

Abstract
Nitric oxide-donating aspirin (NO-ASA) is a promising chemoprevention agent against colon cancer and other cancers. It consists of traditional ASA to which a NO-releasing moiety is bound through a spacer. NO-ASA inhibits colon cancer cell growth several hundred times more potently than does ASA. In Min mice, NO-ASA inhibited intestinal carcinogenesis without affecting cell proliferation. Thus, we examined whether NO-ASA's most important cell kinetic effect is the induction of apoptosis. After confirming induction of apoptosis in Min mice, we studied the underlying mechanism in human colon adenocarcinoma cells. NO-ASA's spacer formed a conjugate with glutathione, depleting glutathione stores. This induced oxidative stress (increased intracellular levels of peroxides and 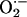 ) leads to apoptosis by activating the intrinsic apoptosis pathway. NO-ASA disrupted adherens junctions by inducing cleavage of β- and γ-catenin, resulting in cell detachment. NO-ASA inhibited Wnt signaling by a dual mechanism: at low concentrations it blocked the formation of β-catenin/Tcf complexes (dominant mechanism), and at higher concentrations it also cleaved β-catenin. These findings provide a mechanism of action by a potent chemopreventive agent, underscore the significance of these pathways in regulating cell death in the context of cancer chemoprevention, and present a paradigm for developing agents with enhanced cancer cell growth inhibitory properties.
) leads to apoptosis by activating the intrinsic apoptosis pathway. NO-ASA disrupted adherens junctions by inducing cleavage of β- and γ-catenin, resulting in cell detachment. NO-ASA inhibited Wnt signaling by a dual mechanism: at low concentrations it blocked the formation of β-catenin/Tcf complexes (dominant mechanism), and at higher concentrations it also cleaved β-catenin. These findings provide a mechanism of action by a potent chemopreventive agent, underscore the significance of these pathways in regulating cell death in the context of cancer chemoprevention, and present a paradigm for developing agents with enhanced cancer cell growth inhibitory properties.
Keywords: NO-aspirin, cell death, Wnt signaling, chemoprevention
Aspirin (acetylsalicylic acid, ASA) prevents colon cancer, but its limited efficacy and side effects preclude its application to cancer prevention. Nitric oxide-donating aspirin (NO-ASA), consisting of a traditional ASA that bears a NO-releasing moiety, appears to be safer than ASA and is currently undergoing clinical evaluation for the prevention of colon cancer (1).
Compared with ASA, NO-ASA is >1,000-fold more potent in inhibiting the growth of colon and other cancer cell lines (2). NO-ASA inhibits proliferation and induces apoptosis in colon cancer cells, and these effects may account for its extraordinary enhancement in potency. However, in Min mice, NO-ASA inhibited intestinal carcinogenesis but failed to affect cell proliferation (3). Thus, we considered the possibility that NO-ASA's most important cell kinetic effect is the induction of apoptosis. After demonstrating that NO-ASA indeed induces apoptosis in the intestinal mucosa of Min mice, we studied in detail the molecular events mediating the induction of apoptosis by NO-ASA. Our data show that NO-ASA induces apoptosis through a series of steps that begins with the generation of an oxidative stress state, which activates the intrinsic apoptosis pathway, accompanied by disruption of adherens junctions and inhibition of Wnt signaling. These steps encompass a unique sequence of events in response to a potent chemopreventive agent.
Materials and Methods
Reagents. NO-ASA [2-(acetyloxy)benzoic acid 4-(nitrooxymethyl)-phenyl ester] was from NicOx (Sophia Antipolis, France). The caspase 3 substrate Ac-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA), the pancaspase inhibitor (benzyloxycarbonyl-Val-Ala-Asp, Z-VAD-FMK), the caspase 3 inhibitor (Z-DEVD-FMK), the proteasome inhibitor (MG-132), dihydroethidium (DHE), and 2′,7′-dichlorofluorescine diacetate (H2DCFDA) were from Calbiochem. All other reagents were from Sigma.
Cell Culture. SW480, HCT-15, and LoVo human colon adenocarcinoma cells (American Type Culture Collection), seeded at 5.5 × 104 cells per cm2 and allowed to attach for 24 h, were treated with NO-ASA as indicated. Cell viability was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (Roche Diagnostics). Apoptosis was determined by DAPI staining, TUNEL assay, and annexin V staining (2). Caspase 3 activity was determined by assaying the cleavage of Ac-DEVD-pNA.
Luciferase Reporter Gene Assay. Cells (2 × 105) plated in 12-well plates were transfected with luciferase reporter constructs TOP-flash or FOPflash (Upstate Biotechnology, Lake Placid, NY) and β-galactosidase reporter plasmid as described (4); 2 h after transfection, cells were treated for 18 h with NO-ASA. Luciferase activity, normalized to β-galactosidase values, was expressed as relative fold compared with the control.
HPLC Analysis of NO-ASA and Its Metabolites. These analyses were performed as described in ref. 5.
Cellular Glutathione (GSH) Levels. GSH levels were determined by the GSH reductase-coupled 5,5′-dithiobis(2-nitrobenzoic acid) assay (6) by using a standard curve generated with each experiment.
Immunoblotting and Immunoprecipitation. These techniques were performed by following standard protocols (4). Nuclear and cytoplasmic extracts were prepared as described (7). For cytochrome c levels, we prepared cytosolic extracts (8), and for cell adhesion molecules, cells were fractionated in Triton X-100 (9). Antibodies to catenins and E-cadherin were from BD Transduction Laboratories, antibodies to cyclin D1 were from Upstate Cell Signaling (Lake Placid, NY), and all others were from Santa Cruz Biotechnology.
Determination of Reactive Oxygen Species (ROS). SW480 cells were pretreated with either 5 μM H2DCFDA or 5 μM DHE in RPMI medium 1640 without FCS or phenol red for 1 h as described (10) and incubated in 80 μM NO-ASA for 1.5 h.
Animal Study. C57BL/6J APCMin/+ mice and the corresponding C57BL/6J+/+ wild-type mice (The Jackson Laboratory), of which the APCMin/+ mice are a congenic derivative, were treated for 21 days with NO-ASA or vehicle as detailed elsewhere (3). Intestinal tissue sections were examined for apoptosis by using the TUNEL assay (Roche Applied Science, Indianapolis). TUNEL-positive cells were counted in ≥50 crypts. The apoptosis index was calculated as the number of positive cells divided by the number of all crypt cells and multiplied by 100.
Results
NO-ASA Induces Apoptosis in APCMin/+ Mice. NO-ASA inhibits intestinal carcinogenesis in APCMin/+ mice by 59% without affecting epithelial cell proliferation (3). We evaluated the effect of NO-ASA on epithelial cell apoptosis in small intestinal tissue samples from the same APCMin/+ mice and the corresponding C57BL/6J+/+ wild-type mice by using the TUNEL assay (Fig. 1 A and B). NO-ASA had no effect on apoptosis in the intestinal epithelium of wild-type mice. However, it increased the apoptosis index 2.9-fold in histologically normal intestinal epithelium [0.81 ± 0.23 (mean ± SEM, for this and subsequent values) in NO-ASA-treated vs. 0.27 ± 0.15 in the control; P < 0.01] and 1.9-fold in adenomas (0.52 ± 0.20 in NO-ASA-treated vs. 0.31 ± 0.16 in the control; P < 0.05). Of note, as expected (11), the histologically normal-appearing intestinal mucosa of APCMin/+ mice has lower levels of apoptosis compared with the wild-type mice. Measurement by immunohistochemistry of the expression of cleaved caspase 3, a marker of apoptosis, gave similar results (data not shown).
Fig. 1.

NO-ASA induces apoptosis in intestinal mucosa of APCMin/+ mice and in SW480 colon cancer cells. (A) TUNEL-stained normal and adenomatous tissues from APCMin/+ mice demonstrate increased apoptosis (arrows) in response to NO-ASA. (B) Apoptosis index from the intestinal mucosa of wild-type controls and APCMin/+ mice, mean ± SEM (n = 10). *, P < 0.05; **, P < 0.01. (C) SW480 cells were grown in the presence of NO-ASA. (D) SW480 cells treated with NO-ASA for 18 h were harvested and stained with DAPI. (Inset) Fluorescence microscopy of DAPI-stained cells; arrows indicate apoptotic cells. (Magnification: ×40.) (E) Poly(ADP-ribose) polymerase (PARP) cleavage in SW480 cells treated with NO-ASA for 18 h. (F) Caspase 3 activity in SW480 cells treated with 0–120 μM NO-ASA. (G) Caspase 8, procaspase 9, and cytochrome c in SW480 cells treated with 80 μM NO-ASA.
NO-ASA Induces Apoptosis by Activating the Intrinsic Apoptosis Pathway. In view of the above results, we pursued the mechanism by which NO-ASA induces apoptosis by using human colon cancer cell lines. NO-ASA profoundly inhibited the growth of SW480 cells (IC50 = 82 μM at 24 h and 34 μM at 48 h) (Fig. 1C). This effect was accompanied by induction of apoptosis, which involved up to 75% of cells (Fig. 1D). NO-ASA induced the cleavage of poly(ADP-ribose) polymerase (Fig. 1E), suggesting activation of caspase 3 (12). Indeed, NO-ASA activated caspase 3, its activity becoming nearly 6-fold greater than the control (Fig. 1F). The activation of caspase 3 was also reflected in the cleavage of procaspase 3, which generated 19- and 17-kDa caspase 3 fragments; the latter is the active caspase 3 (13).
The two major pathways of apoptosis are the intrinsic, characterized by cytochrome c release and caspase 9 activation, and the extrinsic, involving activation of caspase 8 and 10 (14). To determine which pathway is operative, we assayed the levels of cytochrome c and caspase 9 and 8 (Fig. 1G). As indicated by the cleavage of procaspase 9, caspase 9 becomes activated, while, in parallel, cytochrome c is released from mitochondria. In contrast, the extrinsic pathway is not affected by NO-ASA (no procaspase 8 cleavage that would generate caspase 8). HCT-15 and LoVo cells gave similar results (data not shown).
NO-ASA Induces Caspase 3-Mediated Cleavage of β-Catenin, Inhibits β-Catenin/Tcf Transcriptional Activity, and Decreases the Expression of Cyclin D1. β-Catenin translocates to the nucleus, where it interacts with the T cell transcription factor (Tcf) modulating the transcription of specific genes and plays a critical role in colon carcinogenesis (15). Some chemopreventive agents induce caspase 3-mediated β-catenin cleavage, ultimately decreasing the transactivation of this pathway (16). Thus, we investigated whether activation of caspase 3 by NO-ASA affected the catenins as well (Fig. 2).
Fig. 2.

NO-ASA inhibits Wnt signaling in SW460 colon cancer cells. (A) SW480 cells were treated with NO-ASA for 18 h (Left) or with 80 μM NO-ASA for the indicated times (Right). (B) β-Catenin/Tcf transcriptional activity was determined in SW480 cells transfected with luciferase reporter plasmids that have Tcf-4-binding sites (TOPflash) or a mutant site (FOPflash) and the β-galactosidase reporter plasmid as the control. After transfection, cells were treated with NO-ASA. Luciferase activity, normalized to β-galactosidase activity, was expressed as fold activation compared with control. (C) β-Catenin and caspase 3 were determined in cytosolic and nuclear extracts of SW480 cells treated for 18 h with NO-ASA (+) or solvent (-).
Treatment of SW480 cells with NO-ASA led to the cleavage of β-catenin, generating a distinct ≈70-kDa fragment. This effect, evident at 18 h, was at no time complete; densitometric analysis of immunoblots indicated that about half of β-catenin remained intact (data not shown). Of note, the release of caspase 3 from procaspase 3 preceded β-catenin cleavage, suggesting a possible association between the two (Fig. 2 A).
Because β-catenin cleavage was assessed in total cell protein extracts, it was important to clarify whether cleaved β-catenin was present in the cytoplasm, the nucleus, or both. Cleaved β-catenin was present in both the cytoplasm and the nucleus of only NO-ASA-treated cells. Caspase 3 had a similar distribution, in contrast with its precursor procaspase 3 that was present only in the cytoplasm (Fig. 2C). These findings indicate that β-catenin cleavage occurs in the cytoplasm and that these proteolytic products translocate into the nucleus.
The cleavage of nuclear β-catenin suggested that β-catenin nuclear signaling might be attenuated. We evaluated the effect of β-catenin cleavage on its transcriptional activity by transiently transfecting these cells with reporter plasmids bearing Tcf-4 binding sequences. NO-ASA strongly inhibited this binding (Fig. 2B) with an IC50 of 1.0 ± 0.1 μM (mean ± SEM). At 18 h, NO-ASA at 10 μM inhibited β-catenin/Tcf transcriptional activity by 90% compared with the control, essentially abolishing it at 50 μM. [The activity of the mutant construct (negative control) was ≤1% (data not shown).] Interestingly, there was no detectable cleavage of β-catenin at NO-ASA concentrations up to 10 μM, indicating that this effect was inhibited by mechanisms dependent on NO-ASA concentration.
To test the functional consequence of β-catenin cleavage, we determined the expression of cyclin D1 (Fig. 2 A), a Wnt-responsive gene, which is also important to colorectal cancer (17). NO-ASA reduced cyclin D1 levels at concentrations below those that induce β-catenin cleavage; the reduction in cyclin D1 levels preceded the onset of caspase 3 cleavage by 6 h.
NO-ASA Dismantles Adherens Junctions by Cleaving β-Catenin. In addition to its role in Wnt signaling, β-catenin plays a critical role in cell–cell adhesion (18). Cadherins bind β- and γ-catenin, which interact with α-catenin, linking them to actin filaments (see Fig. 6 Inset). Because NO-ASA detaches SW480 cells from the culture dish, we examined its effect on adherens junctions proteins (Fig. 3). NO-ASA led to the cleavage of β- and γ-catenin, but not of α-catenin or E-cadherin (Fig. 3A). Because the function of β-catenin in cell–cell adhesion depended on its subcellular localization, we determined the effect of NO-ASA on its subcellular distribution. After the induction of apoptosis by NO-ASA, total cell proteins were fractionated into Triton X-100-soluble and Triton X-100-insoluble fractions. β-Catenin was present in both fractions, but the extent of its cleavage differed between them (Fig. 3B). NO-ASA-induced cleavage of β-catenin was partial in the Triton X-100-soluble fraction and almost complete in the Triton-X-insoluble fraction. This finding indicates that membrane β-catenin, which is involved in adherens junctions, is extensively cleaved.
Fig. 6.

Induction of cell death by NO-ASA. The spacer part of NO-ASA reacts with GSH, depleting cell GSH levels and thus generating oxidative stress, which activates the intrinsic apoptosis pathway. Activation of caspase 3 cleaves β-catenin, leading to disruption of adherens junctions. NO-ASA also inhibits Wnt signaling; this effect is enhanced by caspase 3-induced cleavage of β-catenin. Changes in the expression of the many Wnt signaling-dependent genes, including cyclin D1, contribute to cell death. (Inset) Cell adherens junctions. Arrows, positive effect; T-shaped arrows, negative effect; dashed arrows, speculative effect.
Fig. 3.

Effect of NO-ASA on adherens junctions in SW480 cells. (A) E-cadherin and α- and γ-catenin levels were determined in cells treated with NO-ASA for 18 h. (B) SW480 cells were treated with 50 μM or 80 μM NO-ASA or solvent control. Cell proteins were separated into Triton X-100-soluble and -insoluble fractions. (C) SW480 cells were treated for 18 h with NO-ASA. E-cadherin or α-catenin was immunoprecipitated by using anti-E-cadherin (Upper) or anti-α-catenin (Lower) antibodies and fractionated by SDS/PAGE.
Because the E-cadherin/β-catenin/α-catenin complex directly controls cell–cell adhesion, we examined whether cleavage of β-catenin impaired its association with α-catenin and E-cadherin and, therefore, the integrity of adherens junctions. After the cells had been treated with NO-ASA for 18 h, cell lysates were immunoprecipitated by using an antibody to E-cadherin. As shown in Fig. 3C, at its higher concentrations (≥50 μM), NO-ASA induced cleavage of β-catenin, whose fragments were still physically associated with E-cadherin. However, the levels of α-catenin were dramatically reduced. Conversely, when we immunoprecipitated these adhesion complexes by using an antibody to α-catenin, we detected no E-cadherin associated with this complex. Therefore, the cleavage of β-catenin disrupted the adherence junctions; this disruption became virtually complete at high NO-ASA concentrations.
Caspase Inhibitors Diminish NO-ASA-Induced β-Catenin Degradation, cyclin D1 Down-Regulation, and Apoptosis. Our data suggest that NO-ASA induces caspase activation, which, in turn, is involved in the degradation of β-catenin, and ultimately culminates in apoptotic cell death. To assess the validity of this notion, we pretreated SW480 cells with a pancaspase inhibitor (Z-VAD-FMK) or a specific caspase 3 inhibitor (Z-DEVD-FMK) for 1 h and 50 μM NO-ASA for 18 h and determined the levels of procaspase 3, caspase 3, β-catenin, and cyclin D1 (Fig. 4). Both caspase inhibitors abrogated caspase 3 activation, β-catenin degradation, and cyclin D1 down-regulation, indicating a direct association between caspase 3 and these changes (Fig. 4A). To exclude the possibility that proteasome activity is involved in β-catenin degradation, we pretreated these cells with the proteasome inhibitor MG-132, which reduces the degradation of ubiquitin-conjugated proteins. Proteasome inhibition failed to reverse β-catenin cleavage; levels of cyclin D1 were greatly increased in cells treated only with the proteasome inhibitor (no NO-ASA), indicating that MG-132 was active.
Fig. 4.

Caspase inhibitors block NO-ASA-induced apoptosis. (A) SW480 cells, pretreated with the pancaspase inhibitor Z-VAD-FMK (50 μM), the caspase 3 inhibitor Z-DEVD-FMK (50 μM), or the proteasome inhibitor MG132 (25 μM), were treated with 50 μM NO-ASA for 18 h. (B) SW480 cells pretreated with Z-VAD-FMK were cotransfected with luciferase reporter plasmids as in Fig. 2. (C) SW480 cells were treated with Z-DEVD-FMK for 1 h before and for 18 h after NO-ASA treatment. Cells were then subjected to TUNEL assay; apoptotic cells stain dark blue. (Magnification: ×20.) (D) SW480 cells were treated with Z-DEVD-FMK for 1 h before treatment with NO-ASA for 18 h, stained with annexin V and propidium iodide (PI), and analyzed by flow cytometry. The percentage of cells in each compartment is shown.
To determine the extent to which cleavage of β-catenin affects β-catenin/Tcf transcriptional activity, we assessed the effect of the pancaspase inhibitor Z-VAD-FMK. SW480 cells were transiently transfected with reporter plasmids, treated for 18 h with 50 μM NO-ASA or vehicle, and also pretreated for 30 min with or without Z-VAD-FMK. NO-ASA strongly inhibited β-catenin/Tcf transcriptional activity (Fig. 4B). In vehicle-treated cells, Z-VAD-FMK had no significant effect on β-catenin/Tcf transcriptional activity. In contrast, in NO-ASA-treated cells, Z-VAD-FMK increased β-catenin/Tcf transcriptional activity from about 0.05-fold to about 0.17-fold. This finding indicates that the contribution of β-catenin cleavage to the inhibition of Wnt signaling is modest.
The biological relevance of these effects was documented by evaluating the extent of apoptosis in response to NO-ASA, with or without pretreatment with the caspase 3 inhibitor Z-DEVD-FMK (Fig. 4D). Concurrent staining of the cells with propidium iodide (PI) distinguishes between viable, early apoptotic (they exclude PI), and necrotic or late apoptotic cells (they stain with PI). NO-ASA induced apoptosis and pretreatment with the caspase 3 inhibitor prevented apoptosis almost completely.
NO-ASA Induces Oxidative Stress in SW480 Cells. Oxidative stress is known to activate the cell death machinery (19). Therefore, we examined the possibility that NO-ASA may induce apoptosis by inducing a state of oxidative stress. We determined the levels of ROS in SW480 cells in response to NO-ASA (Fig. 5). SW480 cells loaded with one of two probes for ROS were treated with NO-ASA 80 μM for 1.5 h. These probes were DHE, which preferentially measures superoxide anion (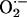 ), and H2DCFDA, which probes for H2O2 and other peroxides. Both DHE- and H2DCFDA-derived fluorescence were increased after treatment with NO-ASA, indicating that NO-ASA increased intracellular levels of H2O2 and perhaps other peroxides and also of
), and H2DCFDA, which probes for H2O2 and other peroxides. Both DHE- and H2DCFDA-derived fluorescence were increased after treatment with NO-ASA, indicating that NO-ASA increased intracellular levels of H2O2 and perhaps other peroxides and also of 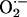 (Fig. 5C).
(Fig. 5C).
Fig. 5.

NO-ASA induces oxidative stress: effects on caspase 3 activation, Wnt signaling, and apoptosis. (A) After treatment of SW480 cells with 50 μM NO-ASA for 1.5 h, NO-ASA and its metabolites were resolved by HPLC. Peak a, conjugate of the NO-ASA spacer moiety with GSH. Peak b, deacetylated NO-ASA. (B) Total cellular GSH levels after a 4-h treatment of SW480 cells with NO-ASA. (C) SW480 cells were treated with H2DCFDA (detects peroxides) or DHE (detects mainly superoxide anion) for 1 h followed by 80 μM NO-ASA for 1.5 h. Fluorescence was measured by flow cytometry in single-cell suspensions of the attached cells. NO-ASA induced both sets of species. (D) SW480 cells treated with 2.5 mM N-acetylcysteine (NAC) for 1 h before treatment with NO-ASA for 18 h. Procaspase 3, caspase 3, β-catenin, cyclin D1, and α-tubulin were determined by immunoblotting. (E) SW480 cells were treated with 2.5 mM NAC for 1 h before cotransfection with luciferase reporter plasmids as in Fig. 2. (F) SW480 cells were either untreated or pretreated with 2.5 mM NAC for 1 h before their treatment with NO-ASA for 18 h. Single-cell suspensions were stained with annexin V and PI and analyzed by flow cytometry. (G) SW480 cells were treated for 24 h with dl-buthionine (S,R)-sulfoximine (BSO), an inhibitor of GSH synthesis, followed by treatment with NO-ASA for 18 h. Data are expressed as percent of the control.
As shown in Fig. 5A, the spacer moiety of NO-ASA formed a conjugate with GSH (5), prompting us to evaluate whether NO-ASA affected intracellular GSH levels. Total GSH levels were determined after a 4-h treatment of these cells with NO-ASA at 0–200 μM. There was a clear concentration-dependent reduction in GSH levels, which became as low as 34% of control levels (Fig. 5B).
To evaluate whether the redox state of the cell plays a role in the activation of caspase 3 and the subsequent cascade of events, we pretreated SW480 cells with N-acetylcysteine (NAC), a GSH-increasing agent (20). NAC almost completely blocked the activation of caspase 3 brought about by 50 μM NO-ASA; no such activation was seen at 10 μM NO-ASA. NAC also partially blocked the cleavage of β-catenin occurring with 50 μM NO-ASA. Cyclin D1 levels were significantly restored at all NO-ASA concentrations (Fig. 5D). NAC also reversed the inhibition of β-catenin/Tcf transcriptional activity in response to NO-ASA (Fig. 5E). Pretreatment of SW480 cells with NAC reduced NO-ASA-induced apoptosis from 38% to 13.9% (Fig. 5F). Conversely, GSH depletion [induced by dl-buthionine (S,R)-sulfoximine (BSO), which inhibits GSH biosynthesis (21)], enhanced the cell growth inhibitory effect of NO-ASA (Fig. 5G). NO-ASA inhibited the growth of SW480 cells (IC50 >80 μM under our experimental protocol), but pretreatment with 10 μM BSO reduced the IC50 to 20 μM; this effect became more pronounced with 100 μM BSO (IC50 = 5–10 μM, on the basis of this and experiments not described).
Discussion
NO-ASA displayed a profound growth inhibitory effect in human colon cancer cells far greater than that of traditional ASA. This effect involved extensive cell death by apoptosis. Our findings indicate that NO-ASA induces apoptosis cells by inducing a state of oxidative stress, which activated the intrinsic apoptosis pathway, by disrupting adherens junctions, and by inhibiting Wnt signaling. This constellation of molecular events may account, at least in part, for the remarkably enhanced potency of NO-ASA.
As summarized in Fig. 6, treatment of SW480 (and other) human colon cancer cells with NO-ASA was associated with activating cleavage of poly(ADP-ribose) polymerase by caspase 3, which cleaves it at a conserved sequence (22). Our data confirmed the activation of caspase 3 by NO-ASA. The role of caspase 3 in NO-ASA-induced apoptosis was further documented by the finding that two caspase inhibitors blocked the induction of apoptosis by NO-ASA. Caspase 3 can be activated only by upstream initiator caspases, which are capable of auto-catalytic activation (23). While procaspase 8 remained intact during NO-ASA treatment of the SW480 cells, procaspase 9 was activated; coincident with its activation was the release of cytochrome c from the mitochondria. These findings establish that NO-ASA activates the intrinsic apoptosis pathway.
A key proximal event in the induction of apoptosis by NO-ASA is the induction of oxidative stress. There was substantial accumulation of ROS (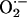 and peroxides, H2O2 being the most important among them) at 1.5 h and at NO-ASA concentrations around the IC50 for cell growth inhibition, which induces apoptosis in nearly half the cells. GSH stores were depleted, at least in part, because NO-ASA's spacer formed a conjugate with GSH. The significance of oxidative stress in this process is evidenced by two manipulations of the system affecting the levels of GSH, the most important antioxidant mechanism in mammalian cells (24). Pretreatment with BSO, which depletes intracellular GSH, reduced the IC50 of NO-ASA for cell growth from 85 μM to 20 μM. Alternatively, supplementing the cells with NAC greatly attenuated the apoptotic effect of NO-ASA. This was accompanied by diminished activation of caspase 3. What underlies the accumulation of ROS is not entirely clear. NO-ASA depleted GSH by forming a conjugate with it. GSH responds directly to intracellular redox changes and is also used as a cofactor for antioxidant enzymes such as GSH peroxidase, which is involved in the reduction of peroxides. NO-ASA may have increased ROS levels, at least in part, through its effect on GSH (19).
and peroxides, H2O2 being the most important among them) at 1.5 h and at NO-ASA concentrations around the IC50 for cell growth inhibition, which induces apoptosis in nearly half the cells. GSH stores were depleted, at least in part, because NO-ASA's spacer formed a conjugate with GSH. The significance of oxidative stress in this process is evidenced by two manipulations of the system affecting the levels of GSH, the most important antioxidant mechanism in mammalian cells (24). Pretreatment with BSO, which depletes intracellular GSH, reduced the IC50 of NO-ASA for cell growth from 85 μM to 20 μM. Alternatively, supplementing the cells with NAC greatly attenuated the apoptotic effect of NO-ASA. This was accompanied by diminished activation of caspase 3. What underlies the accumulation of ROS is not entirely clear. NO-ASA depleted GSH by forming a conjugate with it. GSH responds directly to intracellular redox changes and is also used as a cofactor for antioxidant enzymes such as GSH peroxidase, which is involved in the reduction of peroxides. NO-ASA may have increased ROS levels, at least in part, through its effect on GSH (19).
The cleavage of β-catenin by caspase 3 was the critical event in the disassembly of the adherens junctions during NO-ASA-induced apoptosis; the end molecules of these junctions, α-catenin and E-cadherin, remained intact (although no longer in physical association with each other through β-catenin). Two findings indicate that the cleavage of β-catenin was mediated by caspase 3: caspase 3 activation preceded β-catenin cleavage by at least 6 h, and caspase inhibitors blocked β-catenin cleavage. There was also cleavage of γ-catenin, which participates in the formation of adherens junctions. Our data also documented that neither α-catenin nor E-cadherin was affected. Our immunoprecipitation studies, which assessed the physical association between β-catenin and E-cadherin, further demonstrated that the physical association of this chain of proteins was lost. Immunoprecipitated E-cadherin had greatly reduced amounts of α-catenin attached to it, and this reduction depended on the concentration of NO-ASA; fragments of β-catenin were still attached to it. Conversely, immunoprecipitated α-catenin had fragments of β-catenin associated with it, but had no fragments of E-cadherin associated with it. Finally, caspase inhibitors blocked not only the cleavage of β-catenin but also the progression of the cell to apoptosis. This effect indicates that these findings are internally consistent with the proposed mechanism.
Another consequence of the chain of events set in motion by the induction of apoptosis by NO-ASA was the inhibition of Wnt signaling, a pathway that regulates cell death and also is of great importance to colon carcinogenesis (25). β-Catenin is cleaved by NO-ASA at concentrations >50 μM, and such cleavage became evident after 18 h of treatment with NO-ASA. In contrast, the levels of cyclin D1, whose expression depends on the Wnt pathway and which influences carcinogenesis mainly through its cell cycle effects (17), are suppressed at concentrations of NO-ASA as low as 1 μM and as early as 6 h. Thus, the cleavage of β-catenin contributes to diminished Wnt signaling in response to NO-ASA treatment only later and at higher NO-ASA concentrations.
The suppression in Wnt signaling that occurs before β-catenin cleavage goes into effect must occur by means of a different mechanism. Indeed, most of the suppression of the β-catenin/Tcf transcriptional activity occurs at NO-ASA concentrations that do not cleave β-catenin. This conclusion was confirmed by the finding that a caspase inhibitor, which completely blocked β-catenin cleavage, had only a modest effect on β-catenin/Tcf transcriptional activity. Thus, NO-ASA inhibits Wnt signaling at such low concentrations without affecting β-catenin levels. The functional consequence of the suppressed β-catenin/Tcf transcriptional activity is reflected in the expression of cyclin D1. This dual mechanism of inhibition of Wnt signaling appears unique.
In conclusion, NO-ASA brings about the death of SW480 cells through a series of steps that begin with its reaction with GSH, leading to a state of oxidative stress that, in turn, activates the intrinsic apoptosis pathway. The activation of caspase 3, representing the downstream effector caspases, is followed by disruption of the adherens junctions, which results from the cleavage of β-catenin by caspase 3. NO-ASA has a direct and rapid effect on Wnt signaling; the inhibition of Wnt signaling is further increased once caspase 3 is activated and β-catenin is cleaved. The effect of these changes on the many genes whose transcription depends on Wnt signaling contributes to the demise of the cell. These findings provide a previously unrecognized sequence of events in response to a potent chemopreventive agent, underscore the significance of these pathways in the regulation of cell death in the context of cancer chemoprevention, and may present a paradigm for developing agents with enhanced cancer cell growth inhibitory properties.
Acknowledgments
This work was supported by National Institutes of Health Grant CA92423.
Author contributions: J.G. and B.R. designed research; J.G. and X.L. performed research; J.G., X.L., and B.R. analyzed data; and J.G. and B.R. wrote the paper
Conflict of interest statement: No conflicts declared.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: ASA, aspirin; NO-ASA, NO-donating aspirin; DEVD-FMK, Asp-Glu-Val-Asp fluoromethyl ketone; Z-VAD-FMK, benzyloxycarbonyl-Val-Ala-Asp-FMK; DHE, dihydroethidium; H2DCFDA, 2′,7′-dichlorofluorescine diacetate; GSH, glutathione; ROS, reactive oxygen species; NAC, N-acetylcysteine; PI, propidium iodide; BSO, dl-buthionine (S,R)-sulfoximine.
References
- 1.Rigas, B. & Kashfi, K. (2004) Trends Mol. Med. 10, 324-330. [DOI] [PubMed] [Google Scholar]
- 2.Kashfi, K., Ryann, Y., Qiao, L. L., Williams, J. L., Chen, J., Del Soldato, P., Traganos, F. & Rigas, B. (2002) J. Pharmacol. Exp. Ther. 303, 1273-1282. [DOI] [PubMed] [Google Scholar]
- 3.Williams, J. L., Kashfi, K., Ouyang, N., del Soldato, P., Kopelovich, L. & Rigas, B. (2004) Biochem. Biophys. Res. Commun. 313, 784-788. [DOI] [PubMed] [Google Scholar]
- 4.Nath, N., Kashfi, K., Chen, J. & Rigas, B. (2003) Proc. Natl. Acad. Sci. USA 100, 12584-12589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao, J., Kashfi, K. & Rigas, B. (2005) J. Pharmacol. Exp. Ther. 312, 989-997. [DOI] [PubMed] [Google Scholar]
- 6.Kleinman, W. A., Komninou, D., Leutzinger, Y., Colosimo, S., Cox, J., Lang, C. A. & Richie, J. P., Jr. (2003) Biochem. Pharmacol. 65, 741-746. [DOI] [PubMed] [Google Scholar]
- 7.Natarajan, K., Singh, S., Burke, T. R., Jr., Grunberger, D. & Aggarwal, B. B. (1996) Proc. Natl. Acad. Sci. USA 93, 9090-9095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang, C. Y., Guttridge, D. C., Mayo, M. W. & Baldwin, A. S., Jr. (1999) Mol. Cell. Biol. 19, 5923-5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lampugnani, M. G., Corada, M., Caveda, L., Breviario, F., Ayalon, O., Geiger, B. & Dejana, E. (1995) J. Cell Biol. 129, 203-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Troyano, A., Fernandez, C., Sancho, P., de Blas, E. & Aller, P. (2001) J. Biol. Chem. 276, 47107-47115. [DOI] [PubMed] [Google Scholar]
- 11.Mahmoud, N. N., Carothers, A. M., Grunberger, D., Bilinski, R. T., Churchill, M. R., Martucci, C., Newmark, H. L. & Bertagnolli, M. M. (2000) Carcinogenesis 21, 921-927. [DOI] [PubMed] [Google Scholar]
- 12.Lazebnik, Y. A., Kaufmann, S. H., Desnoyers, S., Poirier, G. G. & Earnshaw, W. C. (1994) Nature 371, 346-347. [DOI] [PubMed] [Google Scholar]
- 13.Jaiswal, A. S., Marlow, B. P., Gupta, N. & Narayan, S. (2002) Oncogene 21, 8414-8427. [DOI] [PubMed] [Google Scholar]
- 14.Watson, A. J. (2004) Gut 53, 1701-1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson, W. J. & Nusse, R. (2004) Science 303, 1483-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steinhusen, U., Badock, V., Bauer, A., Behrens, J., Wittman-Liebold, B., Dorken, B. & Bommert, K. (2000) J. Biol. Chem. 275, 16345-16353. [DOI] [PubMed] [Google Scholar]
- 17.Heinen, C. D., Goss, K. H., Cornelius, J. R., Babcock, G. F., Knudsen, E. S., Kowalik, T. & Groden, J. (2002) Gastroenterology 123, 751-763. [DOI] [PubMed] [Google Scholar]
- 18.Jamora, C. & Fuchs, E. (2002) Nat. Cell Biol. 4, E101-E108. [DOI] [PubMed] [Google Scholar]
- 19.Klaunig, J. E. & Kamendulis, L. M. (2004) Annu. Rev. Pharmacol. Toxicol. 44, 239-267. [DOI] [PubMed] [Google Scholar]
- 20.Atmaca, G. (2004) Yonsei Med. J. 45, 776-788. [DOI] [PubMed] [Google Scholar]
- 21.Griffith, O. W. & Meister, A. (1979) J. Biol. Chem. 254, 7558-7560. [PubMed] [Google Scholar]
- 22.Oliver, F. J., de la Rubia, G., Rolli, V., Ruiz-Ruiz, M. C., de Murcia, G. & Murcia, J. M. (1998) J. Biol. Chem. 273, 33533-33539. [DOI] [PubMed] [Google Scholar]
- 23.Degterev, A., Boyce, M. & Yuan, J. (2003) Oncogene 22, 8543-8567. [DOI] [PubMed] [Google Scholar]
- 24.Meister, A. (1991) Pharmacol. Ther. 51, 155-194. [DOI] [PubMed] [Google Scholar]
- 25.Huls, G., Koornstra, J. J. & Kleibeuker, J. H. (2003) Lancet 362, 230-232. [DOI] [PubMed] [Google Scholar]