

Abstract
Peroxisome biogenesis disorders (PBDs) such as Zellweger syndrome (ZS) and neonatal adrenoleukodystrophy are autosomal recessive diseases caused by defects in peroxisome assembly, for which 13 genotypes have been identified. Expression of the human peroxin Pex3p cDNA encoding a 373-amino-acid peroxisomal membrane protein morphologically and biochemically restored peroxisome biogenesis, including peroxisomal membrane assembly, in fibroblasts from PBDG-02, a patient with complementation group G (CG-G) ZS. Patient PBDG-02 carried a homozygous, inactivating mutation—a 97-bp deletion of nucleotide residues at positions 942–1038—resulting in a 32-amino-acid truncation and in a frameshift inducing both a 3-amino-acid substitution and a termination codon. Genomic PCR analysis revealed mutation of T→G at eight bases upstream of the splicing site at the boundary of intron 10 and exon 11 of PEX3 gene, giving rise to a deletion of all of exon 11. When assessed by expression in a pex3 mutant of Chinese hamster ovary cells and the patient’s fibroblasts, PBDG-02–derived PEX3 cDNA was found to be defective in peroxisome-restoring activity. These results provide evidence that PEX3 is a novel, pathogenic gene responsible for CG-G PBDs.
Peroxisomes function in various metabolic pathways, including β-oxidation of very-long-chain fatty acids and synthesis of ether lipids (van den Bosch et al. 1992). Human fatal genetic peroxisomal biogenesis disorders (PBDs) include Zellweger syndrome (ZS [MIM 214100]), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and rhizomelic chondrodysplasia punctata (RCDP) (Lazarow and Moser 1995). The primary cause of the peroxisome deficiency in PBDs, comprised of 13 complementation groups (CGs) (Kinoshita et al. 1998; Shimozawa et al. 1998; Ghaedi et al. 1999), is a failure in peroxisome biogenesis (Braverman et al. 1995; Subramani 1997; Fujiki 2000). Genetic heterogeneities containing >15 CGs have been identified in mammals, having been determined by use of fibroblasts from patients with PBDs and of peroxisome-deficient Chinese hamster ovary (CHO) cell mutants (Ghaedi et al. 1999; Fujiki 2000) (see table 1). To date, 10 PEX genes have been shown to be pathogenic for 10 CGs of PBDs; several genes have yet to be identified (Fujiki 2000). Much knowledge of peroxisome biogenesis has been gained on the basis of findings of topogenic signals and peroxins required for peroxisomal protein import. However, thoroughly molecular mechanisms involved in assembly of peroxisomal membrane vesicles have yet to be defined. In most CGs, fibroblasts from patients with PBDs contain morphologically recognizable peroxisomal membrane remnants, which implies that membrane biogenesis is normal, despite the impaired import of matrix proteins (Santos et al. 1992; Wendland and Subramani 1993; Shimozawa et al. 1998). In contrast, in three CGs—CG-D (CG9 in the United States), CG-G, and CG-J—fibroblasts from patients with PBDs are absent from peroxisomal remnants, and are thus indicative of apparent defects in peroxisome membrane assembly (Kinoshita et al. 1998; Shimozawa et al. 1998). PEX16 (Honsho et al. 1998; South and Gould 1999) and PEX19 (Matsuzono et al. 1999) were shown to be responsible for PBDs of CG-D and CG-J, respectively. Using ZPG208, a CHO cell mutant deficient in peroxisome assembly, we cloned rat PEX3 cDNA by genetic phenotype-complementation assay (Ghaedi et al. 2000). We now report that human PEX3 (HsPEX3 [MIM 603164]) complements the impaired peroxisome biogenesis in fibroblasts devoid of peroxisomal membrane vesicles from a patient with CG-G PBD.
Table 1.
Complementation Groups of Patients with PBDs and CHO Cell Mutants
|
CG Designation of Patients with PBDs |
|||||
| Japanese | U.S./European | Phenotype | CHO Mutant | Peroxisomal Ghostsa | Gene |
| G | ZS | ZPG208 | − | PEX3 | |
| A | 8 | ZS, NALD, IRD | ZP124 | + | |
| B | 7(5)b | ZS, NALD | + | PEX10 | |
| C | 4 | ZS, NALD | ZP92 | + | PEX6 |
| D | 9 | ZS | − | PEX16 | |
| E | 1 | ZS, NALD, IRD | Z24/ZP107 | + | PEX1 |
| F | 10 | ZS, IRD | Z65 | + | PEX2 |
| H | 13 | ZS, NALD | ZP128 | + | PEX13 |
| J | ZS | ZP119 | − | PEX19 | |
| 2 | ZS, NALD | ZP105/ZP139 | + | PEX5 | |
| 3 | ZS, NALD, IRD | ZP109 | + | PEX12 | |
| 6 | ZS, NALD | + | |||
| R | 11 | RCDP | ZPG207 | + | PEX7 |
| ZP110 | + | PEX14 | |||
| ZP114 | + | ||||
| ZP126 | + | ||||
A plus sign (+) denotes that peroxisomal remnants (ghosts) are present; a minus sign (−) denotes that they are absent.
From the study by Shimozawa et al. (1998)
Fibroblasts from PBDG-02, a patient with CG-G ZS, showed a diffused, cytosolic localization of catalase, which is indicative of peroxisome deficiency, as verified by cell staining with an anticatalase antibody (Shimozawa et al. 1992) (see fig. 1A, subpanel b). When PBDG-02's fibroblasts were fused with the CHO pex3 mutant ZPG208 (Ghaedi et al. 2000), the impaired protein import was not restored (data not shown), which demonstrates that PBDG-02's cells are in the same CG as is ZPG208 (table 1). Peroxisomal remnants, so-called ghosts, were not discernible in PBDG-02's fibroblasts (table 1), as assessed by cell staining with antibody to 70-kD peroxisomal membrane protein (PMP70) (not shown), thereby confirming a defect in peroxisome membrane biogenesis. These results collectively show that PBDG-02's cells are impaired in biogenesis of not only soluble proteins but also membrane polypeptides, an impairment most likely caused by a defect in PEX3. HsPEX3 expression indeed complemented impaired import of catalase (fig. 1A, subpanel a) and of peroxisome-targeting signal type I (PTS1) proteins, as well as 3-ketoacyl-CoA thiolase, a PTS2-protein (data not shown), in PBDG-02's fibroblasts. PMP70-positive peroxisomes were also observed, in a superimposable manner, with peroxisomes stained by use of anticatalase antibody (not shown). Therefore, Pex3p could morphologically restore peroxisome assembly in PBDG-02’s fibroblasts.
Figure 1.

Impaired PEX3 in a patient with CG-G ZS. A, Expression of HsPEX3, which restores peroxisome assembly. HsPEX3 and flag-tagged PEX3delEx11 derived from PBDG-02 (see fig. 1B) were separately expressed in PBDG-02's fibroblasts (a and b); flag-tagged HsPEX3 and flag-tagged PEX3delEx11 were transfected into pex3 ZPG208 cells (c and d). Cell staining was done by use of primary rabbit antibodies to catalase (a and b) and PMP70 (c and d) and of a secondary Texas Red–labeled sheep anti-rabbit immunoglobulin G antibody. In b, the scale bar (which also applies to a) = 40 μm; in d, the scale bar (which also applies to c) = 20 μm. Note that peroxisomes were restored in (a and c) but not in (b and d). B, Mutation analysis of PEX3 from a patient with CG-G ZS, PBDG-02. Partial nucleotide sequence and the deduced amino acid sequence of PEX3 cDNA isolated from a normal control (upper-left panel) and PBDG-02 (lower-left panel) are shown. A 97-bp deletion of nucleotide residues 942–1038 (boxed) was identified. PCR was also done for DNA from a normal control (upper-right panel) and from PBDG-02's fibroblasts (lower-right panel); the nucleotide sequence of PCR products was determined. Only partial sequence at the boundary of intron 10 and exon 11 is shown; a codon for Leu315 is underlined in the sequence of the normal control.
We next verified the biogenesis of peroxisomal proteins in CG-G fibroblasts. PMP70 was not discernible in PBDG-02's fibroblasts (fig. 2, top panel, lane 2), presumably because of a rapid degradation, as seen in the CHO pex19 mutant ZP119 (Kinoshita et al. 1998), although PMP70 was detectable in normal fibroblasts (lane 1). When PBDG-02's fibroblasts were transiently transfected with HsPEX3, PMP70 was detected, as in normal cells (lane 3), indicating that the impaired biogenesis of PMP70 was restored. On transfection of HsPEX3, Pex3p was also detectable, albeit at a low level, when anti-C-terminal Pex3p peptide antibody was used (Ghaedi et al. 2000), thereby confirming the expression of HsPex3p, whereas Pex3p was not discernible in PBDG-02's fibroblasts (data not shown). Peroxisomal 3-ketoacyl-CoA thiolase of fatty acid β-oxidation system is synthesized as a larger, 44-kD precursor with an amino-terminal, cleavable PTS2 and is processed to 41-kD mature form in peroxisomes (Tsukamoto et al. 1990). In normal fibroblasts, only the matured thiolase was detected (fig. 2, middle panel, lane 1), thereby providing evidence for a rapid processing of the precursor form. In PBDG-02's cells, the larger precursor was not detectable (fig. 2, middle panel, lane 2), presumably because of degradation. When PBDG-02's fibroblasts were transiently transfected with HsPEX3, the mature thiolase was clearly discerned (fig. 2, middle panel, lane 3). A PTS1 protein, acyl-CoA oxidase (AOx), is synthesized as a 75-kD polypeptide (A component) and is proteolytically converted into 53- and 22-kD polypeptides (B and C components, respectively) in peroxisomes (Tsukamoto et al. 1990). All three polypeptide components were evident in normal fibroblasts from a control (fig. 2, bottom panel, lane 1), whereas they were barely detectable in PBDG-02's cells (fig. 2, bottom panel, lane 2), possibly reflecting the occasional peroxisome-positive cell and degradation of most of AOx-A (Tsukamoto et al. 1990; Ghaedi et al. 1999). When PBDG-02's cells were transfected with HsPEX3, the three components of AOx were detected at a distinct level, indicative of proper import and proteolytic conversion of AOx (fig. 2, bottom panel, lane 3). These results demonstrate that HsPEX3 can complement the impaired biogenesis of peroxisomal proteins in PBDG-02's cells. HsPEX3 expression restored peroxisome biogenesis only in the CHO pex3 mutants (Ghaedi et al. 2000) (see fig. 1A, subpanel c) in 13 CGs of peroxisome-deficient CHO cell mutants, where the CHO mutants of 10 CGs represent PBD CGs (Tsukamoto et al. 1990; Shimozawa et al. 1992; Kinoshita et al. 1998; Ghaedi et al. 1999; Toyama et al. 1999) (table 1). Taken together, these observations confirm that Pex3p is a peroxisome biogenesis factor for CG-G PBD. Therefore, PEX3 is the 11th gene identified as being responsible for peroxisome-deficiency diseases.
Figure 2.
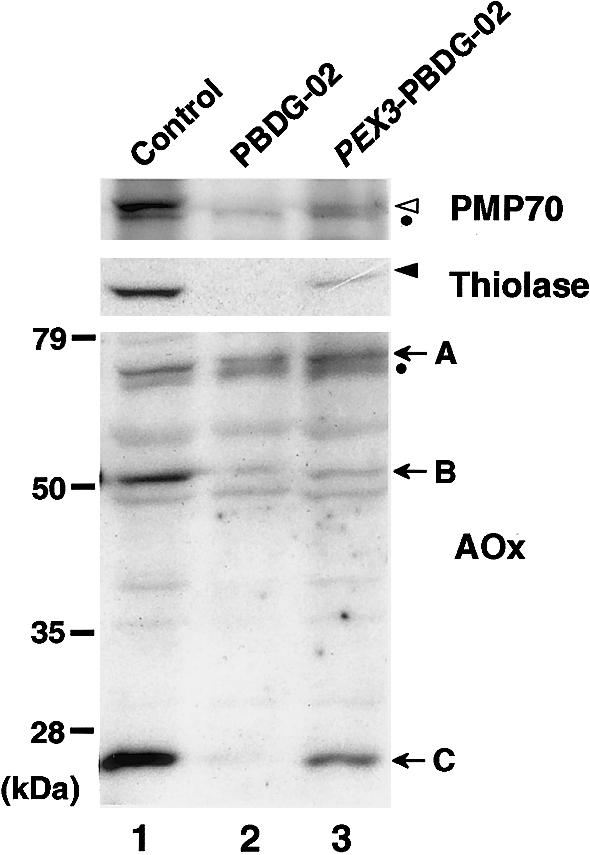
Complementation of biogenesis of peroxisomal proteins. Cell lysates (∼2×105 cells) were subjected to SDS-PAGE and were transferred to polyvinylidene difluoride membrane. Immunoblot analysis was done by use of rabbit antibodies to PMP70 (top), thiolase (middle), and AOx (bottom) and an ECL western-blotting detection reagent. Lane 1, Normal control fibroblasts. Lane 2, PBDG-02's fibroblasts. Lane 3, PBDG-02's fibroblasts transiently transfected with HsPEX3. Unblackened and blackened arrowheads indicate PMP70 and a mature form of thiolase, respectively; arrows indicate AOx components A–C. Dots indicate nonspecific bands (Kinoshita et al. 1998; Tamura et al. 1998).
Both to isolate PEX3 cDNA from PBDG-02's fibroblasts and to determine the PEX3 dysfunction, we conducted reverse transcription–PCR (RT-PCR), using poly(A)+ RNA and a pair of HsPEX3-specific PCR primers: a sense HsPEX3-BamF (5′-TAAGCTTGAGATCTTGAGGTCTGTATGGA-3′) and an antisense HsPEX3-ApaR (5′-GGGTACCGGGCCCTCATTTCTCCAGTTGC-3′) (where the termination codon is underlined), as described elsewhere (Honsho et al. 1998). Subsequent sequencing of cDNA clones by the dideoxy-chain termination method indicated a 97-bp deletion of nucleotide residues at positions 942–1038, resulting in a 32-amino-acid truncation and a frameshift inducing a 3-amino-acid substitution and a termination codon (fig. 1B, left). This truncated part corresponded exactly to exon 11 of the HsPEX3 gene (Muntau et al. 2000). Fifteen of the isolated PEX3 cDNA clones, termed “PEX3delEx11,” all carried the same mutation, thereby suggesting a homozygous event causing a defective splicing. To search for both a mutation site in the intron and zygosity of a PEX3delEx11 mutant allele, genomic PCR with a pair of PEX3-specific primers—forward primer Ex11-104–133F (5′-TCACAGCTAGAGGACAGATTACTGTATAGT-3′) and reverse primer Ex11-711–737R (5′-TGCTATGGTCCGTTATTACTCTCATGG-3′)—was used to amplify the sequence between residues 104 and 737, encompassing 361 nucleotide residues on the 3′ side of intron 10, the 97 nucleotides of exon 11, and 176 nucleotides on the 5′ side of intron 11 in the HsPEX3 gene (GenBank accession number AJ009873) (Muntau et al. 2000). In seven independent clones isolated from PCR products, we identified a single type of nucleotide sequence, which, apparently, gave rise to a deletion of exon 11: T was mutated to G at eight bases upstream of the splicing site at the boundary of intron 10 and exon 11 (fig. 1B, right). These observations were interpreted to mean that patient PBDG-02, who has ZS, was a homozygote for the T→G mutation, causing the deletion of exon 11. This homozygosity may be explained by this patient's history, since PBDG-02 was the third child of consanguineous Italian parents (Poulos et al. 1995). This mutation inactivated the function of PEX3, as verified by expression of flag-PEX3delEx11 in PBDG-02's fibroblasts and in pex3 ZPG208 cells of the same CG, where peroxisomes were not formed, as assessed by staining of catalase and PMP70, respectively (fig. 1A, subpanels b and d). Normal flag-HsPEX3 restored peroxisome assembly in ZPG208 (fig. 1A, subpanel c), as well as in PBDG-02's fibroblasts (not shown), indicating that flag tagging did not interfere with Pex3p function. The data collectively indicate that mutation of PEX3 is the genetic-related cause of CG-G PBDs. Moreover, these findings show the importance that the C-terminal area of Pex3p has for biological activity.
We also investigated the kinetics of peroxisome assembly, with respect to both membrane vesicle formation and soluble-protein import. PBDG-02's fibroblasts were transfected with HsPEX3 and were monitored for peroxisome biogenesis, at various time points, by our visualizing of several endogenous peroxisomal proteins, including the membrane peroxin Pex14p—a convergent component of a potential import machinery (Shimizu et al. 1999; Otera et al. 2000)—and PMP70, as well as matrix proteins PTS1 and catalase. We observed that peroxisomal membrane vesicles were apparently formed prior to the import of matrix proteins (data not shown), in agreement with the findings for the CHO pex3 mutant ZPG208 (Ghaedi et al. 2000). Therefore, Pex3p is most likely to be involved in peroxisome assembly at the early stage of peroxisomal membrane vesicle formation. Mammalian Pex3p interacts with Pex19p (Soukupova et al. 1999; Ghaedi et al. 2000; Sacksteder et al. 2000). It is likely that Pex3p functions as a peroxisome biogenesis factor, possibly by interacting with other PEX proteins as well (Soukupova et al. 1999; Ghaedi et al. 2000; Sacksteder et al. 2000). The precise mechanisms by which Pex3p functions in peroxisome membrane assembly remain to be defined.
We had previously cloned mammalian PEX3 cDNAs by functional phenotype-complementation assay, using CHO cell mutants (Ghaedi et al. 2000). In that report, we had concluded that PEX3 expression could not complement fibroblasts from PBDG-02 (who, in that report, was designated “CG-G patient 2”) (Ghaedi et al. 2000). We observed that PBDG-02's fibroblasts are unstable with respect to peroxisome deficiency: peroxisomes, including peroxisomal remnants, emerge either occasionally or readily, depending on cell-culture conditions such as consecutive cell-culture passages. Moreover, cell growth rate varies significantly. The main reason for the conclusion in our earlier report appears to have been the following: cell-doubling time was >4 d, and phenotype complementation was verified 4 d after PEX3 transfection (Ghaedi et al. 2000)—whereas the cell-doubling time in the present study was 1.5 d, and peroxisome restoration was mostly assessed 5 d after HsPEX3 transfection. In the present work, we selected, from several different lots of stocks that had been prepared at various times of cell passage, one cell stock showing a better, typical mutant phenotype. For a more prolonged time after transfection of HsPEX3, we observed cells for phenotype complementation; for cell mutants, including patients’ fibroblasts, we did not find that cell-culture conditions resulted in such a difference in morphological phenotype. Possibly because of differences in cell growth rate, the kinetics of peroxisome assembly apparently vary, depending on the genotype of >15 CGs of impaired peroxisome biogenesis in mammals. To what extent such differentiated peroxisome assembly is relevant to the severity of PBDs is, at present, unclear.
Acknowledgments
We thank N. Matsumoto and Y. Nakajo for technical assistance, S. Tamura for comments, and other members of the Fujiki laboratory for discussions. This work was supported, in part, by a CREST grant (to Y.F.) from the Japan Science and Technology Corporation and by Grants-in-Aid for Scientific Research (to Y.F.) from The Ministry of Education, Science, Sports and Culture. M. Ohara provided language assistance.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Web/Genbank (for human PEX3 cDNA sequence [accession number AB035307])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for ZS [MIM 214100] and for PEX3 [MIM 603164])
References
- Braverman N, Dodt G, Gould SJ, Valle D (1995) Disorders of peroxisome biogenesis. Hum Mol Genet 4:1791–1798 [DOI] [PubMed] [Google Scholar]
- Fujiki Y (2000) Peroxisome biogenesis and peroxisome biogenesis disorders. FEBS Lett 476:42–46 [DOI] [PubMed] [Google Scholar]
- Ghaedi K, Kawai A, Okumoto K, Tamura S, Shimozawa N, Suzuki Y, Kondo N, Fujiki Y (1999) Isolation and characterization of novel peroxisome biogenesis-defective Chinese hamster ovary cell mutants using green fluorescent protein. Exp Cell Res 248:489–497 [DOI] [PubMed] [Google Scholar]
- Ghaedi K, Tamura S, Okumoto K, Matsuzono Y, Fujiki Y (2000) The peroxin Pex3p initiates membrane assembly in peroxisome biogenesis. Mol Biol Cell 11:2085–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honsho M, Tamura S, Shimozawa N, Suzuki Y, Kondo N, Fujiki Y (1998) Mutation in PEX16 is causal in the peroxisome-deficient Zellweger syndrome of complementation group D. Am J Hum Genet 63:1622–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita N, Ghaedi K, Shimozawa N, Wanders RJA, Matsuzono Y, Imanaka T, Okumoto K, Suzuki Y, Kondo N, Fujiki Y (1998) Newly identified Chinese hamster ovary cell mutants are defective in biogenesis of peroxisomal membrane vesicles (peroxisomal ghosts), representing a novel complementation group in mammals. J Biol Chem 273:24122–24130 [DOI] [PubMed] [Google Scholar]
- Lazarow PB, Moser HW (1995) Disorders of peroxisome biogenesis. In: Scriver CR, Beaudet AI, Sly WS, Valle D (eds) The metabolic basis of inherited disease. McGraw-Hill, New York, pp 2287–2324 [Google Scholar]
- Matsuzono Y, Kinoshita N, Tamura S, Shimozawa N, Hamasaki M, Ghaedi K, Wanders RJA, Suzuki Y, Kondo N, Fujiki Y (1999) Human PEX19: cDNA cloning by functional complementation, mutation analysis in a patient with Zellweger syndrome and potential role in peroxisomal membrane assembly. Proc Natl Acad Sci USA 96:2116–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntau AC, Holzinger A, Mayerhofer PU, Gaertner J, Roscher AA, Kammerer S (2000) The human PEX3 gene encoding a peroxisomal assembly protein: genetic organization, positional mapping, and mutation analysis in candidate phenotypes. Biochem Biophys Res Commun 268:704–710 [DOI] [PubMed] [Google Scholar]
- Otera H, Harano T, Honsho M, Ghaedi K, Mukai S, Tanaka A, Kawai A, Shimizu N, Fujiki Y (2000) The mammalian peroxin Pex5pL, the longer isoform of mobile PTS1-transporter, translocates Pex7p-PTS2 protein complex into peroxisomes via its initial docking site Pex14p. J Biol Chem 275:21703–21714 [DOI] [PubMed] [Google Scholar]
- Poulos A, Christodoulou J, Chow CW, Goldblatt J, Paton BC, Orii T, Suzuki Y, Shimozawa N (1995) Peroxisomal assembly defects: clinical, pathologic, and biochemical findings in two patients in a newly identified complementation group. J Pediatr 127:596–599 [DOI] [PubMed] [Google Scholar]
- Sacksteder KA, Jones JM, South ST, Li X, Liu Y, Gould SJ (2000) PEX19 binds multiple peroxisomal membrane proteins, is predominantly cytoplasmic, and is required for peroxisome membrane synthesis. J Cell Biol 148:931–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MJ, Hoefler S, Moser AB, Moser HW, Lazarow PB (1992) Peroxisome assembly mutations in humans: structural heterogeneity in Zellweger syndrome. J Cell Physiol 151:103–112 [DOI] [PubMed] [Google Scholar]
- Shimizu N, Itoh R, Hirono Y, Otera H, Ghaedi K, Tateishi K, Tamura S, Okumoto K, Harano T, Mukai S, Fujiki Y (1999) The peroxin Pex14p: cDNA cloning by functional complementation on a Chinese hamster ovary cell mutant, characterization, and functional analysis. J Biol Chem 274:12593–12604 [DOI] [PubMed] [Google Scholar]
- Shimozawa N, Suzuki Y, Zhang Z, Imamura A, Kondo N, Kinoshita N, Fujiki Y, Tsukamoto T, Osumi T, Imanaka T, Orii T, Beemer F, Mooijer P, Dekker C, Wanders RJ (1998) Genetic basis of peroxisome-assembly mutants of humans, Chinese hamster ovary cells, and yeast: identification of a new complementation group of peroxisome-biogenesis disorders apparently lacking peroxisomal-membrane ghosts. Am J Hum Genet 63:1898–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimozawa N, Tsukamoto T, Suzuki Y, Orii T, Fujiki Y (1992) Animal cell mutants represent two complementation groups of peroxisome-defective Zellweger syndrome. J Clin Invest 90:1864–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukupova M, Sprenger C, Gorgas K, Kunau W-H, Dodt G (1999) Identification and characterization of the human peroxin PEX3. Eur J Cell Biol 78:357–374 [DOI] [PubMed] [Google Scholar]
- South ST, Gould SJ (1999) Peroxisome synthesis in the absence of preexisting peroxisomes. J Cell Biol 144:255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramani S (1997) PEX genes on the rise. Nat Genet 15:331–333 [DOI] [PubMed] [Google Scholar]
- Tamura S, Okumoto K, Toyama R, Shimozawa N, Tsukamoto T, Suzuki Y, Osumi T, Kondo N, Fujiki Y (1998) Human PEX1 cloned by functional complementation on a CHO cell mutant is responsible for peroxisome-deficient Zellweger syndrome of complementation group I. Proc Natl Acad Sci USA 95:4350–4355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama R, Mukai S, Itagaki A, Tamura S, Shimozawa N, Suzuki Y, Kondo N, Wanders RJA, Fujiki Y (1999) Isolation, characterization, and mutation analysis of PEX13-defective Chinese hamster ovary cell mutants. Hum Mol Genet 8:1673–1681 [DOI] [PubMed] [Google Scholar]
- Tsukamoto T, Yokota S, Fujiki Y (1990) Isolation and characterization of Chinese hamster ovary cell mutants defective in assembly of peroxisomes. J Cell Biol 110:651–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bosch H, Schutgens RBH, Wanders RJA, Tager JM (1992) Biochemistry of peroxisomes. Annu Rev Biochem 61:157–197 [DOI] [PubMed] [Google Scholar]
- Wendland M, Subramani S (1993) Presence of cytoplasmic factors functional in peroxisomal protein import implicates organelle-associated defects in several human peroxisomal disorders. J Clin Invest 92:2462–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]