

Abstract
Heme oxygenases (HOs) catalyze the oxidation of heme to biliverdin, carbon monoxide (CO), and free iron. Iron acquisition is critical for invading microorganisms to enable survival and growth. Here we report the crystal structure of ChuS, which displays a previously uncharacterized fold and is unique compared with other characterized HOs. Despite only 19% sequence identity between the N- and C-terminal halves, these segments of ChuS represent a structural duplication, with a root-mean-square deviation of 2.1 Å between the two repeats. ChuS is capable of using ascorbic acid or cytochrome P450 reductase-NADPH as electron sources for heme oxygenation. CO detection confirmed that ChuS is a HO, and we have identified it in pathogenic Escherichia coli O157:H7. Based on sequence analysis, this HO is present in many bacteria, although not in the E. coli K-12 strain. The N- and C-terminal halves of ChuS are each a functional HO.
Keywords: ChuS, carbon monoxide, structural repeat, heme degredation
The incorporation of iron into proteins as a biocatalyst or electron carrier enables organisms to fulfill many vital biological processes, including photosynthesis, N2 fixation, methanogenesis, H2 production and consumption, respiration, the tricarboxylic acid cycle, oxygen transport, gene regulation, and DNA biosynthesis (1). The importance of iron for bacterial survival and pathogenesis is evident by the variety of iron-acquisition and transport systems that have evolved, such as the secretion of low-molecular-weight (low-MW) siderophores capable of binding free iron and transporting it into the cell via specific membrane receptors (2). As part of a coordinated immune response against invading microorganisms, mammals specifically limit iron availability by producing the iron-binding proteins transferrin and lactoferrin, which depress free extracellular iron to levels insufficient to sustain bacterial growth (3). Alternatively, invading pathogens may acquire iron directly from host iron sources such as heme, hemoglobin, or hemopexin either through the production of specific outer membrane receptors that bind directly to host heme-sequestering proteins or through the secretion of heme-binding proteins (hemophores) that then deliver the heme-protein complex to bacterial cell-surface receptors (4). Enterohemorrhagic pathogens, such as Escherichia O157:H7, for which isolates from human patients have been shown to have stimulated growth in the presence of heme and hemoglobin, may induce hemolytic lesions to gain access to these serum sources of iron (5).
By using a laboratory strain of Escherichia coli [1017 (ent:Tn5)] that was unable to import heme into the cytoplasm, it was established that the outer membrane receptor ChuA (E. coli heme-utilization gene A) is required for heme uptake along with TonB, an energy-transducing protein associated with ExbB and ExbD that uses the proton motive force of the cytoplasmic membrane for the passage of ligands into the periplasm (6). Four other members of the heme uptake operon from Yersinia enterocolitica are required to complete heme uptake, transport, and use as an iron source by E. coli K-12 (7, 8), including the periplasmic heme-binding protein HemT, the heme permease protein HemU, the ATP-binding hydrophilic protein HemV, and the protein HemS, whose role may be to regulate the amount of non-protein-associated heme in the cell (8).
Restriction mapping and sequence analysis of selected regions of E. coli O157:H7 DNA, the serotype responsible for outbreaks of hemorrhagic colitis and hemolytic uremic syndrome, has revealed that the organization of the heme transport locus of this strain is strikingly similar to that of other enteric bacteria, such as Y. enterocolitica (9). This organization is especially true with respect to the region encoding heme outer membrane receptors such as ChuA, which are followed by a short gap, and then by ChuS or one of its homologues. In Shigella dysenteriae the intergenic region between ShuA and ShuS are separated by a 48-nt region with sequence homology to the consensus Fur box but no obvious -10 and -35 promoter elements (7). Furthermore, a promoter was likely to be present within the intergenic region because a mini-Tn10 insertion in ShuA was not polar with respect to ShuS expression in minicells (10).
Although many reports have focused on the proteins involved in heme transport across the outer membrane and periplasm, the fate of heme once it has reached the bacterial cytoplasm has only recently received attention. Investigations of eukaryotes revealed that release of iron stores requires the heme porphyrin ring to be degraded by monooxygenases known as heme oxygenases (HOs). HOs are enzymatically unique, as they use heme as both a substrate and a cofactor in the binding of oxygen for intramolecular degradation of the porphyrin macromolecule to biliverdin, CO, and free iron (11). Several bacterial HOs have been identified, including HemO (12), HmuO (13), IsdG/I (14), cyanobacterial HO-1 and HO-2 (15), and PigA/BphO (16). All of these were reviewed (17), and the structures of HemO (18), HmuO (19), and PigA (16) have been determined, as well as a partial structural characterization for BphO (20). The crystal structures of HemO, HmuO, and PigA all share a strong structural similarity to mammalian HOs in that they are mainly α-helical and sandwich heme between two helices. At present, no sequence comparison or structural analysis has identified a HO in an E. coli strain.
Here we report the crystal structure of ChuS, which has a fold with two tandem repeats and does not show structural similarity to known mammalian or bacterial HOs. ChuS is capable of binding heme and can break down heme using ascorbic acid or cytochrome P450 reductase-NADPH (CPR) as electron sources for oxygenation. CO detection by gas chromatography confirmed that ChuS is a HO, and we identified it in E. coli O157:H7. In addition, HO activity occurs in the independently expressed N- and C-terminal halves of ChuS.
Materials and Methods
Sequence Analysis, Protein Expression, and Purification. Sequence analysis was carried out with blast (21). Full-length ChuS and the N- and C-terminal halves of ChuS were subcloned into pET expression vectors, yielding fusion proteins to either an N- or C-terminal His tag or an N-terminal GST tag. Based on the results of small-scale expression trials, we selected the following constructs: ChuS fused to an N-terminal His-6 tag (ChuS), the N-terminal half of ChuS fused to a C-terminal His-6 tag (N-ChuS), and the C-terminal half of ChuS fused to an N-terminal GST tag (C-ChuS). In each case, 1-liter cultures of BL21(DE3) cells carrying plasmid for the respective recombinant protein were grown at 37°C in Terrific Broth (Bioshop, Burlington, Canada) supplemented with 100 μg/ml ampicillin. Protein expression was induced by using 0.4 mM isopropyl β-d-thiogalactoside for 5 h. ChuS protein substituted with selenomethionine was expressed in the metA- E. coli strain DL41 in LeMaster medium (22).
All proteins were purified by using standard methods. Briefly, His-6-tagged proteins were purified by nickel nitrilotriacetate agarose batch purification in phosphate buffer (pH 8.0), followed by purification by Resource Q and Hi-Trap metal-chelating columns on an ΔTKA Explorer FPLC. Collected fractions were monitored by SDS/PAGE, for which those that contained pure protein were pooled together and dialyzed against 50 mM Tris·HCl buffer, pH 8.0/150 mM NaCl for subsequent crystallization, or 100 mM sodium phosphate buffer (pH 7.0) for HO spectral and activity measurements. C-ChuS was purified on a GSTrap FF column. For more details, see Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Crystallization of Full-Length ChuS. Crystals were obtained by the hanging-drop vapor diffusion method by mixing 2.5 μl of 20 mg/ml ChuS with 2.5 μl of reservoir solution consisting of 20% (wt/vol) polyethylene glycol 3350 and 100 mM Bis-Tris (pH 6.5). Crystals appeared after 1-3 d. ChuS crystals were further improved by microseeding using 12-20% (wt/vol) polyethylene glycol 3350. Selenomethionine derivative crystals were obtained in the same way. For cryoprotection, crystals were soaked in the reservoir solution containing ethylene glycol [concentration increased stepwise to 30% (vol/vol)], picked up using a nylon loop, and flash-cooled in the N2 cold stream at 100 K. Two nonisomorphous crystal forms belonging to space group P21 were obtained. Crystals having the larger unit cell contain three molecules in the asymmetric unit, whereas those with the smaller unit cell contain only one molecule.
Data Collection and Structure Determination. Native and multiple-wavelength anomalous dispersion (MAD) data sets were collected, respectively, for the two different crystal forms at beamlines 17-ID and 19-BM of the Advance Photon Source, Argonne National Laboratory (University of Chicago, Argonne, IL). The native data were collected for 261° in total, with 1.0° oscillation. Selenomethionine MAD data were collected at peak and inflection wavelengths for 360° and remote wavelength for 180°, all with 1.0° oscillation. Data were processed with hkl2000 (23). The initial structure of the three-molecule form was determined by using the MAD data with solve (24). Density modification, including 3-fold noncrystallographic symmetry averaging, was carried out with resolve (25). Automatic chain tracing was performed by using resolve (25), and the remainder of the model was built by manual fitting using xtalview/xfit (26). When the model of one of the three molecules was ≈80% built, it was used as a search model in molecular replacement using data obtained from the single-molecule form. This technique resulted in an unambiguous solution that was subsequently refined using cns (27).
Reconstitution with Heme and Absorbance Measurements. Heme complexes of ChuS, N-ChuS, and C-ChuS were prepared by dissolving heme into 0.5% (vol/vol) ethanolamine and adding small volumes of this mixture to protein until a final ratio of 4:1 (mol/mol) was obtained for ChuS and 2:1 for N-ChuS and C-ChuS. ChuS-heme and N-ChuS-heme were then applied to Resource Q or Superdex 200 Prep Grade columns, respectively, to remove noncoordinated heme. Reconstituted protein-heme complex concentrations were determined by adapting the reported extinction coefficient for ShuS (ε410) of 79.5 and 159 mM-1·cm-1 for ChuS and N-ChuS, respectively (28). Pure fractions of C-ChuS were concentrated and used for spectrophotometric analysis and CO detection in which 2× heme was added to protein before measurements. Absorbance data were then collected by using a microplate spectrophotometer (Bio-Tek). As a control in separate experiments, 10 μM catalase and 10 μM superoxide dismutase were added to the reaction wells, followed by ascorbic acid or CPR-NADPH, and the difference spectra were recorded. Quadruplicate series of ChuS-heme reconstitution titrations were also conducted to examine the ChuS-heme interaction.
CO Activity Assay and Inhibition. Briefly, in 2-ml vials, 250-μl reaction volumes of 20 μM heme and 10 μM ChuS, N-ChuS, or C-ChuS in 100 mM phosphate buffer (pH 7.0) were incubated with constant shaking at room temperature for 6 min, at which time enzymatic heme degradation was initiated by adding either CPR and NADPH or ascorbic acid at final concentrations of 17.7 nM and 135 μM or 50 μM, respectively. Vials were then sealed with screw caps and blue silicone rubber septa, and the headspace above each was immediately purged with CO-free air, and incubated for 45 min at 24°C with constant shaking. The reaction was stopped by placing the vials on powdered dry ice (-78°C), where they remained for ≈30 min. The amount of liberated CO in the headspace of each vial was measured using a gas chromatograph equipped with an HgO reduction detector (Trace Analytical, Newark, DE). The amount of CO resulting from ChuS-catalyzed breakdown of heme was determined by comparing peak area measurements for CO against linear CO standard curves (n = 20 determinations; average correlation coefficient, 0.993) (Fig. 5). Technical details are described in refs. 29 and 30. In a parallel experiment, Sn(IV) mesoporphyrin IX dissolved in 0.5% (vol/vol) ethanolamine was also added during initial mixing, which acted as a competitive inhibitor.
Fig. 5.

Compiled average CO detected for ChuS, N-ChuS, and C-ChuS, with either ascorbic acid (red) or CPR-NADPH (blue) as an electron source for heme oxygenation. Error bars represent standard errors.
Dynamic Light Scattering (DLS) of C-ChuS and N-ChuS. DLS was conducted for ChuS, ChuS-heme, and N-ChuS each at a 1 mg/ml concentration in 100 mM phosphate buffer (pH 7.0) using a DynaPro99 instrument (Protein Solutions, Charlottesville, VA). DLS was also conduced on samples of 1 mg/ml ChuS and ChuS-heme in 50 mM Tris/150 mM NaCl, pH 8.0.
EPR Experiments. EPR spectra were obtained on a Bruker EMX spectrometer at X-band fitted with a high spectral quality cavity equipped with a liquid helium flow cryostat (Oxford Instruments, Oxon, U.K.). The standard instrument conditions were temperature, 10 K; modulation amplitude, 20 G; and power, 2 mW at a gain of 1 × 104.
Results
Expression and Purification. ChuS overexpression resulted in cells exhibiting a blue-green pigment characteristic of biliverdin accumulation (11, 20, 31). After nickel nitrilotriacetate agarose purification and dialysis in low-ionic-strength Tris buffer, fractions containing ChuS appeared purple and turned to a straw color over time. EPR of ChuS fractions after following nickel nitrilotriacetate agarose purification did not reveal the presence of any coordinated metal ion, and spectral data did not reveal the source of the pigmentation. This pigmentation was not observed in ChuS grown in LeMaster medium in DL41(DE3) cells or for the N-ChuS or C-ChuS constructs. The purple coloration of ChuS was lost after the subsequent FPLC anion exchange step. Expression of full-length ChuS and N-ChuS yielded >60 mg of pure protein from 1 liter of culture. Recombinant C-ChuS was insoluble in pilot expression trials for N- or C-terminal His-tagged versions and could only be expressed in soluble form as a GST-C-ChuS construct. Average yields of 2.6 mg of pure GST-C-ChuS per liter of culture were obtained.
Structure Determination and Analysis. Two different crystal forms were obtained from the same crystallization conditions, that shared the same overall morphology and space group (P21), but differed in unit cell dimensions and number of molecules in the asymmetric unit. We collected MAD data for the three-molecule crystal form and native data for the single-molecule crystal form, and the crystal structure of ChuS was solved by using a combination of MAD and molecular replacement with the two crystal forms. The higher resolution (2.15-Å) structure from the single-molecule form is presented here, although the final structures for ChuS from the two crystal forms are essentially identical, with an average rms deviation of 0.92 Å. The final structure of ChuS contains residues 1-337 of a total 342 aa with no ligand. Because of an absence of electron density, a gap exists in the structure between residues 12 and 23 (12-QNPGKYARDI-23), and between residues 165 and 173 (165-KAVDAPV-173). In total, 99.7% of residues are within the allowed region of the Ramachandran plot. Only one residue, Asp-114, was in the disallowed region and resides within a surface-exposed β-turn. Final refinement statistics are summarized in Table 1.
Table 1. Diffraction data and refinement statistics.
| Native | Peak | Inflection | Remote | |
|---|---|---|---|---|
| Space group | P21 | P21 | ||
| Cell dimensions | ||||
| a, Å | 41.683 | 46.072 | ||
| b, Å | 57.878 | 194.172 | ||
| c, Å | 59.575 | 60.461 | ||
| β, ° | 96.02 | 100.383 | ||
| No. of molecules, ASU | 1 | 3 | ||
| Wavelength, Å | 0.9795 | 0.9803 | 0.9804 | 0.9642 |
| Resolution range, Å | 50–2.15 | 50–2.60 | 50–2.60 | 50–2.40 |
| Total reflections | 53,596* | 170,485† | 176,691† | 149,352† |
| Unique reflections | 14,606* | 32,694† | 32,966† | 40,453† |
| Completeness‡, % | 96.9 (82.8) | 99.8 (99.6) | 99.7 (98.4) | 96.3 (76.5) |
| Rsym(I), % | 7 | 9.7 | 9.1 | 7.8 |
| I/σI‡ | 15.0 (2.4) | 17.4 (2.4) | 16.6 (2.0) | 11.4 (1.0) |
| Refinement (single-molecule form) | ||||
| Resolution range, Å | 50–2.15 | |||
| Rwork, % | 20.1 | |||
| Rfree, % | 26.3 | |||
| No. reflections total/Rfree | 12,315/744 | |||
| 2,428/331/2,75 | ||||
| No. atoms, protein/solvent (H2O)/total | 9 | |||
| β factors Å2, | 29.7/48.6/32.0 | |||
| protein/solvent (H2O)/total | ||||
| rms deviation for bond length, Å/bond angle, ° | 0.009/1.44 |
Number of reflections after merging
Number of reflections without merging of Bijovet pairs
Values in parentheses are for the outermost shell (2.23–2.15 Å)
The overall structure of ChuS comprises a central core of two large pleated β-sheets, each consisting of nine antiparallel β-strands sandwiched together and bowing outward in a saddle motif (Fig. 1). Each β-sheet is flanked at its N terminus by one pair of parallel α-helices and at its C terminus by a set of three α-helices in an α-loop-α-loop-α configuration. Two large clefts are on opposite sides of the central core of β-pleated sheets, with the third side of the clefts delineated by the flanking sets of three α-helices. A long stretch of coil connecting the N- and C-terminal halves runs from I156 to V183, in which the density gap occurs (Fig. 1). Structural comparison of ChuS against structures within the SCOP (Structural Classification of Proteins) database (32) was performed with the program ssm (33). This search did not reveal any significant match; hence, ChuS represents a previously uncharacterized fold.
Fig. 1.

Ribbon diagram of ChuS. The core of nine antiparallel β-strands is visible in the front face. The gaps in the structure are shown with dashed lines.
ChuS Contains a Structural Duplication. On close inspection, the two halves of ChuS were found to be structurally similar. Including side chains, the two halves superimpose to an rms deviation of 2.1 Å (Fig. 2). Sequence alignment of the two halves reveals only 19.0% identity. Nevertheless, at the 3D level, many residues are well conserved structurally, including Arg-29 and Arg-209, Phe-127 and Phe-304, and Tyr-138 and Tyr-315. The two halves of ChuS associate across the central portion of the β-pleated sheets. At the interface, residues from the C-terminal half are hydrophobic (Val-244, Val-250, Phe-274, Lys-276), point toward the interior of the C-terminal subunit, and expose regions of the backbone across the interface. In contrast, most interface residues from the N-terminal half are hydrophilic (Asn-66, Tyr-70, His-73, Asn-75, Arg-95) and point across the interface between the two halves. The side chains of the N-terminal-interacting residues are in contact with the backbone region of the C-terminal half. This structural similarity suggests that the two halves of ChuS may be functionally redundant or that they have dual functions. Furthermore, the second gap in the structure is exactly at the midpoint of the ChuS sequence in a flexible/disordered linker connecting the two halves.
Fig. 2.

The two halves of ChuS superimposed, with an overall rms deviation of 2.1 Å. The N-terminal portion of ChuS is shown in red, and the C-terminal portion is shown in blue.
Spectral Properties of the Heme-ChuS Complex. Based on difference spectra comparing free heme to full-length ChuS, N-ChuS, and C-ChuS, we determined that all three formed complexes when reconstituted with heme (Figs. 3). The spectra of ChuS resemble those of other HOs in that a Soret maximum is evident at 408 nm, with a smaller set of peaks for the β-band at 545 nm and for the α-band at 580 nm, suggesting that the ChuS-heme complex formed is ferric hexacoordinate in the high-spin state at neutral pH (34). The spectrum for N-ChuS has a peak at 402 nm (which is smaller than for ChuS), a smaller peak at 530 nm, and a shoulder at 625 nm. Although the spectrum for N-ChuS with heme did not show definitive heme coordination, it remained unchanged after gel filtration, indicating a specific interaction between the two. C-ChuS with heme showed a broad peak centered at 384 nm, a smaller peak at 540 nm, and two small shoulders at 585 and 625 nm.
Fig. 3.

Spectral analysis. Full-length ChuS and the N- and C-terminal halves of ChuS all form complexes when reconstituted with heme, with peaks at 408, 402, and 384 nm, respectively (black lines). (A) After addition of ascorbic acid as an electron donor, ChuS and its two halves were all capable of heme oxygenation, as evident from the spectral shifts (gray lines). Absorbance values corresponding to wavelengths >500 nm have been multiplied by 10 to amplify absorbance signals corresponding to the β- and α-bands. (B) Similar to A, but NADPH and the P450 reductase system were used as the electron source.
Despite structural similarity between the two halves, the spectra for C-ChuS and N-ChuS differed, perhaps indicating different environments for heme coordination. Based on absorbance at 408 nm during ChuS-heme reconstitution and assuming two binding sites, the estimated Kd is 1.0 ± 0.3 μM (n = 3) based on the following expression.
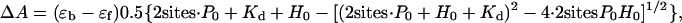 |
where P0 is the amount of protein, H0 is the concentration of heme, and (εb - εf) is the difference in the Soret absorption spectra between the bound and free states (see Fig. 6, which is published as supporting information on the PNAS web site). This estimated Kd suggests a slightly weaker association than that characterized for the mammalian hHO-1 (0.84 ± 0.2 μM) (35, 36) but tighter than that for other bacterial heme oxygenases: HmuO (2.5 ± 1.0 μM) (37), IsdG (5.0 ±1.5 μM), and IsdI (3.5 ± 1.4 μM) (14).
Heme Oxygenation by ChuS. Bacterial and mammalian HOs use the CPR-NADPH system, ferredoxin, flavodoxin, or ascorbic acid as reducing partners in vitro (11, 34, 38-41). To effectively monitor and quantify the HO activity of ChuS, 50 μM ascorbic acid was used. This concentration is between 100-fold (42) and 350-fold (34) less than that used for characterizing other HOs. The use of this lower concentration of ascorbic acid demonstrates highly efficient catalysis of ChuS with this electron donor. By using small amounts of ascorbic acid, heme degradation through coupled oxidation was minimized (43). To examine the heme-degrading activity of ChuS, we monitored the reaction by UV-visible spectroscopy after the addition of ascorbic acid (Fig. 4). As the reaction proceeded, the Soret peak at 408 nm decreased, the small broad peak at 560 nm increased, and the absorption at 680 nm initially increased then decreased. Because the addition of catalase and superoxide dismutase did not affect the trends of the spectral change, this effect can be attributed to specific heme degradation by ChuS and its variants. These spectral shifts are similar to those observed for other HOs, which suggests the formation of biliverdin and free iron rather than the Fe3+-biliverdin complex formed by HO-1. Furthermore, similar spectral shifts were also observed for heme reconstituted to N-ChuS and C-ChuS, indicating that both halves of ChuS are capable of degrading heme independently (Fig. 3A). Similar end-point spectra were also observed for ChuS, N-ChuS, and C-ChuS when CPR was used as the electron donor (Fig. 3B).
Fig. 4.

Spectral change of ChuS reconstituted with heme, monitored over time after ascorbic acid addition. Absorbance measurements between 500 and 700 nm have been multiplied by 5 to amplify the change in the β- and α-bands. Arrows follow changes in absorption at 408, 545, and 680 nm during reaction progression. The spectral change corresponds with the formation of the product, biliverdin.
Detection of CO Release. All three constructs were capable of releasing CO (Fig. 5). When ascorbic acid was used as the electron source, C-ChuS was 1.5 times more effective at CO production than ChuS and 10 times more effective then N-ChuS, whose release was only slightly above the background level of heme breakdown. When N-ChuS and C-ChuS were added to the same reaction vessel, the amount of CO evolved was greater than the levels observed for ChuS alone, but not as great as those observed for C-ChuS. When CPR was used as the electron source, the amount of CO produced by ChuS and C-ChuS was five times less than when ascorbic acid was used. In contrast, N-ChuS produced more than four times as much CO as when ascorbic acid was used and was slightly more effective at producing CO than either the C-ChuS or full-length ChuS.
Organization of the Heme Uptake Operon. Given the similarity between the heme uptake operon of Y. enterocolitica and E. coli O157:H7, it is likely that homologues of HemR, HemT, HemU, HemV, and HemS would be required for heme use in E. coli. Sequence comparison between the heme uptake loci of these organisms revealed the homologous genes ChuA, ChuT, ChuU, various putative ATP-binding components of the heme transport system, and ChuS. Sequence identities ranged between 38% and 68%, and similarities were between 58% and 80%. The highest identity was conserved between HemS and ChuS, suggesting conservation of function in iron acquisition and prevention of heme toxicity. The lower identity with the other proteins may reflect differences between the periplasmic and membrane environments of the two organisms. Furthermore, sequence comparison revealed the following ChuS orthologs with strong sequence similarity: ChuS (CFT073), ShuS (Shigella), EhuS (Enterobacter and Erwinia), HemS (Yersinia), plu2633 (Photorhabdus), HmuS (Rhizobium and Sinorhizobium), and BhuS (Bordetella), suggesting that this HO is present in many other bacteria (For sequence alignments, see Fig. 7, which is published as supporting information on the PNAS web site).
DLS. The derived molecular radii of N-ChuS, ChuS, and ChuS-heme in phosphate buffer and ChuS and ChuS-heme in Tris buffers were all ≈2.90 nm, corresponding to a MW of 41 kDa for an elongated protein. This is in agreement with that expected for an N-ChuS dimer or ChuS monomer. The longest dimension of ChuS from the crystal structure corresponds to a radius of 2.88 nm, indicating that the N-ChuS homodimer is similar in shape to full-length ChuS. The monomeric organization of ChuS was confirmed by using a gel filtration column calibrated with a set of MW standards (data not shown). This finding is contradictory to a previous report showing that a homologue of ChuS, ShuS (98% identity), forms an oligomer with an estimated MW of 650 kDa (28). When ChuS or ChuS reconstituted with heme were examined in the Tris buffer system after 4 d of incubation at 4°C, DLS results suggested the formation of various high-MW aggregates without any detection of a ChuS monomer. In contrast, the protein remained monomeric after 1 week in phosphate buffer.
Inhibition by Sn(IV) Mesoporphyrin. In the presence of Sn(IV) mesoporphyrin IX, a competitive inhibitor for heme binding (44), a dose-dependent decrease in HO activity was observed from 2- to 7-fold. This level of inhibition is similar to that characterized for other HOs in the presence of competitive inhibitors (44).
Discussion
For pathogenic organisms that cause hemolytic lesions, such as the E. coli O157:H7 and CFT073 strains, heme degradation by oxygenation may be a significant mechanism for iron acquisition and/or prevention of heme toxicity. Monitoring the spectral changes of ChuS reconstituted with heme when ascorbic acid or NADPH-CPR were used as electron donors provided direct evidence that ChuS catalyzes the breakdown of heme to biliverdin, CO, and free iron. In vivo, E. coli may use reductants, such as ferredoxins and flavodoxin, as electron sources for heme oxygenation (20). The liberation of CO and inhibition of CO release by addition of the prototypical HO inhibitor Sn(IV) mesoporphyrin IX support ChuS as a HO. Although the spectra of N-ChuS and C-ChuS does not definitively suggest HO activity, unequivocal detection of CO release indicates that both halves of ChuS possess the ability to break down heme. Based on the significant sequence similarity of proteins between Enterobacter, Erwinia, Shigella, and Yersinia to E. coli ChuS, we can extend our identification of ChuS as an HO to these other bacterial genera. Further work must be done to characterize heme breakdown products resulting from the action of N-ChuS and C-ChuS and the underlying mechanisms.
Although the overall structure of all bacterial and mammalian HOs characterized to date share the same mainly α-helical fold, the structure of ChuS is unique in that it is comprised of two central sets of antiparallel β-sheets, each flanked by two pairs of α-helices. The absorption spectra of the two halves of ChuS after reconstitution with heme are notably different from each other and the full-length protein. This observation is especially true for the C-terminal half, whose broad peak centered at 384 nm is unique compared with the Soret range of 402-412 nm observed for other HOs identified to date (17).
The structure of ChuS consists of a structural duplication, despite the fact that the two halves share only 19% sequence identity. This unique structural feature may be explained in part by the expression profiles of recombinant full-length ChuS, N-ChuS, and C-ChuS. Recombinant forms of full-length ChuS and N-ChuS were highly expressed, whereas the expression of C-ChuS was at least 30 times lower and required a large GST tag to increase protein solubility. Considering its structure, this poorer solubility of C-ChuS is not surprising. In contrast to the N-terminal half, the interface of the C-terminal half consists of hydrophobic residues (Val-244, Val-250, Phe-274, and Lys-276), exposure of which would certainly lead to poor solubility. CO release experiments provide further insights as to why an enzyme having a structural repeat should exist when the N-terminal half alone would suffice. In the presence of ascorbic acid as an electron donor, C-ChuS produced over 18 times more CO than the weakly acting N-ChuS. When CPR was used as the electron source, N-ChuS was almost twice as reactive as C-ChuS. Together, the two halves of ChuS would enable pathogenic E. coli to use different electron donors while stabilizing the less-soluble C-terminal half of the molecule. This versatility, in turn, would increase the chance of survival of E. coli strains in the host, thereby maximizing their virulence.
In addition to having an essential role in the acquisition of iron from host sources, ChuS may also protect E. coli against heme toxicity. There is growing evidence that heme accumulation can result in cell damage and tissue injury, because heme catalyzes the formation of reactive oxygen species, resulting in oxidative stress (45, 46). Furthermore, because heme has a low MW and is lipophilic, it can easily intercalate into the membrane and impair lipid bilayers and organelles, such as mitochondria and nuclei, and destabilize the cytoskeleton (47-49). Therefore, the HO activity of ChuS may be required to convert heme to biliverdin, a precursor to the less-reactive species bilirubin. This role for ChuS was also suggested through the appearance of a blue-green pigment during ChuS expression, characteristic of biliverdin accumulation. The ORF YhhX has been identified in E. coli K12, O157:H7, and CFT073. YhhX is downstream of two Fur binding sites (50), shares 19% sequence identity to human biliverdin reductase chain A, and may be the oxidoreductase that converts biliverdin to bilirubin. The structure of a homologue to YhhX (41% identity) has recently been deposited in the Protein Data Bank, and publication of its structural and functional analysis could confirm the identity of the second enzyme in the heme degradation pathway.
Finally, our results from DLS demonstrated that ChuS and ChuS-heme both form high-MW aggregates after 4 d of incubation in Tris buffer at 4°C. This aggregation could be the cause of the absence of HO activity previously reported for ShuS, a homologue of ChuS (28).
In summary, we determined the structure of ChuS, which represents a previously uncharacterized fold and is different from that of known mammalian and bacterial HOs. The structure contains tandem structural repeats, which are both functional in terms of heme oxygenation activity, albeit with somewhat different properties. Through heme spectral analysis and CO quantification, we identified ChuS as an HO present in pathogenic E. coli O157:H7.
Supplementary Material
Acknowledgments
We thank Dr. Gerald Marks for support, Dr. B. McLaughlin for help with gas chromatography, Dr. B. Hill for EPR experimentation and interpretation, other members of the Z.J. laboratory (especially M. Adams) for help with synchrotron data collection, and Jim Blonde for help with cloning. We also thank Dr. Mike Nesheim for expert help and Advanced Photon Source Beamlines 17-ID and 19-BM for diffraction data. This work was supported by the Canadian Institutes of Health Research. M.D.L.S. was supported by an E. G. Bauman Fellowship. Z.J. is a Canada Research Chair in Structural Biology and a Natural Sciences and Engineering Research Council Steacie Fellow.
Author contributions: M.D.L.S. and Z.J. designed research; M.D.L.S. and G.P.P. performed research; K.N., A.M., and M.C. contributed new reagents/analytical tools; M.D.L.S. and G.P.P. analyzed data; and M.D.L.S. and Z.J. wrote the paper.
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HO, heme oxygenase; N-ChuS, N-terminal half of ChuS fused to a C-terminal His-6 tag, C-ChuS, C-terminal half of ChuS fused to an N-terminal GST tag; CPR, cytochrome P450 reductase; DLS, dynamic light scattering; MW, molecular weight.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1U9T).
References
- 1.McHugh, J. P., Rodriguez-Quinones, F., Abdul-Tehrani, H., Svistunenko, D. A., Poole, R. K., Cooper, C. E. & Andrews, S. C. (2003) J. Biol. Chem. 278, 29478-29486. [DOI] [PubMed] [Google Scholar]
- 2.Crosa, J. H. (1989) Microbiol. Rev. 53, 517-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrews, S. C., Robinson, A. K. & Rodriguez-Quinones, F. (2003) FEMS Microbiol. Rev. 27, 215-237. [DOI] [PubMed] [Google Scholar]
- 4.Genco, C. A. & Dixon, D. W. (2001) Mol. Microbiol. 39, 1-11. [DOI] [PubMed] [Google Scholar]
- 5.Law, D. & Kelly, J. (1995) Infect. Immun. 63, 700-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Torres, A. G. & Payne, S. M. (1997) Mol. Microbiol. 23, 825-833. [DOI] [PubMed] [Google Scholar]
- 7.Stojiljkovic, I. & Hantke, K. (1994) Mol. Microbiol. 13, 719-732. [DOI] [PubMed] [Google Scholar]
- 8.Stojiljkovic, I. & Hantke, K. (1992) EMBO J. 11, 4359-4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wyckoff, E. E., Duncan, D., Torres, A. G., Mills, M., Maase, K. & Payne, S. M. (1998) Mol. Microbiol. 28, 1139-1152. [DOI] [PubMed] [Google Scholar]
- 10.Mills, M. & Payne, S. M. (1995) J. Bacteriol. 177, 3004-3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu, W., Wilks, A. & Stojiljkovic, I. (2000) J. Bacteriol. 182, 6783-6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu, W., Hunt, D. J., Richardson, A. R. & Stojiljkovic, I. (2000) J. Bacteriol. 182, 439-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmitt, M. P. (1997) J. Bacteriol. 179, 838-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skaar, E. P., Gaspar, A. H. & Schneewind, O. (2004) J. Biol. Chem. 279, 436-443. [DOI] [PubMed] [Google Scholar]
- 15.Cornejo, J., Willows, R. D. & Beale, S. I. (1998) Plant J. 15, 99-107. [DOI] [PubMed] [Google Scholar]
- 16.Friedman, J., Lad, L., Li, H., Wilks, A. & Poulos, T. L. (2004) Biochemistry 43, 5239-5245. [DOI] [PubMed] [Google Scholar]
- 17.Frankenberg-Dinkel, N. (2004) Antioxid. Redox. Signal. 6, 825-834. [DOI] [PubMed] [Google Scholar]
- 18.Schuller, D. J., Zhu, W., Stojiljkovic, I., Wilks, A. & Poulos, T. L. (2001) Biochemistry 40, 11552-11558. [DOI] [PubMed] [Google Scholar]
- 19.Hirotsu, S., Chu, G. C., Unno, M., Lee, D. S., Yoshida, T., Park, S. Y., Shiro, Y. & Ikeda-Saito, M. (2004) J. Biol. Chem. 279, 11937-11947. [DOI] [PubMed] [Google Scholar]
- 20.Wegele, R., Tasler, R., Zeng, Y., Rivera, M. & Frankenberg-Dinkel, N. (2004) J. Biol. Chem. 279, 45791-45802. [DOI] [PubMed] [Google Scholar]
- 21.Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hendrickson, W. A., Horton, J. R. & LeMaster, D. M. (1990) EMBO J. 9, 1665-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otwinowski, Z. & Minor, W. (1997) in Methods in Enzymology (Academic, New York), pp. 307-326. [DOI] [PubMed]
- 24.Terwilliger, T. C. & Berendzen, J. (1999) Acta Crystallogr. D 55, 849-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terwilliger, T. C. (2000) Acta Crystallogr. D 56, 965-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McRee, D. E. (1999) J. Struct. Biol. 125, 156-165. [DOI] [PubMed] [Google Scholar]
- 27.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta. Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 28.Wilks, A. (2001) Arch. Biochem. Biophys. 387, 137-142. [DOI] [PubMed] [Google Scholar]
- 29.Vreman, H. J., Kwong, L. K. & Stevenson, D. K. (1984) Clin. Chem. 30, 1382-1386. [PubMed] [Google Scholar]
- 30.Cook, M. N., Nakatsu, K., Marks, G. S., McLaughlin, B. E., Vreman, H. J., Stevenson, D. K. & Brien, J. F. (1995) Can. J. Physiol. Pharmacol. 73, 515-518. [DOI] [PubMed] [Google Scholar]
- 31.Maki, H., Matsuura, K., Shimada, K. & Nagashima, K. V. (2003) J. Biol. Chem. 278, 3921-3928. [DOI] [PubMed] [Google Scholar]
- 32.Murzin, A. G., Brenner, S. E., Hubbard, T. & Chothia, C. (1995) J. Mol. Biol. 247, 536-540. [DOI] [PubMed] [Google Scholar]
- 33.Krissinel, E. & Henrick, K. (2004) Acta Crystallogr. D 60, 2256-2268. [DOI] [PubMed] [Google Scholar]
- 34.Chu, G. C., Katakura, K., Zhang, X., Yoshida, T. & Ikeda-Saito, M. (1999) J. Biol. Chem. 274, 21319-21325. [DOI] [PubMed] [Google Scholar]
- 35.Wilks, A., Black, S. M., Miller, W. L. & Ortiz de Montellano, P. R. (1995) Biochemistry 34, 4421-4427. [DOI] [PubMed] [Google Scholar]
- 36.Wilks, A., Ortiz de Montellano, P. R., Sun, J. & Loehr, T. M. (1996) Biochemistry 35, 930-936. [DOI] [PubMed] [Google Scholar]
- 37.Wilks, A. & Schmitt, M. P. (1998) J. Biol. Chem. 273, 837-841. [DOI] [PubMed] [Google Scholar]
- 38.Wilks, A. & Ortiz de Montellano, P. R. (1993) J. Biol. Chem. 268, 22357-22362. [PubMed] [Google Scholar]
- 39.Matera, K. M., Zhou, H., Migita, C. T., Hobert, S. E., Ishikawa, K., Katakura, K., Maeshima, H., Yoshida, T. & Ikeda-Saito, M. (1997) Biochemistry 36, 4909-4915. [DOI] [PubMed] [Google Scholar]
- 40.Ishikawa, K., Takeuchi, N., Takahashi, S., Matera, K. M., Sato, M., Shibahara, S., Rousseau, D. L., Ikeda-Saito, M. & Yoshida, T. (1995) J. Biol. Chem. 270, 6345-6350. [DOI] [PubMed] [Google Scholar]
- 41.Blattner, F. R., Plunkett, G., III, Bloch, C. A., Perna, N. T., Burland, V., Riley, M., Collado-Vides, J., Glasner, J. D., Rode, C. K., Mayhew, G. F., et al. (1997) Science 277, 1453-1474. [DOI] [PubMed] [Google Scholar]
- 42.Caignan, G. A., Deshmukh, R., Wilks, A., Zeng, Y., Huang, H. W., Moenne-Loccoz, P., Bunce, R. A., Eastman, M. A. & Rivera, M. (2002) J. Am. Chem. Soc. 124, 14879-14892. [DOI] [PubMed] [Google Scholar]
- 43.Crusats, J., Suzuki, A., Mizutani, T. & Ogoshi, H. (1998) J. Org. Chem. 63, 602-607. [DOI] [PubMed] [Google Scholar]
- 44.Vreman, H. J., Cipkala, D. A. & Stevenson, D. K. (1996) Can. J. Physiol. Pharmacol. 74, 278-285. [PubMed] [Google Scholar]
- 45.Vercellotti, G. M., Balla, G., Balla, J., Nath, K., Eaton, J. W. & Jacob, H. S. (1994) Artif. Cells Blood Substitutes, Immobilization Biotechnol. 22, 207-213. [DOI] [PubMed] [Google Scholar]
- 46.Jeney, V., Balla, J., Yachie, A., Varga, Z., Vercellotti, G. M., Eaton, J. W. & Balla, G. (2002) Blood 100, 879-887. [DOI] [PubMed] [Google Scholar]
- 47.Balla, G., Jacob, H. S., Eaton, J. W., Belcher, J. D. & Vercellotti, G. M. (1991) Arterioscler. Thromb. 11, 1700-1711. [DOI] [PubMed] [Google Scholar]
- 48.Beri, R. & Chandra, R. (1993) Drug Metab. Rev. 25, 49-152. [DOI] [PubMed] [Google Scholar]
- 49.Ryter, S. W. & Tyrrell, R. M. (2000) Free Radic. Biol. Med. 28, 289-309. [DOI] [PubMed] [Google Scholar]
- 50.Vassinova, N. & Kozyrev, D. (2000) Microbiology 146, 3171-3182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.