

Abstract
The IBD2 locus on chromosome 12 has been linked to both Crohn disease (CD) and ulcerative colitis (UC) but has not been detected in some CD-dominated data sets. In the present study, we genotyped 581 relative pairs with inflammatory bowel disease (252 from CD-only families, 138 from UC-only families, and 191 from mixed families containing cases of both CD and UC), using 12 markers spanning the IBD2 locus. A GENEHUNTER-PLUS multipoint LOD score of 3.91 was detected for pairs from UC-only families, compared with 1.66 for CD-only and 1.29 for mixed families. The difference between the LOD scores for UC and CD was significant in two different tests for heterogeneity (P=.0057 for one test and P=.0375 for the other). IBD2 thus appears to make a major contribution to UC susceptibility but to have only a relatively minor effect with regard to CD, for which there may be substantially more locus heterogeneity.
Crohn disease (CD [MIM 266600]) and ulcerative colitis (UC [MIM 191390]), the two common forms of inflammatory bowel disease (IBD), have a combined prevalence, in populations of northern European origin, of 150–200/100,000. Evidence for a strong genetic contribution to IBD comes from twin studies, which demonstrate a substantially higher rate of disease concordance in MZ compared with DZ twins, and from family studies, which demonstrate consistently high λs scores (sibling risk/population prevalence). This is particularly true for CD (λs=20–35; twin concordance MZ:DZ = 36%:12%) and, to a lesser extent, for UC (λs=8–15; twin concordance MZ:DZ = 12%:3%) (Tysk et al. 1988; Probert et al. 1993; Satsangi et al. 1994; Subhani et al. 1998).
Although genetic susceptibility is clearly important, the inheritance of IBD is complex. Furthermore, the etiologic basis of the relationship between CD and UC is as yet unexplained. Some phenotypic features are shared; up to 10% of cases are classified as indeterminate; and the two forms of IBD run together within a single pedigree in ∼30% of multiply affected families (Kirsner 1973; Satsangi et al. 1994). However, there are also some notable epidemiological differences—for example, in sibling recurrence risk and in twin concordance rates (Tysk et al. 1988; Probert et al. 1993; Subhani et al. 1998)—and CD and UC remain distinct clinical entities with separate patterns of behavior. The genetic model that best fits is therefore one in which CD and UC are related polygenic diseases that may share some susceptibility loci but differ at others.
Genome scanning in IBD has established a number of replicated regions of linkage, including loci on chromosomes 3, 6, 12, 14, and 16. The candidate region on chromosome 12 (the IBD2 locus [MIM 601458]) has been reported for both CD and UC (Satsangi et al. 1996; Akolkar et al. 1998; Curran et al. 1998; Duerr et al. 1998; Hampe et al. 1999; Ma et al. 1999), whereas the region on chromosome 16 (the IBD1 locus [MIM 266600]) appears to be linked exclusively to CD (Hugot et al. 1996; Ohmen et al. 1996; Parkes et al. 1996; Brant et al. 1998; Cavanaugh et al. 1998; Cho et al. 1998; Curran et al. 1998; Annese et al. 1999; Hampe et al. 1999; IBD Genetics Consortium 2000).
Although the chromosome 12 linkage has been replicated in a number of data sets (Satsangi et al. 1996; Akolkar et al. 1998; Curran et al. 1998; Duerr et al. 1998; Hampe et al. 1999; Ma et al. 1999), it has not been found in all, and three studies claimed to exclude this region for a locus-specific λs>2 (Brant et al. 1998; Rioux et al. 1998; Vermeire et al. 2000). There are many precedents for failure to replicate linkages in complex diseases—and many possible explanations, particularly relating to power (Suarez et al. 1994; Mandal et al. 1999), but we were struck by the fact that the studies that have failed to detect linkage between IBD and the chromosome 12 locus have contained few relative pairs with UC. The aim of the current study, therefore, was to evaluate further the IBD2 locus in a large panel of multiply affected families and, specifically, to determine its relative contribution to CD susceptibility and UC susceptibility.
Ethical approval for this work was given by the Central Oxford Research and Ethics Committee and the University of Pittsburgh Institutional Review Board for Biomedical Research. The panel of U.K. and U.S. families studied are described in table 1. A total of 581 IBD-affected relative pairs were genotyped, including 396 affected sib pairs. There were 252 affected relative pairs from CD-only families, 138 pairs from UC-only families, and 191 pairs from families in which cases of UC and CD coexisted (“mixed” families). Case notes were reviewed to confirm the diagnoses. All individuals in the panel were white European in origin. Ashkenazi Jewish ethnicity was reported in 43 (11.7%) of the 367 families. The U.K. and U.S. panels each included families used and described in earlier studies, in addition to families more recently recruited, particularly with regard to the U.S. panel (Satsangi et al. 1996; Duerr et al. 1998).
Table 1.
Number of Families and Number of Genotyped Affected Relative Pairs
| Category | All IBD | CD Only | Mixed | UC Only |
| U.K. families | 171 | 79 | 30 | 62 |
| Genotyped affected relative pairs | 222 | 100 | 45 | 77 |
| Sib pairs | 199 | 93 | 35 | 71 |
| Half-sib pairs | 1 | 0 | 0 | 1 |
| Avuncular pairs | 18 | 6 | 7 | 5 |
| Grandparent-grandchild pairs | 1 | 0 | 1 | 0 |
| First-cousin pairs | 3 | 1 | 2 | 0 |
| U.S. families | 196 | 86 | 73 | 37 |
| Genotyped affected relative pairs | 359 | 152 | 146 | 61 |
| Sib pairs | 197 | 106 | 59 | 32 |
| Half-sib pairs | 6 | 5 | 1 | 0 |
| Avuncular pairs | 67 | 23 | 32 | 12 |
| Grandparent-grandchild pairs | 4 | 0 | 2 | 2 |
| First-cousin pairs | 46 | 12 | 23 | 11 |
| Other, more distant pairs | 39 | 6 | 29 | 4 |
| Total families | 367 | 165 | 103 | 99 |
| Total genotyped affected relative pairs | 581 | 252 | 191 | 138 |
Twelve markers spanning the peak of the chromosome 12 candidate interval, as defined in previous reports by both our group and others (Satsangi et al. 1996; Akolkar et al. 1998; Curran et al. 1998; Duerr et al. 1998; Hampe et al. 1999; Ma et al. 1999), were studied in the combined U.K./U.S. panel. Each microsatellite was amplified by PCR using fluorescence-labeled oligoprimers. The fluorescently labeled PCR amplimers were electrophoresed and detected on ABI 373 and 377 DNA sequencers (Applied Biosystems) and were genotyped by either GENESCAN/GENOTYPER (Applied Biosystems) (U.K. panel) or TrueAllele (Cybergenetics) (U.S. panel) software. A control DNA, CEPH 1347-02, was run on each genotyping gel, and the results were used to standardize allele calls between gels and between the two genotyping labs. Genotyping data were checked for Mendelian inconsistencies, by PEDCHECK (O'Connell and Weeks 1998 [also see the University of Pittsburgh Division of Statistical Genetics Web page]).
Marker order was determined by radiation-hybrid mapping using Genebridge 4 and Stanford TNG panels (both from Research Genetics). Map distances were calculated by the ILINK function of the VITESSE package (O'Connell and Weeks 1995 [also see the University of Pittsburgh Division of Statistical Genetics Web page]), on the basis of genotyping data from the family panel and of calculation of multipoint distances between overlapping sets of four markers.
Single- and multipoint linkage analyses were performed by GENEHUNTER-PLUS (Kong and Cox 1997 [also see those authors' GENEHUNTER-PLUS Web page, for GENEHUNTER-PLUS version 1.2]). This modified version of the GENEHUNTER program (Kruglyak et al. 1996) uses a simple one-parameter linear model to compute a nonparametric LOD score, analogous to a parametric LOD score, which can be used for construction of support regions and for comparison of loci. Our analysis used the Spairs statistic, which measures identity-by-descent allele sharing between all pairs of affected relatives. The panel was considered as a whole (IBD overall) and then was divided into UC-only, CD-only, and mixed family subsets.
The results of single-point linkage analysis of the 12 markers studied are presented in table 2. The highest single-point LOD score for IBD overall occurred at D12S83 (LOD score 5.26). Most of the support for linkage comes from the 138 affected relative pairs from UC-only families, in whom the peak single-point LOD score was 3.78 at D12S1586, compared with a peak LOD score of 1.59 at D12S90 in the 252 affected pairs from CD-only families and a peak LOD score of 2.09 at D12S83 in the 191 affected pairs from mixed families.
Table 2.
Single-Point GENEHUNTER-PLUS LOD Scores for 12 Markers Studied in the Combined U.K./U.S. Panel
|
GENEHUNTER-PLUS Single-Point LOD Score for |
|||||||
| Map Positiona(cM) | Microsatellite Locus | Heterozygosity(%) | No. of Alleles Observed | All IBD | CD Only | Mixed | UC Only |
| 0.0 | D12S85 | 68.2 | 13 | 2.31 | .28 | 1.23 | 1.13 |
| 5.3 | D12S368 | 82.5 | 9 | 2.62 | .38 | 1.59 | 1.11 |
| 6.8 | D12S1586 | 84.6 | 13 | 3.11 | .25 | .52 | 3.78 |
| 8.1 | D12S1724 | 78.5 | 13 | 5.21 | 1.52 | 1.45 | 2.49 |
| 10.9 | D12S90 | 76.2 | 11 | 4.20 | 1.59 | 1.23 | 1.41 |
| 11.6 | D12S305 | 66.8 | 10 | 2.09 | .54 | .88 | .80 |
| 12.5 | D12S1700 | 78.7 | 14 | 2.63 | .30 | 1.57 | 1.43 |
| 13.1 | D12S1056 | 70.0 | 6 | 3.25 | .97 | .71 | 1.87 |
| 13.6 | D12S1662 | 71.6 | 9 | 4.41 | 1.01 | .77 | 3.48 |
| 14.4 | D12S83 | 77.4 | 13 | 5.26 | 1.35 | 2.09 | 2.05 |
| 15.1 | D12S1655 | 70.4 | 9 | 2.02 | .56 | .65 | .95 |
| 25.1 | D12S335 | 80.7 | 14 | 1.18 | .01 | .12 | 2.58 |
Marker order was determined by radiation-hybrid mapping, and genetic distance was computed by the VITESSE package.
Results of the multipoint linkage analysis are presented in figure 1. The peak GENEHUNTER-PLUS LOD score for IBD overall was 5.19. A maximum LOD score of 3.91 was observed in the UC-only families, compared with 1.66 in the CD-only families and 1.29 in the mixed families. The discrepancy of 7 cM between the peaks of the linkage curves for UC and CD, reflected in the two peaks of the IBD curve, probably reflects the limited resolution of haplotype-sharing methods of linkage analysis for fine mapping in polygenic diseases (Kruglyak and Lander 1996). However, it is also possible that there is more than one IBD-susceptibility gene in this region, with these exerting differential effects in UC and CD. A precedent for detection of more than one gene per linkage interval comes from fine mapping in mouse models of type 1 diabetes (Podolin et al. 1998).
Figure 1.

Results of multipoint nonparametric linkage analysis for markers across the 25-cM candidate interval on chromosome 12. Results are given for IBD overall and for subsets containing UC-only, CD-only, and mixed families. LOD scores derived from GENEHUNTER-PLUS (Y-axis) are plotted against distance (in cM) from D12S85.
To formally test for heterogeneity between the UC-only and CD-only families, two simulation studies were employed. In the first simulation study, we examined the probability of obtaining the GENEHUNTER-PLUS LOD-score difference that we observed under the null hypothesis of no linkage. For the second simulation study, we examined the probability of randomly dividing our sample into two groups similar in size to our UC-only and CD-only families and achieving a similarly high difference in GENEHUNTER-PLUS LOD scores. Taken together, these two studies show that our division of families is unique—and that the LOD-score difference that we observed would not be obtained very often by chance.
For the first simulation study, we used Morton’s M test (Morton 1956):
 |
This test divides a sample into n groups and then compares the LOD scores in the individual groups versus the combined LOD score in the entire sample. In the equation above, Zi(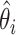 ) is the total LOD score for the families in the ith class (i=1,…,n), and
) is the total LOD score for the families in the ith class (i=1,…,n), and 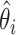 is the maximum-likelihood estimate of the recombination fraction in the ith class. The null hypothesis of no heterogeneity between groups would be represented by the case in which the sum of the individual groups' LOD scores would be exactly equal to the combined sample LOD score (i.e.,
is the maximum-likelihood estimate of the recombination fraction in the ith class. The null hypothesis of no heterogeneity between groups would be represented by the case in which the sum of the individual groups' LOD scores would be exactly equal to the combined sample LOD score (i.e., 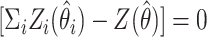 ). We simulated the distribution of this statistic to determine the P value associated with our observed statistic value. In the computer simulation, random genotype data for all the markers in the region were generated 10,000 times each for the group of CD-only families and for the group of UC-only families, and, each time, Morton’s M test was performed using the GENEHUNTER-PLUS LOD scores computed on the basis of the simulated data. The P value for our observed heterogeneity statistic value was determined by counting how often the simulated data produced a heterogeneity statistic value as high as our observed heterogeneity statistic value. The simulations showed that the heterogeneity statistic value that we observed between the UC-only families and the CD-only families is unlikely to occur by chance (P=.0057) and that it therefore represents true evidence of heterogeneity between these two sets of families.
). We simulated the distribution of this statistic to determine the P value associated with our observed statistic value. In the computer simulation, random genotype data for all the markers in the region were generated 10,000 times each for the group of CD-only families and for the group of UC-only families, and, each time, Morton’s M test was performed using the GENEHUNTER-PLUS LOD scores computed on the basis of the simulated data. The P value for our observed heterogeneity statistic value was determined by counting how often the simulated data produced a heterogeneity statistic value as high as our observed heterogeneity statistic value. The simulations showed that the heterogeneity statistic value that we observed between the UC-only families and the CD-only families is unlikely to occur by chance (P=.0057) and that it therefore represents true evidence of heterogeneity between these two sets of families.
The second simulation study that we performed looked at the significance of the division of samples. For this study, we compared the distribution of LOD scores obtained when the available families (367) were randomly split into two groups of sizes similar to those of the UC-only and CD-only groups (99 and 165 families, respectively). To do this, we randomly selected (without replacement) 99 pedigrees from our sample and then another 165 pedigrees and then calculated the maximum GENEHUNTER-PLUS LOD scores for the new samples. This procedure was repeated 10,000 times, to generate a distribution of GENEHUNTER-PLUS LOD-score differences. A difference greater than or equal to our observed UC-CD LOD-score difference (2.25) was observed only 375 times in 10,000 replications (P=.0375). Together, then, these two simulation studies indicate that we have significant heterogeneity in our sample and that clinical diagnosis is a significant factor influencing the LOD-score difference.
With the LOD score of 3.91 for UC-only families, the results of this study provide the first evidence of a “significant” linkage result for UC when the established genomewide criteria of Lander and Kruglyak (1995) are used. The multipoint LOD score of 5.19 for IBD is certainly also respectable in the context of a polygenic disease. Although there is likely to be some positive bias in these results, given that both the U.K. and the U.S. panels contained affected relative pairs known to show increased haplotype sharing in this region, it is also true that the number of affected relative pairs has expanded by a total of 39% beyond those reported originally, and the intuitive expectation that LOD scores from different data sets should be additive can be confounded by heterogeneity in the study populations.
Of interest was the impact that increasing the size of the data set had on the LOD scores, both for IBD overall and for the CD and UC subgroups. In the original U.K. data set (208 affected relative pairs), peak multipoint LOD scores were 3.87 for IBD, 1.08 for CD, and 1.54 for UC (M.P. and D.P.J., unpublished data), and in the U.S. data set (208 affected relative pairs) they were 2.79 for IBD, 1.79 for CD, and 1.82 for UC (Duerr et al. 1998); for the two expanded data sets combined (581 affected relative pairs) the peak multipoint LOD scores are 5.19 for IBD, 1.66 for CD, and 3.91 for UC. Thus, although the evidence for linkage of this region to IBD overall has increased, it can be seen that this is predominantly due to an increase in the strength of linkage to UC, with the LOD score for CD essentially unchanged despite the increase in genotyped families. This suggests relatively more heterogeneity within the CD population, at least with regard to the IBD2 locus, as well as a stronger contribution of this region to UC susceptibility. The latter is corroborated both by the stronger evidence for linkage to UC compared with that to CD, despite the substantially smaller size of the UC group (138 affected relative pairs from UC-only families, compared with 252 affected relative pairs from CD-only families), and, in particular, by the fact that the difference between the LOD score for UC and that for CD is significant in the heterogeneity tests.
The apparently stronger effect that we have observed for UC is likely to have an impact on interpretation of future linkage reports in this region and might, in part, explain the failure of some (CD-dominated) data sets to show significant linkage at markers within the chromosome 12 candidate interval (Brant et al. 1998; Rioux et al. 1998; Vermeire et al. 2000). If the evidence for linkage heterogeneity between the disease subgroups at IBD2 is indicating a major gene effect for UC but a relatively minor one for CD, and if there truly is more linkage heterogeneity at IBD2 within the CD population, then clearly this linkage will be more difficult to detect in panels of families containing a majority of CD relative pairs.
The picture emerging for the IBD2 locus is therefore one of a major contribution with regard to UC susceptibility but probably a relatively minor effect for CD—with the likelihood of substantially more heterogeneity, with regard to IBD2, for CD. Given the findings of the current study, attempts at fine mapping the IBD2 locus should probably focus, at least in the first instance, on individuals and families with UC. The increasing availability of sequence data, single-nucleotide polymorphisms, and information regarding positional candidate genes, together with the accumulation of ever larger IBD subsets, can only help this process—and, thereby, help us to understand the contribution that this locus makes to the pathogenesis of IBD.
Acknowledgments
We thank the families who have taken part in this study, and we thank their clinicians for providing clinical data. The National Association for Crohn’s and Colitis (United Kingdom) and the Crohn’s & Colitis Foundation of America, Inc. (United States) helped in recruitment of study subjects. We also acknowledge Jody Brown, Katherine Kim, April Studinski, Beth Kladny, Ellen Sanders, and Rinesty Sumargo (all of the University of Pittsburgh) and Daphne Lever and Heather Holt (both of the University of Oxford) for their assistance with study-subject recruitment. We also thank Leilei Zhang, Lara Chensny, John Jaminet, and Michael Ford (all of the University of Pittsburgh), David van Heel and Paulomi Vyas (both of the University of Oxford), and the staff at Oxagen (United Kingdom) for their technical expertise, and we thank Mark Lathrop and John Bell (both of the University of Oxford) for their help and advice. M.P. and J.S. hold Medical Research Council awards, and R.H.D. received support from the Crohn’s & Colitis Foundation of America, Inc., National Institute of Health grant DK-50993, and the Scaife Family Foundation.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- GENEHUNTER-PLUS Web page, ftp://galton.uchicago.edu/pub/kong (for GENEHUNTER-PLUS version 1.2)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for CD [MIM 266600], UC [MIM 191390], IBD1 [MIM 266600], and IBD2 [MIM 601458])
- University of Pittsburgh Division of Statistical Genetics Web page, http://watson.hgen.pitt.edu/pub/register (for PEDCHECK and VITESSE programs)
References
- Akolkar P, Gulwani-Akolkar B, Lin XY, Danzi SE, Pergolizzi R, Katz S, Terwilliger J, Silver J (1998) Fine mapping of regions linked to Crohn's disease on chromosomes 12 and 16. Am J Hum Genet Suppl 63:A279 [Google Scholar]
- Annese V, Latiano A, Bovio P, Forabosco P, Piepoli A, Lombardi G, Andreoli A, Astegiano M, Gionchetti P, Riegler G, Sturniolo GC, Clementi M, Rappaport E, Fortina P, Devoto M, Gasparini P, Andriulli A (1999) Genetic analysis in Italian families with inflammatory bowel disease supports linkage to the IBD1 locus—a GISC study. Eur J Hum Genet 7:567–573 [DOI] [PubMed] [Google Scholar]
- Brant SR, Fu Y, Fields CT, Baltazar R, Ravenhill G, Pickles MR, Rohal PM, Mann J, Kirschner BS, Jabs EW, Bayless TM, Hanauer SB, Cho JH (1998) American families with Crohn's disease have strong evidence for linkage to chromosome 16 but not chromosome 12. Gastroenterology 115:1056–1061 [DOI] [PubMed] [Google Scholar]
- Cavanaugh JA, Callen DF, Wilson SR, Stanford PM, Sraml ME, Gorska M, Crawford J, Whitmore SA, Shlegel C, Foote S, Kohonen-Corish M, Pavli P (1998) Analysis of Australian Crohn's disease pedigrees refines the localization for susceptibility to inflammatory bowel disease on chromosome 16. Ann Hum Genet 62:291–298 [DOI] [PubMed] [Google Scholar]
- Cho JH, Nicolae DL, Gold LH, Fields CT, Labuda MC, Rohal PM, Pickles MR, Qin L, Fu Y, Mann JS, Kirschner BS, Jabs EW, Weber J, Hanauer SB, Bayless TM, Brant SR (1998) Identification of novel susceptibility loci for inflammatory bowel disease on chromosomes 1p, 3q and 4q: evidence for epistasis between 1p and IBD1. Proc Natl Acad Sci USA 95:7502–7507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran ME, Lau KF, Hampe J, Schreiber S, Bridger S, Macpherson AJS, Cardon LR, Sakul H, Harris TJR, Stokkers P, van Deventer SJH, Mirza M, Raedler A, Kruis W, Meckler U, Theuer D, Herrmann T, Gionchetti P, Lee J, Mathew C, Lennard-Jones J (1998) Genetic analysis of inflammatory bowel disease in a large European cohort supports linkage to chromosomes 12 and 16. Gastroenterology 115:1066–1071 [DOI] [PubMed] [Google Scholar]
- Duerr RH, Barmada MM, Zhang L, Davis S, Preston RA, Chensny LJ, Brown JL, Ehrlich GD, Weeks DE, Aston CE (1998) Linkage and association between inflammatory bowel disease and a locus on chromosome 12. Am J Hum Genet 63:95–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampe J, Schreiber S, Shaw SH, Lau KF, Bridger S, Macpherson AJS, Cardon LR, Sakul H, Harris TJR, Buckler A, Hall J, Stokkers P, van Deventer SJH, Nürnberg P, Mirza MM, Lee JCW, Lennard-Jones JE, Mathew CG, Curran ME (1999) A genomewide analysis provides evidence for novel linkages in inflammatory bowel disease in a large European cohort. Am J Hum Genet 64:808–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugot JP, Laurent-Puig P, Gower-Rousseau C, Olson JM, Lee JC, Beaugerie L, Naom I, Dupas JL, Van Gossum A, Groupe d’Etude Thérapeutique des Affections Inflammatories Digestives, Orholm M, Bonaiti-Pellie C, Weissenbach J, Mathew CG, Lennard-Jones JE, Cortot A, Colombel JF, Thomas G (1996) Mapping of a susceptibility locus for Crohn's disease on chromosome 16. Nature 379:821–823 [DOI] [PubMed] [Google Scholar]
- IBD Genetics Consortium, The (2000) The international IBD genetics consortium confirms linkage of Crohn's disease to a locus on chromosome 16 (IBD1). Gastroenterology 118 Suppl 2:A708 [Google Scholar]
- Kirsner JB (1973) Genetic aspects of inflammatory bowel disease. Clin Gastroenterol 2:557–576 [Google Scholar]
- Kong A, Cox NJ (1997) Allele-sharing models: LOD scores and accurate linkage tests. Am J Hum Genet 61:1179–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Lander ES (1996) Limits on fine mapping of complex traits. Am J Hum Genet 58:1092–1093 [PMC free article] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed] [Google Scholar]
- Ma Y, Ohmen JD, Li Z, Bentley LG, McElree C, Pressman S, Targan SR, Fischel-Ghodsian N, Rotter JI, Yang H (1999) A genome-wide search identifies potential new susceptibility loci for Crohn's disease. Inflamm Bowel Dis 5:271–278 [DOI] [PubMed] [Google Scholar]
- Mandal DM, Sorant AJ, Pugh EW, Marcus SE, Klein AP, Mathias RA, O'Neill J, Temiyakarn LF, Wilson AF, Bailey-Wilson JE (1999) Environmental covariates: effects on the power of sib-pair linkage methods. Genet Epidemiol 17 Suppl 1:S643–S648 [DOI] [PubMed] [Google Scholar]
- Morton N (1956) The detection and estimation of linkage between the genes for elliptocytosis and the Rh blood type. Am J Hum Genet 8:80–96 [PMC free article] [PubMed] [Google Scholar]
- O'Connell JR, Weeks DE (1995) The VITESSE algorithm for rapid exact multilocus linkage analysis via genotype set-recoding and fuzzy inheritance. Nat Genet 11:402–408 [DOI] [PubMed] [Google Scholar]
- ——— (1998) PedCheck: a program for identification of genotyping incompatibilites in linkage analysis. Am J Hum Genet 63:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmen JD, Yang H-Y, Yamomoto KK, Zhao H-Y, Ma Y, Bentley LG, Huang Z, Gerwehr S, Pressman S, McElree C, Targan S, Rotter JI, Fischel-Ghodsian N (1996) Susceptibility locus for inflammatory bowel disease on chromosome 16 has a role in Crohn's disease, but not in ulcerative colitis. Hum Mol Genet 5:1679–1683 [DOI] [PubMed] [Google Scholar]
- Parkes M, Satsangi J, Lathrop GM, Bell JI, Jewell DP (1996) Susceptibility loci in inflammatory bowel disease. Lancet 348:1588 [DOI] [PubMed] [Google Scholar]
- Podolin PL, Denny P, Armitage N, Lord CJ, Hill NJ, Levy ER, Peterson LB, Todd JA, Wicker LS, Lyons PA (1998) Localization of two insulin-dependent diabetes (Idd) genes to the Idd10 region on mouse chromosome 3. Mamm Genome 9:283–286 [DOI] [PubMed] [Google Scholar]
- Probert CS, Jayanthi V, Hughes AO, Thompson JR, Wicks AC, Mayberry JF (1993) Prevalence and family risk of ulcerative colitis and Crohn's disease: an epidemiological study among Europeans and south Asians in Leicestershire. Gut 34:1547–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioux JD, Daly MJ, Green T, Stone V, Lander ES, Hudson TJ, Steinhart AH, Bull S, Cohen Z, Greenberg G, Griffiths A, McLeod R, Silverberg M, Williams CN, Siminovitch KA (1998) Absence of linkage between inflammatory bowel disease and selected loci on chromosomes 3, 7, 12 and 16. Gastroenterology 115:1062–1065 [DOI] [PubMed] [Google Scholar]
- Satsangi J, Parkes M, Louis E, Hashimoto L, Kato N, Welsh K, Terwilliger JD, Lathrop GM, Bell JI, Jewell DP (1996) Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet 14:199–202 [DOI] [PubMed] [Google Scholar]
- Satsangi J, Rosenberg WMC, Jewell DP (1994) The prevalence of inflammatory bowel disease in relatives of patients with Crohn’s disease. Eur J Gastroenterol Hepatol 6:413–416 [Google Scholar]
- Suarez BK, Hampe CL, van Eerdwegh P (1994) Problems of replicating linkage claims in psychiatry. In: Gershon ES, Cloninger CR (eds) Genetic approaches to mental disorders. American Psychiatric Press, Washington, DC, pp 23–46 [Google Scholar]
- Subhani J, Montgomery SM, Pounder RE, Wakefield AJ (1998) Concordance rates of twins and siblings in inflammatory bowel disease (IBD). Gastroenterology 114(4, pt 2): A1093 [Google Scholar]
- Tysk C, Lindberg E, Järnerot G, Flodérus-Myrhed B (1988) Ulcerative colitis and Crohn's disease in an unselected population of monozygotic and dizygotic twins. A study of heritability and the influence of smoking. Gut 29:990–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeire S, Peeters M, Vlietinck R, Parkes M, Satsangi J, Jewell D, Rutgeerts P (2000) Exclusion of linkage of Crohn's disease to previously reported regions on chromosomes 12, 7, and 3 in the Belgian population indicates genetic heterogeneity. Inflamm Bowel Dis 6:165–170 [DOI] [PubMed] [Google Scholar]