .55 | .6 | .5 | .55 | .8 | .3 | .35 | .35 | .3 | .35 |
| 3 | .65 | .35 | .65 | .5 | .5 | .5 | .65 | .5 | .55 | .6 | .6 | .6 |
| 4 | .35 | .4 | .55 | .35 | .3 | .35 | .45 | .35 | .35 | .35 | .4 | .35 |
| q=.4: | ||||||||||||
| 2 | .6 | .5 | .5 | .4 | .45 | .4 | .7 | .55 | .6 | .65 | .6 | .6 |
| 3 | .45 | .65 | .5 | .45 | .5 | .5 | .45 | .35 | .35 | .35 | .3 | .35 |
| 4 | .7 | .4 | .55 | .6 | .6 | .6 | .65 | .5 | .4 | .45 | .4 | .45 |
Power Comparisons
Here we describe the results from our power study using samples from a homogeneous population. We also assume a three-marker system, with each marker having two alleles. Among the eight possible haplotypes, haplotypes 111 and 222 are the high-risk haplotypes with the same haplotype frequency, and the other six haplotypes have equal frequencies and the same risk. The high-risk haplotype frequency can be calculated by the formula reported by Schaid (1996). We assume that 300 families are ascertained from this population, through an affected child, and that the significance level is set at .001. As mentioned above, this level of significance is most appropriate for finding genes via candidate regions, and it may introduce too many false-positive results for a genomewide search of disease genes. However, our main purpose here is to compare the performance of different testing procedures, and we note that the results are similar when other significance levels are chosen. We present the power comparisons, with attributable risk of 20%, in figures 1 and 2. The relative performance of these tests is similar when the attributable risk is 10% or 15% (data not shown).
Figure 1.

Power comparison among different testing procedures, under the dominant disease model. The attributable risk at the locus is 20%, and the sample consists of 300 families.
Figure 2.

Power comparison among different testing procedures, under the recessive disease model. The attributable risk at the locus is 20%, and the sample consists of 300 families.
For the dominant disease model (fig. 1), we vary the relative risk for the high-risk genotype (with one or two copies of either haplotype 111 or haplotype 222) versus other genotypes, at 2, 4, and 10. We can see that we would achieve the best power if we knew the true haplotypes in the parents (i.e., Th). Among the five other tests that do not require known parental haplotypes, Ts and Td have the lowest power. All three multilocus tests (Tu, Tc, and Tml) that are based on reconstruction of the transmission/nontransmission table have better power, with Tml having the highest power, Tc having the lowest power, and Tu having power intermediate between Tml and Tc.
The power of different statistical tests under the recessive disease model is plotted in figure 2. As in the dominant-model case, the relative risk for the high-risk genotype versus other genotypes is varied at 2, 4, and 10. The test that analyzes each marker separately (Ts) has the lowest power, and the test that assumes known haplotype information for all families (Th) has the largest power. The other four tests show similar patterns, with the dominant model.
DRD2 and Alcoholism
In this section, we apply the statistical methods that we have discussed, to study genetic linkage between the DRD2 locus and alcoholism. Among the 77 family trios included in this study, there were 55 German families and 22 Hungarian families. Three biallelic polymorphisms spanning 30 kb within the DRD2 locus were genotyped: TaqIB, TaqID, and TaqIA (Kidd et al. 1998). A full description of this data set and analyses that are more comprehensive will be described elsewhere. All the significance levels were estimated by simulations as described above. When markers are analyzed separately, the TDT yields markerwise P values of .41 for TaqIB, .12 for TaqID, and .04 for TaqIA. When we adjust these P values to take multiple comparisons into account, using the method described by Lazzeroni and Lange (1998), the adjusted P values for these three markers are .90 for TaqIB, .71 for TaqID, and .23 for TaqIA. When the three RFLPs are analyzed jointly, there are 32 families with ambiguous haplotypes. The P values are .053, .018, .032, and .025, for Td, Tu, Tc, and Tml, respectively, for the combined sample from the two populations. For each of the four multilocus methods, the estimated counts that a particular haplotype is transmitted and not transmitted are summarized in table 3. The results for the 55 German families are summarized in table 4, and the results for the 22 Hungarian families are summarized in table 5. The general transmission patterns are similar in the two populations, although they are more extreme in the Hungarian families.
Table 3.
Statistical Tests of Genetic Linkage between the DRD2 Locus and Alcoholism, in 77 Combined German and Hungarian Families
|
No. of Transmissionsa |
||||||||
|
Td (P=.053) |
Tu (P=.018) |
Tc (P=.032) |
Tml (P=.025) |
|||||
| Haplotype | Transmitted | Not Transmitted | Transmitted | Not Transmitted | Transmitted | Not Transmitted | Transmitted | Not Transmitted |
| 111 | 2 | 4 | 9.3 | 6.8 | 9.5 | 7.9 | 9.0 | 7.4 |
| 112 | 8 | 17 | 11.8 | 26.6 | 13.2 | 25.8 | 13.0 | 26.0 |
| 121 | 5 | 5 | 17.7 | 11.4 | 17.4 | 10.6 | 18.0 | 11.0 |
| 122 | 3 | 6 | 8.3 | 10.3 | 9.0 | 10.8 | 9.1 | 10.6 |
| 211 | 8 | 2 | 15.1 | 8.9 | 14.8 | 9.0 | 15.0 | 9.2 |
| 212 | 54 | 39 | 68.0 | 55.8 | 70.6 | 57.3 | 71.1 | 57.3 |
| 221 | 0 | 0 | 0 | 0 | 2.5 | 3.5 | 2.0 | 3.3 |
| 222 | 10 | 17 | 18 | 28.3 | 17.2 | 29.2 | 17.0 | 29.1 |
Details of the four multilocus TDTs are discussed in the text and also are summarized in table 1. Data are the number of families in which the eight haplotypes are transmitted and not transmitted from the parents to the affected offspring.
Table 4.
Statistical Tests of Genetic Linkage between the DRD2 Locus and Alcoholism, in 55 German Families
|
No. of Transmissionsa |
||||||||
|
Td (P=.556) |
Tu (P=.270) |
Tc (P=.169) |
Tml (P=.201) |
|||||
| Haplotype | Transmitted | Not Transmitted | Transmitted | Not Transmitted | Transmitted | Not Transmitted | Transmitted | Not Transmitted |
| 111 | 1 | 4 | 8.1 | 6.1 | 8.1 | 7.2 | 7.9 | 6.9 |
| 112 | 4 | 6 | 5.8 | 11.8 | 6.0 | 10.9 | 5.9 | 11.0 |
| 121 | 5 | 4 | 15.4 | 8.9 | 15.4 | 8.4 | 15.7 | 8.6 |
| 122 | 2 | 4 | 4.7 | 7.3 | 4.4 | 7.6 | 4.4 | 7.4 |
| 211 | 6 | 2 | 12.5 | 8.0 | 12.3 | 7.2 | 12.6 | 7.3 |
| 212 | 35 | 29 | 44.7 | 41.1 | 47.5 | 42.8 | 47.6 | 42.8 |
| 221 | 0 | 0 | 0 | 0 | 2.2 | 2.3 | 1.8 | 2.2 |
| 222 | 9 | 13 | 14.9 | 22.8 | 14.0 | 23.8 | 14.1 | 23.8 |
Data are as described in the footnote to table 3.
Table 5.
Statistical Tests of Genetic Linkage between the DRD2 Locus and Alcoholism, in 22 Hungarian Families
|
No. of Transmissionsa |
||||||||
|
Td (P=.038) |
Tu (P=.052) |
Tc (P=.150) |
Tml (P=.063) |
|||||
| Haplotype | Transmitted | Not Transmitted | Transmitted | Not Transmitted | Transmitted | Not Transmitted | Transmitted | Not Transmitted |
| 111 | 1 | 0 | 1.2 | .5 | 1.5 | .8 | 1.1 | .2 |
| 112 | 4 | 11 | 5.8 | 15.0 | 7.1 | 14.9 | 6.1 | 15.0 |
| 121 | 0 | 1 | 2.3 | 2.4 | 1.7 | 2.2 | 2.3 | 2.5 |
| 122 | 1 | 2 | 3.7 | 3.1 | 4.7 | 3.2 | 5.6 | 3.4 |
| 211 | 2 | 0 | 2.5 | 1.0 | 2.5 | 2.3 | 2.5 | 2.3 |
| 212 | 19 | 10 | 23.5 | 14.5 | 23.0 | 14.5 | 24.4 | 14.5 |
| 221 | 0 | 0 | 0 | 0 | .4 | 1.1 | .1 | 1.0 |
| 222 | 1 | 4 | 3.0 | 5.4 | 3.2 | 5.5 | 2.0 | 5.2 |
Data are as described in the footnote to table 3.
Discussion
The rapid progress in the identification of polymorphic markers in the human genome has been driving the developments of powerful and robust statistical methods for finding the genes underlying complex traits. The TDT, proposed by Spielman et al. (1993), has proved to be one powerful approach. The TDT using multiple tightly linked markers may further increase the statistical power. However, to apply existing methods, we need to either discard families with ambiguous haplotypes or analyze the markers separately, resulting in potential loss of power. In this article, we have proposed that the TDT be extended to multiple markers. Our simulation studies demonstrate that this multimarker approach can extract more information on genetic linkage than can single-marker tests that examine markers separately.
There are basically three classes of TDTs when there are more than two alleles at the locus of interest: (1) analysis of all of the alleles simultaneously, without specific genetic models being assumed (e.g., see Sham and Curtis 1995; Spielman and Ewens 1996); (2) analysis of each allele separately and use of the maximal TDT as the test statistic, an approach called “max-TDT” (Schaid 1996; Ewens and Spielman 1997); and (3) analysis of all the alleles under specific genetic models (Schaid 1996). In this article, we have focussed on the first approach, by treating all alleles equally. The second or the third approach may offer better power under certain circumstances. Another alternative, which is similar to the max-TDT, is to group alleles before the TDT is performed. The effects that allele grouping has on the power to detect linkage disequilibrium have been studied by Zouros et al. (1977) and Weir and Cockerham (1978). Those investigators found that, depending on the levels of linkage disequilibrium, allelic frequencies, and degrees of freedom, the power can either increase or decrease after grouping. The group-TDT is expected to be more powerful than either the TDT or max-TDT, if several marker alleles are associated with the disease mutation; however, when only one marker allele is associated with the disease mutation, or when the degree of association is relatively uniform across all marker alleles, the group-TDT may be less powerful than either the TDT or the max-TDT.
Although we have considered only three biallelic markers in our simulation studies and in the application to the alcoholism data set, our methods have also been found to be more powerful than existing methods, for genetic systems involving more biallelic markers and/or microsatellite markers (authors' unpublished results). However, the gain in statistical power may be compromised by the existence of many haplotypes if the genetic system under study has many biallelic markers and/or if certain microsatellite markers have many alleles. For such genetic systems, methods similar to those proposed by Templeton et al. (1987) and Clayton and Jones (1999) can be employed to reduce the complexities, by formation of haplotype groups on the basis of their similarities. Both theoretical and empirical studies are needed to develop and evaluate statistical methods that can reduce the complexity of such multisite systems.
Of the three counting schemes for estimation of haplotype frequencies, the Tml, which estimates haplotype frequencies by assuming that the parents consist of a random sample of individuals from a population having Hardy-Weinberg equilibrium, and Tu, which estimates haplotype frequencies by using unambiguous families, have similar power, and both are more powerful than the third counting scheme, Tc. For the real data on alcoholism, the estimated P values are also similar for Tml and Tu. This is because unambiguous families make a substantial contribution to the haplotype-frequency estimates in the derivation of the Tml for the genetic systems considered in this article; thus, the haplotype frequencies estimated by the two approaches are similar. However, the similarity between the two testing procedures may not hold for other genetic systems. When the number of markers is increased, a higher proportion of the families will become ambiguous with respect to the resolution of haplotypes, and fewer families can be used to estimate haplotype frequencies. Therefore, of the three counting schemes discussed in this article, we recommend the use of the Tml.
In this article, we have assumed that both parents are available for genotyping. In the case of a single marker, the TDT has been extended both to families consisting of sibships without parents (Curtis 1997; Boehnke and Langefeld 1998; Horvath and Laird 1998; Spielman and Ewens 1998; Teng and Risch 1999) and to families consisting of one affected child and only one parent (Sun et al. 1999). The same ideas may be used to extend our methods to either sibships without parents or sibships with only one parent. In addition, the availability of additional children may help to reduce the number of compatible haplotype groups in the parents and may eliminate ambiguity altogether. The other assumption in our methods is that there is no recombination among the tightly linked markers under study. This assumption can be relaxed to allow for recombinations among the markers, but more parameters are needed to define the recombination fractions among the markers, and extra computations are required. Overall, there may be little benefit in considering the recombinations for tightly linked markers. If linkage disequilibrium exists across the region for a nonadmixture population, then recombination must be quite infrequent and probably can be safely ignored.
Although the proposed methods are a valid test for the null hypothesis of no linkage, they are conservative, because, in the construction of table 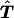 , the assignment of haplotype groups on the basis of the genotypes of the individual sites is carried out under the assumption of no linkage. This will diminish the linkage evidence present in the original sample. An alternative approach, which may be more powerful, is to assume a parametric model and to compare the fit of the observed data under the null and alternative hypotheses. Following Zhao (1999), we can write the probability of a given set of genotypes g as
, the assignment of haplotype groups on the basis of the genotypes of the individual sites is carried out under the assumption of no linkage. This will diminish the linkage evidence present in the original sample. An alternative approach, which may be more powerful, is to assume a parametric model and to compare the fit of the observed data under the null and alternative hypotheses. Following Zhao (1999), we can write the probability of a given set of genotypes g as
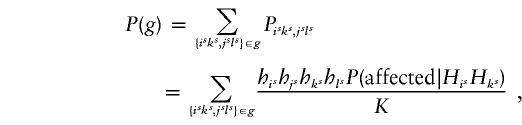 |
where K is the disease prevalence in the population, P(affected|HisHks) is the penetrance for the genotype comprised of haplotypes HisHks, and the his are the haplotype frequencies. Under the null hypothesis of no linkage all the P(affected|HisHks) are the same, whereas under the alternative hypothesis they may take on different values. Denote the maximum likelihood under the null and alternative hypotheses by L0 and La, respectively. Then the likelihood-ratio statistic 2log(La/L0) can be used to assess the statistical significance against the null hypothesis. However, this approach makes the implicit assumption that the underlying population is homogeneous. Thus, unlike the TDT approach, this parametric approach may fail in the presence of population stratification, as does the method of Clayton (1999).
Acknowledgments
We thank Dr. Michael Knapp for his comments on a previous version of this article, and we thank two anonymous reviewers for their constructive comments. This work was supported in part by National Institutes of Health grants GM59507 and HD36834 (both to H.Z.) and AA09379 (to K.K.K.).
Appendix A :
Proposition 1. The expected transmission/nontransmission table 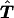 reconstructed as described in the text is symmetrical under the null hypothesis of no linkage.
reconstructed as described in the text is symmetrical under the null hypothesis of no linkage.
Proof.
-
1.
Let haplotype group {ik,jl} denote the event that, in the father, the transmitted haplotype is Hi and the nontransmitted haplotype is Hj and that, in the mother, the transmitted haplotype is Hk and the nontransmitted haplotype is Hl. Suppose that its corresponding set of genotypes g is compatible only with {ik,jl}. Denote the set of genotypes corresponding to {jl,ik} by g′. In fact, g′ consists of parents with the same set of genotypes and of offspring with the nontransmitted genotype at each site. It is easy to see that {jl,ik} is the only haplotype group compatible with g′. Denote all sets of genotypes that have only one compatible haplotype group by U. We have established that, if g∈U, then g′∈U. For such {ik,jl},
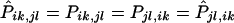 .
. -
2. Suppose that a family with the set of genotypes g has ambiguities and that {ik,jl} is one haplotype group that is compatible with g. Denote the set of genotypes corresponding to {jl,ik} by g′. For every haplotype group {isks,jsls} compatible with g, haplotype group {jsls,isks} must be compatible with g′. Therefore, under the null hypothesis of no linkage, g and g′ have the same probability, because Pisks,jsls=Pjsls,isks. For an arbitrary set of haplotype frequencies hi,
From the above relationships, we get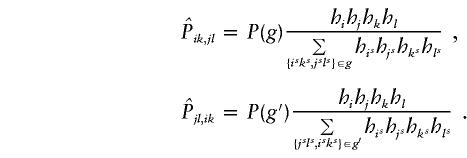
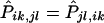 .
When the two cases above are combined, the expected matrix
.
When the two cases above are combined, the expected matrix is symmetrical, because
is symmetrical, because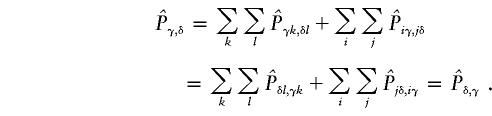
Appendix B :
Proposition 2. The expected transmission/nontransmission table constructed by use of only unambiguous families is symmetrical.
Proof. Suppose that the observed set of genotypes g is compatible with only one haplotype group {ik,jl}. Let g′ and U be as defined in the proof of Proposition 1. Denote the transmission/nontransmission table using only unambiguous families by 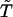 and denote the expected entries in this table by
and denote the expected entries in this table by 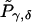 . This table is symmetrical, because g∈U⇔g′∈U, and
. This table is symmetrical, because g∈U⇔g′∈U, and
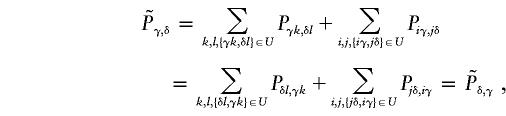 |
under the null hypothesis of no linkage.
Appendix C:
Proposition 3. The expected transmission/nontransmission table constructed by assigning to each ambiguous family its most likely haplotype group is symmetrical.
Proof. Suppose that the observed set of genotypes g has ambiguities. Denote the set of genotypes corresponding to {jl,ik} by g′. For every {isks,jsls} compatible with g, {jsls,isks} must be compatible with g′. Therefore, under the null hypothesis of no linkage, g and g′ have the same probability, because Pisks,jsls=Pjsls,isks. Suppose that, in the set of haplotype groups compatible with g, {imkm,jmlm} is the most likely haplotype group when Hardy-Weinberg equilibrium and random mating are assumed. Then, {jmlm,imkm} must be the most likely haplotype group compatible with g′. Therefore, 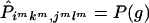 and
and 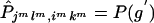 . For all other {isks,jsls} and {jsls,isks},
. For all other {isks,jsls} and {jsls,isks}, 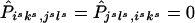 . We can now see that the expected
. We can now see that the expected 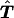 is symmetrical, because
is symmetrical, because
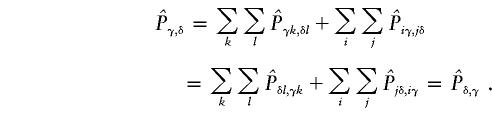 |
References
- Boehnke M, Langefeld CD (1998) Genetic association mapping based on discordant sib pairs: the discordant-alleles test. Am J Hum Genet 62:950–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton DG (1999) A generalization of the transmission/disequilibrium test for uncertain-haplotype transmission. Am J Hum Genet 65:1170–1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton DG, Jones H (1999) Transmission/disequilibrium tests for extended marker haplotypes. Am J Hum Genet 65:1161–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis D (1997) Use of siblings as control in case-control association studies. Ann Hum Genet 61:319–333 [DOI] [PubMed] [Google Scholar]
- Dudbridge F, Koeleman BPC, Todd JA, Clayton DG (2000) Unbiased application of the transmission/disequilibrium test to multilocus haplotypes. Am J Hum Genet 66:2009–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens WJ, Spielman RS (1997) Disease associations and the transmission/disequilibrium test. In: Dracopoli NC (ed) Current protocols in human genetics. Suppl 15. Wiley, New York, pp 1.12.1–1.12.13 [Google Scholar]
- Falk CT, Rubinstein P (1987) Haplotype relative risk: an easy way to construct a proper control sample for risk calculations. Ann Hum Genet 51:227–233 [DOI] [PubMed] [Google Scholar]
- Hawley ME, Kidd KK (1995) HAPLO: a program using the EM algorithm to estimate frequencies of multi-site haplotypes. J Hered 86:409–411 [DOI] [PubMed] [Google Scholar]
- Horvath S, Laird NM (1998) A discordant-sibship test for disequilibrium and linkage: no need for parental data. Am J Hum Genet 63:1886–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd KK, Morar B, Castiglione CM, Zhao H, Pakstis AJ, Speed WC, Bonne-Tamir B, Lu R-B, Goldman D, Lee C, Nam YS, Grandy DK, Jenkins T, Kidd JR (1998) A global survey of haplotype frequencies and linkage disequilibrium at the DRD2 locus. Hum Genet 103:211–227 [DOI] [PubMed] [Google Scholar]
- Lazzeroni LC, Lange K (1998) A conditional inference framework for extending the transmission/disequilibrium test. Hum Hered 48:67–81 [DOI] [PubMed] [Google Scholar]
- Ott J (1989) Statistical properties of the haplotype relative risk. Genet Epidemiol 6:127–130 [DOI] [PubMed] [Google Scholar]
- Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 [DOI] [PubMed] [Google Scholar]
- Rubinstein P, Walker M, Carpenter C, Carrier C, Krassner J, Falk C, Ginsberg F (1981) Genetics of HLA disease associations: the use of the haplotype relative risk (HRR) and the “haplo-delta” (Dh) estimates in juvenile diabetes from racial groups. Hum Immunol 3:384 [Google Scholar]
- Schaid DJ (1996) General score tests for associations of genetic markers with disease using cases and their parents. Genet Epidemiol 13:423–449 [DOI] [PubMed] [Google Scholar]
- Sethuraman B (1997) Topics in statistical genetics. PhD diss, University of California, Berkeley [Google Scholar]
- Sham P (1997) The transmission/disequilibrium tests for multiallelic loci. Am J Hum Genet 61:774–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham PC, Curtis D (1995) An extended transmission/disequilibrium test (TDT) for multi-allele marker loci. Ann Hum Genet 59:323–336 [DOI] [PubMed] [Google Scholar]
- Spielman RS, Ewens WJ (1996) The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet 59:983–989 [PMC free article] [PubMed] [Google Scholar]
- ——— (1998) A sibship test for linkage in the presence of association: the sib transmission/disequilibrium test. Am J Hum Genet 62:450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman RS, McGinnis RE, Ewens WJ (1993) The transmission test for linkage disequilibrium: the insulin gene and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52:506–516 [PMC free article] [PubMed] [Google Scholar]
- Sun F, Flanders WD, Yang Q, Khoury MJ (1999) Transmission disequilibrium test (TDT) when only one parent is available: the 1-TDT. Am J Epidemiol 150:97–104 [DOI] [PubMed] [Google Scholar]
- Templeton AR, Boerwinkle E, Sing CF (1987) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. I. Basic theory and an analysis of alcohol dehydrogenase activity in drosophila. Genetics 117:343–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng J, Risch N (1999) The relative power of family-based and case-control designs for linkage disequilibrium studies of complex human diseases. II. Individual genotyping. Genome Res 9:234–241 [PubMed] [Google Scholar]
- Terwilliger JD, Ott J (1992) A haplotype-based “haplotype relative risk” approach to detecting allelic associations. Hum Hered 42:337–346 [DOI] [PubMed] [Google Scholar]
- Thomson G (1995) Mapping disease genes: family-based association studies. Am J Hum Genet 57:487–498 [PMC free article] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC (1978) Testing hypotheses about linkage disequilibrium with multiple alleles. Genetics 88:633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SR (1997) On extending the transmission/disequilibrium test (TDT). Ann Hum Genet 61:151–161 [DOI] [PubMed] [Google Scholar]
- Zhao H (1999) The interpretation of the parameters in the transmission/disequilibrium test. Am J Hum Genet 64:326–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zouros E, Golding GB, MacKay TFC (1977) The effect of combining alleles into electrophoretic classes on detecting linkage disequilibrium. Genetics 85:543–556 [DOI] [PMC free article] [PubMed] [Google Scholar]