

Summary
One strategy for localization of a quantitative-trait locus (QTL) is to test whether the distribution of a quantitative trait depends on the number of copies of a specific genetic-marker allele that an individual possesses. This approach tests for association between alleles at the marker and the QTL, and it assumes that association is a consequence of the marker being physically close to the QTL. However, problems can occur when data are not from a homogeneous population, since associations can arise irrespective of a genetic marker being in physical proximity to the QTL—that is, no information is gained regarding localization. Methods to address this problem have recently been proposed. These proposed methods use family data for indirect stratification of a population, thereby removing the effect of associations that are due to unknown population substructure. They are, however, restricted in terms of the number of children per family that can be used in the analysis. Here we introduce tests that can be used on family data with parent and child genotypes, with child genotypes only, or with a combination of these types of families, without size restrictions. Furthermore, equations that allow one to determine the sample size needed to achieve desired power are derived. By means of simulation, we demonstrate that the existing tests have an elevated false-positive rate when the size restrictions are not followed and that a good deal of information is lost as a result of adherence to the size restrictions. Finally, we introduce permutation procedures that are recommended for small samples but that can also be used for extensions of the tests to multiallelic markers and to the simultaneous use of more than one marker.
Introduction
The transmission/disequilibrium test (TDT) introduced by Spielman et al. (1993) has become a popular family-based test of linkage between a marker and a susceptibility locus. The attractiveness of the TDT is a result of both its validity in structured populations and its power, which can be much greater than that of conventional linkage tests. The TDT has been extended to multiallelic markers (Bickeboller and Darpoux 1995; Sham and Curtis 1995; Schaid 1996; Spielman and Ewens 1996), to families without parental genotype information (Curtis 1997; Boehnke and Langefeld 1998; Monks et al. 1998; Schaid and Rowland 1998; Spielman and Ewens 1998), and to quantitative traits (Allison 1997; Rabinowitz 1997; Schaid and Rowland 1999). Although the TDT and its extensions that use parental information were designed as tests of linkage in the presence of association, they are also tests of association in the presence of linkage, for samples of unrelated parent/child trios. The advantage of the TDT, in this context, is that it is not sensitive to population stratification in the parental population, which can be a problem for the usual case-control test. For extensions to families without parental-genotype information, the TDT is a valid test of association in the presence of linkage only if samples contain unrelated sibships with exactly two children (one that is affected and one that is unaffected, for tests involving a susceptibility locus). If samples contain larger sibships, then these tests are valid only as tests of linkage.
Tests of association are often used for a candidate gene. Also, once a chromosomal region has been designated, through use of a linkage test, as being of interest, association tests done with the use of markers in the region may be useful for further localization of the susceptibility locus or QTL, since association is thought to exist in human populations for small distances—typically, <2 cM. When the TDT and its extensions are used to test for association, data sets must contain unrelated families of minimal size (one child for families with parental genetic information and two children for families without this information). If families with arbitrary numbers of children have been sampled, then strategies must be used to reduce the data set. One is always reluctant to discard data, because of the probable loss of power; however, if methods are not used to adjust for the correlation between siblings that results from the marker and the QTL being linked, then the false-positive rate for tests of association will be unknown and will be larger than expected.
Martin et al. (1997) generalized the TDT as a test of association in the presence of linkage for a susceptibility gene, for families with an arbitrary number of affected children and with available parental marker–genotype information. More recently, Horvath and Laird (1998), using sibships of arbitrary size, developed a test of association for a susceptibility gene when parental data are missing. In both cases, the authors used the family as the independent sampling unit, to avoid the elevated false-positive rate caused by correlation between sibs. In the present study, we extend the ideas of Martin et al. (1997) and Horvath and Laird (1998), and we propose three tests that can be used for quantitative-trait data and that use information from all children. These tests are valid tests of linkage and association, regardless of the number of children sampled. Throughout the present study, we assume that linkage is present and focus on the test for association in the presence of linkage. For the TQP test, we use genotype information for parents and for all of their children, whereas, for the TQS test, we use genotypes for all siblings (with no parental information). Finally, for the TQPS test, we use a combination of these types of family information. Similar to its treatment in the study by Martin et al. (1997) and Horvath and Laird (1998), the family is treated as the independent sampling unit in all three tests.
In the Methods section below, we introduce each of the three test statistics and derive their distributional properties. Specifically, we show that the test statistics are asymptotically standard normal under the null hypothesis of no linkage or no association. We derive their distribution on the basis of the alternative hypothesis of linkage and association, and, assuming that there is Hardy-Weinberg equilibrium at the marker and at the QTL, we provide formulas to be used for sample-size calculations. Additionally, permutation procedures are given that are recommended for small samples but that can also be used for extensions of the three tests to multiallelic markers and to the simultaneous use of more than one marker.
We then compare the TQP and TQS tests with two other nonparametric tests. The first test, which was introduced by Rabinowitz (1997), uses parental-transmission information. The second test, which was developed by Allison et al. (1999), is a permutation test for which only sibship information is used. We will denote these tests as TR and TA, respectively. Through simulation, we demonstrate that the false-positive rate increases for both of these tests, when nonminimal families are used. We also compare the power of our tests, in which information from all children is used, to the power of the TR and TA tests, in which data sets composed of families of minimal size are used. These comparisons demonstrate the validity of our tests as well as what is gained by use of this additional information. We then provide evidence that our permutation procedures for TQP and TQS are valid. Finally, we demonstrate that the validity of the TQP and TQS tests is not affected by population stratification.
Methods
Notation
Consider a diallelic-marker locus with alleles A1 and A2, with population frequencies p1 and p2, and a diallelic QTL with alleles Q1 and Q2, with population frequencies q1 and q2. Let θ denote the recombination fraction between the marker locus and the QTL. Association—also referred to as linkage disequilibrium—is measured with the use of the disequilibrium coefficient for A1 and Q1, denoted as D, where D=Pr(A1Q1)-p1q1 (Weir 1996). Define μrs as the trait mean for individuals with QTL genotype QrQs, and define σ2E as the trait-distribution variance within one QTL genotype class. The parameter σ2E represents all phenotypic variation not attributable to the QTL. We will use the parameterization in which the trait mean of the homozygotes is centered at zero, so that μ11=a, μ12=d, and μ22=-a, where a>0. The phenotypic variance resulting from the QTL can be written as 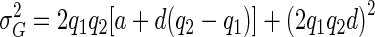 . As a measure of the amount of variation caused by the QTL, we use broad-sense heritability, denoted as H2, which is the proportion of the phenotypic variance caused by the QTL—that is, H2=σ2G/(σ2G+σ2E) (Falconer and Mackay 1996).
. As a measure of the amount of variation caused by the QTL, we use broad-sense heritability, denoted as H2, which is the proportion of the phenotypic variance caused by the QTL—that is, H2=σ2G/(σ2G+σ2E) (Falconer and Mackay 1996).
Suppose that there are F families indexed by i. Let the ti children in the ith family be indexed by j. Unless otherwise noted, all families have the same number of children—that is, ti=t for all families. Let Yij denote the trait value for the jth child in the ith family, and let  denote the mean over all children in all families, as computed by
denote the mean over all children in all families, as computed by  . Let X*iM (X*iF) = 1 if the mother (father) is heterozygous at the marker locus, and let X*iM (X*iF) = 0 otherwise; denote X*iM+X*iF as hi. Let XijM (XijF) indicate whether marker allele A1 was transmitted to the jth child by the mother (father), and define
. Let X*iM (X*iF) = 1 if the mother (father) is heterozygous at the marker locus, and let X*iM (X*iF) = 0 otherwise; denote X*iM+X*iF as hi. Let XijM (XijF) indicate whether marker allele A1 was transmitted to the jth child by the mother (father), and define  as the mean of the ti values of XijM+XijF. See figure 1 for an example of notation.
as the mean of the ti values of XijM+XijF. See figure 1 for an example of notation.
Figure 1.

Example of notation for a family with three children
The null hypothesis is Ho:no linkage or no association, whereas the alternative hypothesis is Ha:linkage and association. The part of the hypotheses concerning linkage is straightforward; however, the part concerning association requires further detail, because of the possibility of population stratification. If there is no population stratification, then the null hypothesis is that there is no linkage or association in the parental population. Alternatively, if there is population stratification, then the null hypothesis is that there is no linkage or association in any of the subpopulations from which parental chromosomes might originate. For clarity of presentation, it is assumed, throughout the present study, that the parental population is homogeneous; however, computations can be extended to a more-complicated population structure. An example is given in Appendix A. For a more-detailed discussion, the reader is referred elsewhere (Ewens and Spielman 1995). When the hypotheses are expressed in terms of parameters, we have Ho:θ=.5 or D=0 versus Ha:θ<.5 and D≠0. In the present study, we assume that the marker and the QTL are in tight linkage and that only the test of association is of interest. For our theoretical derivations and simulations, we assume that the marker and the QTL are both in Hardy-Weinberg equilibrium. This assumption is made for computational convenience and is required only for power calculations.
Test Statistics
We begin with the case for which parental-genotype information is available. If there is no association between the marker allele A1 and the QTL allele Q1, then what is transmitted, at the marker locus, to a child from a marker-heterozygous parent neither affects that child’s quantitative trait nor is related to anything that affects the quantitative trait, regardless of whether the marker and QTL are linked. One measure of the marker/QTL relationship is the covariance between the quantitative trait and a variable representing transmissions at the marker locus (Rabinowitz 1997). Consider a family with one parent that is heterozygous at the marker locus—that is, hi=1. Without loss of generality, suppose that the mother is heterozygous. For each family, the covariance between XijM and Yij should be zero under the null hypothesis. Since the expectation of XijM is .5, an estimate of the covariance is  . Likewise, for a family with hi=2, the covariance between XijM+XijF and Yij should be zero. The expectation of XijM+XijF is 1; therefore, an estimate of the covariance is
. Likewise, for a family with hi=2, the covariance between XijM+XijF and Yij should be zero. The expectation of XijM+XijF is 1; therefore, an estimate of the covariance is 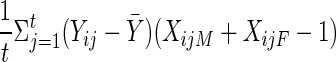 . For the ith family, these estimates of covariance can be written, in our notation, as random variable Ui:
. For the ith family, these estimates of covariance can be written, in our notation, as random variable Ui:
 |
Suppose that our data set contains families with at least one parent that is heterozygous at the marker locus; in this instance, for a random family, we have Ui corresponding to hi=1 or hi=2. In Appendix B, it is shown that the expectation of Ui for such families is
 |
Hence, we see that the expectation of Ui will be zero, if at least one of the following four scenarios is true:
-
1.
The marker and QTL are unlinked (i.e., θ=0.5).
-
2.
There is no association between the marker and the QTL (i.e., D=0).
-
3.
The QTL has no effect on the trait (i.e., a=d=0).
-
4.
a+d(q2-q1) is 0 without a=d=0.
Scenario (4), although possible, is not a likely scenario. Scenario (3) contradicts the definition of a QTL so that only scenarios (1) and (2) are pertinent. Furthermore, scenarios (1) and (2) jointly compose the null hypothesis; therefore, discounting scenario (4), the null hypothesis will be true if and only if E(Ui)=0. Although we have assumed, throughout this discussion, that the population is homogeneous—that is, there is no stratification—it is not difficult to show that, if there is population stratification, the expectation of Ui will be zero if there is no association or no linkage within each subpopulation (see Appendix A).
If alleles A1 and Q1 are positively associated—that is, if D>0—then, since Q1 causes high quantitative-trait values (because a>0), we would expect the covariance to be positive. In other words, high trait values will often occur with transmissions of allele A1, and, therefore, the expectation of Ui will be positive. Similarly, if the A1 and Q1 alleles are negatively associated (D<0), we would expect the covariance to be negative. From equation (1), we can see that this is, indeed, often true; however, it is possible to see a negative covariance resulting from a positive association or a positive covariance resulting from a negative association. Nonetheless, in all of these situations, the expectation of Ui will not be equal to zero.
It is thus reasonable to construct a test of association and linkage, on the basis of Ui. We will denote the number of families with one heterozygous parent as F1 and the number of families with two heterozygous parents as F2. When the family is noted as the independent unit, a reasonable test statistic is
 |
where 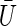 is the mean of the F1+F2 values of Ui and where Var0(Ui) is the variance of the random variable Ui, under the null hypothesis. Knowledge of the underlying genetic model would be needed to compute Var0(Ui) exactly. This type of information will rarely be available; therefore, an estimate must be obtained from the data. Since the expectation of Ui is 0 under the null hypothesis, an estimate of Var0(Ui) is given as
is the mean of the F1+F2 values of Ui and where Var0(Ui) is the variance of the random variable Ui, under the null hypothesis. Knowledge of the underlying genetic model would be needed to compute Var0(Ui) exactly. This type of information will rarely be available; therefore, an estimate must be obtained from the data. Since the expectation of Ui is 0 under the null hypothesis, an estimate of Var0(Ui) is given as 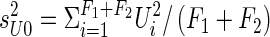 . It is noted that the Ui do not have to be identically distributed, as long as their expectation is 0. An example of such a situation arises when sampling families with information on different numbers of children. Using this estimate in our statistic from equation (2), we have
. It is noted that the Ui do not have to be identically distributed, as long as their expectation is 0. An example of such a situation arises when sampling families with information on different numbers of children. Using this estimate in our statistic from equation (2), we have
 |
Under the alternative hypothesis, the expectation of Ui will be nonzero, and the estimate of variance will no longer be correct. If the number of families sampled is large, then the distribution of TQP can be approximated by a normal random variable with a nonzero mean and with unit variance multiplied by a factor that is a function of VarA(Ui), which is the variance of Ui under the alternative hypothesis, and the expectation of s2U0 under the alternative hypothesis:
 |
where  and
and 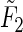 are the expected values of F1 and F2, respectively. That is,
are the expected values of F1 and F2, respectively. That is,
and
From this approximation, we can compute the power for a given model as well as the average sample size needed to achieve a specified power. Let
 |
and let
 |
so that TQP≈WQP×γQP. Substituting equations (1), (3), and (4) into the expectation of WQP from equation (5), we get
 |
Suppose that we are interested in testing the alternative hypothesis that there is positive association (D>0) between the marker allele A1 and the QTL allele that causes high trait values, and suppose that we are assuming that this corresponds to the expectation of Ui being positive. If we let zα denote the value such that Pr(Z⩾zα)=α, where Z is a standard normal random variable, then power is given by
 |
Given a marker and QTL model, along with the type I error rate (α), power can be computed by use of a standard normal distribution.
Of greater interest is the calculation of the sample size needed for a specified power of 1-β—that is, calculation of F for which Pr(TQP⩾zα)=1-β. When the above approximation is used, F must satisfy:
 |
Only E(WQP) is a function of F. Solving equation (6) for F, we get:
 |
It is noted that, although t does not appear explicitly in the formula for F, it will affect the variance of Ui and the expectation of s2U0. Formulas for these are not shown; however, a program that calculates these quantities—along with power or sample size—is available from the authors (University of Washington School of Public Health and Community Medicine Biostatistics).
Next, consider the case for which no parental-genotype information is available. Since inference of parental genotypes in an unknown mixture of populations is not straightforward, we chose not to infer parental genotypes. We instead used informative families to indicate the presence of at least one parent that is heterozygous at the marker. A family is informative if there are at least two children with differing marker genotypes. The probability of an informative family with t children is
 |
We define the following random variable for the ith family:  . This is analogous to Ui with X*iM(XijM-.5)+X*iF(XijF-.5) replaced by an estimate. Conditional on the family being informative, the expectation of Vi is
. This is analogous to Ui with X*iM(XijM-.5)+X*iF(XijF-.5) replaced by an estimate. Conditional on the family being informative, the expectation of Vi is
 |
See Appendix C for the derivation. It is interesting to note that
 |
and, so, as t increases, the expected value of Vi approaches that of Ui. Following the same reasoning used in the construction of TQP, we define a statistic on the basis of an estimate of the variance of Vi under the null hypothesis. Without loss of generality, suppose that the first FI of the F families sampled are informative. Under the null hypothesis, the expectation of Vi is zero, so that an estimate of the null variance is s2V0=(ΣFIi=1V2i)/FI. Let  be the mean of the Vi for the FI informative families. Our statistic is
be the mean of the Vi for the FI informative families. Our statistic is
 |
TQS is asymptotically standard normal under the null hypothesis. As was the case for Ui, under the alternative hypothesis, the expectation of Vi will be nonzero, and, for large samples, an approximation can be used to compute power and sample size. Denote the variance of Vi, under the alternative hypothesis, as σ2VA, and denote the expected number of informative families as  . Then the approximate power of the TQS test, for a test of positive association with type I error α, is given by
. Then the approximate power of the TQS test, for a test of positive association with type I error α, is given by
 |
where Z is a standard normal random variable. For power equal to 1-β, we have a required sample size of
 |
It is straightforward to combine the two types of family data. Let FP represent the number of families for which there is parental-genotype information and in which at least one of the parents is heterozygous. Let FS represent the number of informative families that do not have parental-genotype information. Define the following test statistic as
 |
Our use of this statistic combines the two types of families by giving more weight to the most-sampled type. As before, TQPS is asymptotically standard normal under the null hypothesis and will have the same properties as TQP and TQS. Following the preceeding work for TQP and TQS, it is straightforward to compute either the power for a given model or the sample size required for a specified power. The only additional information needed for these computations is what fraction of the families will (or will not) have parental information.
Permutation Procedures
For small samples, permutation procedures can be used to determine significance levels for the TQP, TQS, and TQPS tests. These procedures can also be used to determine either the significance of extensions to multiallelic markers or extensions that utilize more than one marker.
Under the null hypothesis, the probability that a heterozygous parent transmits marker allele A1 to a child with trait value Y is equal to .5. Thus, if the mother is heterozygous, then, for child j with trait value Yij, XijM is equally likely to be 0 or 1. If there is only one child, then a permutation procedure can be based on random assignment of XijM as being equal to 0 or 1 with equal probability. Complications arise when more than one child in the family has been sampled. These complications are a result of linkage between the marker and the QTL. In the presence of linkage, children with shared marker alleles will have similar quantitative traits, even in the absence of association. This can be taken into account by simultaneous randomization of XijM (and, similarly, of XijF), for heterozygous parents across the sibship. Consider the XijM for family i. Given the vectors TiM=[Xi1M,Xi2M,⋅⋅⋅,XitM]′ and 1-TiM=[1-Xi1M,1-Xi2M,⋅⋅⋅,1-XitM]′, a permutation procedure can be constructed by randomization between TiM and 1-TiM. It is easy to show that this procedure is equivalent to randomization of the sign of UiM. An analogous procedure holds for UiF with vectors TiF and 1-TiF. If the parental contributions to Ui cannot be determined, then the sign of Ui can instead be randomized.
A similar scenario arises when parental-genotype information is not available. First, consider the following form of Vi
 |
where  (
( ) is the fraction of
) is the fraction of  contributed by the mother (father). Noting that Vi will be nonzero only if at least one of the parents is heterozygous, suppose that only the mother is heterozygous. Randomization between the TiM and 1-TiM, for the mother, will be valid under the null hypothesis. While we cannot determine these vectors, this randomization is equivalent to randomization of the sign of Vi. An analogous case exists if only the father is heterozygous. If both parents are heterozygous, then there are four equally likely permutations:
contributed by the mother (father). Noting that Vi will be nonzero only if at least one of the parents is heterozygous, suppose that only the mother is heterozygous. Randomization between the TiM and 1-TiM, for the mother, will be valid under the null hypothesis. While we cannot determine these vectors, this randomization is equivalent to randomization of the sign of Vi. An analogous case exists if only the father is heterozygous. If both parents are heterozygous, then there are four equally likely permutations:
-
1.
TiM and TiF
-
2.
TiM and 1-TiF
-
3.
1-TiM and TiF
-
4.
1-TiM and 1-TiF.
Since we do not know the values for these vectors, we cannot randomize among the four permutations; however, we can randomize between permutations 1 and 4. This is again equivalent to randomization of the sign of Vi.
This results in a unified permutation procedure. Given UiM, UiF, Ui, or Vi for a family, a permutation procedure is based on randomization of the sign of the observed value. We suggest use of a Monte Carlo approximation to measure significance. Details are given elsewhere (Monks et al. 1998).
Simulation Parameters
We considered 18 QTL models. Heritability (H2) was equal to .1, .3, or .5. The QTL allele Q1 had a population frequency of .1 or .5. We studied QTLs with additive, dominant, and recessive modes of inheritance. By setting these parameters, the means for the trait distributions for individuals with QTL genotypes Q1Q1, Q1Q2, and Q2Q2 were uniquely determined. Conditional on an individual’s genotype, the trait distribution is normal, with the appropriate mean and variance, σ2E, equal to 1.
Marker allele A1 had a population frequency of .5 or .8 and was completely linked to the QTL. The disequilibrium coefficient was set to 0 for simulations under the null hypothesis, and it was set to its maximum value for simulations under the alternative hypothesis.
We derived formulas for the power of the TQP and TQS tests. The powers for the TR test with t=1 and for the TA test with t=2 can be computed from the formulas for the TQP and TQS tests, respectively. For estimates of significance level, we simulated 1,000,000 data sets, to achieve precision to three decimal places. For estimates of the significance level for the permutation procedures for TQP and TQS, we used 10,000 simulated data sets with 99 permutations. For estimates of significance level within a stratified population, we also used 10,000 simulated data sets. All estimates correspond to a one-sided test of positive association, with a significance level of α=.01.
Results
Significance Levels of TR and TA for Increasing t
Table 1 contains estimates of the significance level for the TR test, for a QTL with additive mode of inheritance. Estimates are based on 500 families that all have the same number of children t. We show estimates for t=1,⋅⋅⋅,6. For t=1, the TR test is valid as a test of association, and, so, our estimates are equal to the actual significance level of 0.01. As t increases, the actual level of significance increases as a result of the nonvalidity of the TR test. The increase becomes more extreme as heritability, H2, increases. In particular, consider the QTL/marker model with Pr(Q1)=.1 and Pr(A1)=.5, for sibships of size t=6. The estimate of significance level at H2=.1 is .014, which is much less than the estimate for H2=.3, which is .024. When heritability is .5, the estimate of significance level, .034, is even larger. Estimates of significance are also given, for our TQP test, for families with t=6. All estimates are equal to .01 and thus support the validity of the TQP test. The results for a QTL with dominant and recessive modes of inheritance were similar to those presented (data not shown).
Table 1.
Estimates of Significance Level for the TR Test
|
Estimates of Significance Level fora |
|||||||||
|
TR |
TQP |
||||||||
| H2 | Pr(Q1) | Pr(A1) | t = 1 | t = 2 | t = 3 | t = 4 | t = 5 | t = 6 | t = 6 |
| .1 | .1 | .5 | .010 | .011 | .011 | .012 | .014 | .014 | .010 |
| .1 | .1 | .8 | .010 | .011 | .011 | .012 | .013 | .014 | .010 |
| .1 | .5 | .5 | .010 | .011 | .012 | .012 | .013 | .014 | .010 |
| .1 | .5 | .8 | .010 | .011 | .012 | .012 | .013 | .014 | .010 |
| .3 | .1 | .5 | .010 | .012 | .015 | .018 | .021 | .024 | .010 |
| .3 | .1 | .8 | .010 | .012 | .015 | .018 | .021 | .024 | .010 |
| .3 | .5 | .5 | .010 | .012 | .015 | .018 | .021 | .024 | .010 |
| .3 | .5 | .8 | .010 | .012 | .015 | .018 | .021 | .024 | .010 |
| .5 | .1 | .5 | .010 | .014 | .018 | .024 | .029 | .034 | .010 |
| .5 | .1 | .8 | .010 | .014 | .018 | .024 | .029 | .034 | .010 |
| .5 | .5 | .5 | .010 | .014 | .019 | .023 | .028 | .034 | .010 |
| .5 | .5 | .8 | .010 | .014 | .019 | .024 | .028 | .034 | .010 |
Estimates are based on 1,000,000 simulated samples of 500 families with t children, for a QTL with additive mode of inheritance. Estimates for the TQP test, with t=6, are also given.
Table 2 contains estimates of the significance level for the TA test, for a QTL with additive mode of inheritance. Estimates are based on 500 families, all of which have the same number of children. We give estimates for t=2,⋅⋅⋅,6. For t=2, the TA test is a valid test of association, as our estimates confirm. As t increases, the actual level of significance also increases, as a result of the nonvalidity of TA. As was the case for TR, this increase becomes greater as heritability increases. For the same model mentioned above, estimates of significance are .014, .024, and .037 for heritability of .1, .3, and .5, respectively. Estimates of significance are also given, for our TQS test, for families with t=6; in all cases, the estimates are equal to .01. The results for a QTL with dominant and recessive modes of inheritance were similar to those presented (data not shown).
Table 2.
Estimates of Significance Level for the TA Test
|
Estimates of Significance Level fora |
||||||||
| TA |
TQS |
|||||||
| H2 | Pr(Q1) | Pr(A1) | t = 2 | t = 3 | t = 4 | t = 5 | t = 6 | t = 6 |
| .1 | .1 | .5 | .010 | .011 | .012 | .013 | .014 | .010 |
| .1 | .1 | .8 | .010 | .011 | .012 | .013 | .014 | .010 |
| .1 | .5 | .5 | .010 | .011 | .012 | .013 | .014 | .010 |
| .1 | .5 | .8 | .010 | .011 | .012 | .012 | .014 | .010 |
| .3 | .1 | .5 | .010 | .014 | .017 | .020 | .024 | .010 |
| .3 | .1 | .8 | .010 | .014 | .017 | .020 | .024 | .010 |
| .3 | .5 | .5 | .010 | .014 | .017 | .020 | .024 | .010 |
| .3 | .5 | .8 | .010 | .013 | .017 | .020 | .024 | .010 |
| .5 | .1 | .5 | .010 | .017 | .024 | .031 | .037 | .010 |
| .5 | .1 | .8 | .009 | .017 | .023 | .030 | .037 | .010 |
| .5 | .5 | .5 | .010 | .017 | .024 | .030 | .037 | .010 |
| .5 | .5 | .8 | .010 | .017 | .024 | .030 | .037 | .010 |
Estimates are based on 1,000,000 simulated samples of 500 families with t children, for a QTL with additive mode of inheritance. Estimates for the TQS test, with t=6, are also given.
The amount of increase from the expected level of significance, for the TR and TA tests, will depend on more than heritability. The QTL model, marker model, number of children, expected level of significance (in this case, .01), and sample size will all affect the increase. Unfortunately, we will not usually know the parameters of our model and, therefore, will not know the impact of nonvalidity. All that can be stated is that the level of significance will be larger than that which is expected.
Comparison of the TQP and TR Tests
We have established that the TQP test is a valid test of association, regardless of the number of children in the family that have been sampled (see the Methods section above). Although it is expected that the TQP test based on information for all children will be a more-powerful test of association than will the TR test based on information for a single child, to what extent this will occur is unknown. To investigate this, we compared the power of the TQP test done with samples of families with t children with the power of the TR test done with samples of families with one child. Figure 2 contains power curves for the QTL/marker models with additive mode of inheritance and heritability equal to .1, for the TR test and for the TQP test, with t = 2–5. It is noted that the TQP statistic reduces to the TR statistic when t=1. The four models provide power comparisons over a range of values for the disequilibrium coefficient. Specifically, figure 2A–D shows disequilibrium coefficients equal to .02, .05, .1, and .25, respectively. Panels A–D demonstrate that, as the disequilibrium increases, the sample size required for reasonable power decreases. However, despite the level of disequilibrium, the relationship between the TQP and TR tests is clear. Significant information is lost by use of only families of minimal size—that is, families where t=1. As an example, consider figure 2B, for which Pr(A1)=.5 and Pr(Q1)=.1. For 80% power, the TQP test requires 404 families with five children, whereas the TR test requires 1,813 families or, with respect to genotyping, 2,828 genotypes, compared with 5,439 genotypes, respectively. From a planning standpoint, more than four times as many singleton families, compared with families with five children, need to be collected for 80% power. While it is clear that a good deal of power is gained by use of more than one child per family, it is also apparent that, with each additional child used, there is a diminishing gain in power. The largest gain in power is obtained by use of two children per family rather than one. The increase in power gained from use of three—rather than two—children is also substantial; however, information gained from an increase beyond three children per family continues to diminish. For model 2C, the number of families required for 80% power are 808, 417, 287, 221, and 182 for families with one, two, three, four, and five children, respectively. Approximately half as many families with t=2 are needed, compared with families with t=1. This is a sizable decrease, compared with the ∼20% fewer families required with families with five children compared with those with four children. Although most of the gain in power is derived from increasing t from 1 to 2 and from 2 to 3, it is striking to note that, by use of five children—rather than just one child—per family, only 23% as many families are needed.
Figure 2.

Estimated power for the TQP and TR tests of association, for a QTL with additive mode of inheritance and heritability equal to .1. For figures A–D, 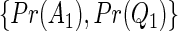 was set equal to
was set equal to 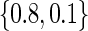 ,
, 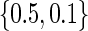 ,
, 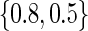 and
and  , respectively. The TQP (dashed lines) test is based on samples of families with two, three, four, or five children (power curves are indicated by increasing power with t), whereas the TR (dotted line) test is based on families with one child. A solid line indicates power equal to 0.8.
, respectively. The TQP (dashed lines) test is based on samples of families with two, three, four, or five children (power curves are indicated by increasing power with t), whereas the TR (dotted line) test is based on families with one child. A solid line indicates power equal to 0.8.
Comparison of the TQS and TA Tests
We also compared the TQS test, in which sibships of size three, four, and five were used, with the TA test, in which sibships of size two were used. The tests were compared with regard to the QTL/marker models in figure 2 (results not shown). Table 3 contains the values of the ratio Ft/F2, where Ft is the number of families required for the TQS test, with samples of sibships of size t, to have 80% power, and where F2 is the number of families required for the TA test to have 80% power. Ratios are given for the four models of figure 2, with t = 2–5. The TQS statistic is equal to the TA test statistic, when t=2. Conclusions reached from these results are identical to those achieved when TQP is compared with TR: considerable information is obtained by use of more than two children per family, and the largest gains in power result from an increase from t=2 to t=3 or from t=3 to t=4. We see that, for each of the models, approximately half as many families are required for TQS with t=3 as are required for TA. Approximately one-third as many families are required for TQS with t=4 as are required for TA. Both of these decreases in required sample size are considerable; however, we again see that the increase in power and, therefore, the decrease in sample size, diminish as t increases.
Table 3.
Sample-Size Ratios for the TQS and TA Tests, at 80% Power
|
Ratioa for Models in Figure 2 |
|||||
| t | A | B | C | D | |
| 2 | 1.000 | 1.000 | 1.000 | 1.000 | |
| 3 | .506 | .510 | .511 | .522 | |
| 4 | .343 | .348 | .350 | .364 | |
| 5 | .261 | .268 | .270 | .286 | |
Ratio of the sample size required for the TQS test, with samples of families with t children, to achieve 80% power to the sample size required for the TA test, with samples of families with two children. Models correspond to those of figure 2.
Comparison of the TQP and TQS Tests
The question arises, with the use of any family-based test, as to how much information is gained by genotyping of parents. To answer this question, we sampled families with a constant number of children t, with t = 2–6. We calculated the number of families, FP, required for the TQP test to have power equal to 80%. We then calculated the number of sibships, FS, needed for the TQS test to have 80% power. Table 4 contains the FP/FS ratio for the 12 marker/QTL models with an additive mode of inheritance for t = 2–6. Now the statistics TQP and TQS are composed of random variables U and V, respectively. Since V is an estimate of U, the statistic TQS is expected to be more variable than is TQP and, thus, is expected to result in a less-powerful test. The FP/FS ratio should therefore be <1. As t increases, the estimate V will improve, and, so, the ratio should increase to one. The results support this. For each of the 12 models, the ratio is smallest for t=2, and it increases with t. Consider, as an example, the model with H2= .1, q1= .5, and p1=.5. For t=2, the ratio is .524. In other words, the TQP test requires only 52.4% of the families that are required for the TQS test. However, if families with six children were sampled, the TQP test would require 86.3% of the families required for the TQS test, thus bringing into question how much effort should be given toward collection of parental genotypes. A few ratios were >1. After further investigation, we have found that, for higher values of heritability, it is possible to have the ratio >1 (results not shown). We also computed the FP/FS ratio for the same 12 marker/QTL models with dominant and recessive modes of inheritance (data not shown). Identical conclusions were reached.
Table 4.
Sample-Size Ratios for the TQP and TQS Tests, at 80% Power
|
Sample-Size Ratioa |
|||||||
| H2 | Pr(Q1) | Pr(A1) | t = 2 | t = 3 | t = 4 | t = 5 | t = 6 |
| .1 | .1 | .5 | .526 | .705 | .795 | .849 | .885 |
| .1 | .1 | .8 | .522 | .699 | .788 | .841 | .876 |
| .1 | .5 | .5 | .524 | .700 | .792 | .850 | .863 |
| .1 | .5 | .8 | .525 | .707 | .795 | .847 | .881 |
| .3 | .1 | .5 | .580 | .783 | .880 | .931 | .967 |
| .3 | .1 | .8 | .571 | .770 | .868 | .924 | .957 |
| .3 | .5 | .5 | .573 | .762 | .844 | .889 | .957 |
| .3 | .5 | .8 | .578 | .777 | .870 | .926 | .971 |
| .5 | .1 | .5 | .636 | .858 | .956 | 1.000 | 1.040 |
| .5 | .1 | .8 | .630 | .850 | .951 | 1.005 | 1.035 |
| .5 | .5 | .5 | .604 | .793 | .870 | .900 | .944 |
| .5 | .5 | .8 | .636 | .847 | .938 | .981 | 1.021 |
Ratio of the sample size required for the TQP test, with samples of t children, to achieve 80% power to the sample size required for the TQS test. All models are for a QTL with additive mode of inheritance.
Permutation Procedure for TQP and TQS
We provide evidence that our permutation procedures for TQP and TQS are valid, by estimation of the significance levels for the TQP and TQS tests for the 36 marker/QTL models. Estimates were based on data from 200 families with a constant number of children t. The range of the t value was 1–5, with the exception that TQS was not applicable for t=1. The 36×5 marker/QTL/sampling models were numbered from 1–180. Figure 3 contains a plot of the estimates of significance levels. Two SD lower and upper bounds are indicated. The significance-level estimates fall satisfactorily within two SDs of .01. This is the case for both the TQP test (fig. 3A) and the TQS test (fig. 3B).
Figure 3.

Estimates of the significance level for the TQP and TQS tests for the 36 marker/QTL models. Estimates are given for samples of 200 families of constant size t, where t = 1–5. The 36×5 models were numbered 1–180. Note that TQS is not applicable for t=1.
Validity of the TQP and TQS Tests under Stratification
To demonstrate that the TQP and TQS tests are valid when there is population stratification, we simulated a population that is a mixture of two homogenous subpopulations. We considered the scenarios where .5 and .75 of our sampled families are from subpopulation 1. For the 12 QTL/marker models with heritability of .1, we simulated samples of 500 families with five children, for all possible assignments of one of these models to subpopulation 1 and of another of the models to subpopulation 2 (132 possibilities). Table 5 contains the mean estimate of the significance level, across these 132 models, for both the TQP test and the TQS test, when subpopulation 1 comprises .5 and .75 of the population. We have also given 95% confidence intervals for each mean. Two of the confidence intervals do not contain the expected significance level of .01. We attribute this to our χ2 approximation. The estimates deviate from .01 by very little. Furthermore, the deviation is in the opposite direction of that which would be expected as a result of problems with stratification.
Table 5.
Estimates of Significance Level for the TQP and TQS Tests in a Stratified Population
| Proportion of Sample from | Meana Estimated Significance Level ( 95% CI) for | ||
| Subpopulation 1 | Subpopulation 2 | TQP | TQS |
| .50 | .50 | .0097 (.00957, .00991) | .0097 (.00955, .00989) |
| .75 | .25 | .0099 (.00975, .01009) | .0100 (.00981, .01015) |
The mean estimated significance level for the 132 marker/QTL models (see text for details). Estimates are based on 10,000 simulated samples of 500 families with five children, for a QTL with heritability of .1.
Discussion
Family-based methods have previously been introduced for testing association of markers or candidate genes with a QTL. These tests avoid the increase in the false-positive rate that occurs in the typical case-control test if there is population stratification. However, the current family-based association tests have sampling restrictions that result in a loss of information. However, if these sampling restrictions are not followed, then the false-positive rate of the tests will increase. Furthermore, we have demonstrated, through simulation, that the amount of increase in the false-positive rate will be model dependent. Since the true underlying model is rarely known, a researcher will be unaware of the true effect of nonvalidity. This problem will be compounded as more and more markers or candidate genes are tested, since a researcher will have no sense of how many of their positive results may be in error.
We have developed three tests that are valid tests of association (and linkage) without the sampling restrictions of current tests. In addition, we have shown that a great deal of power is acquired by use of all children. In fact, for all three of our tests, the power increases when more children are used. If association tests are to be performed with the use of a previously obtained data set, then this would imply that all children should be used in the test of association. If a study is being designed, then other considerations, such as ascertainment and genotyping costs, will determine whether fewer large families or more small families should be sampled. For the majority of the models we have considered, the use of families with parental-genotype information will allow for a more-powerful test than will tests in which only sibship information is used. The size of this increase in power is largely dependent on how many children have been sampled. As the number of children in the family increases, the power gained by having parental information diminishes. In practice, a data set will contain families with and without parental genetic information. While one strategy would be to ignore the parental information and to use the TQS test, we would recommend the use of our TQPS test. Scenarios do exist where use of sibship information only can result in a more-powerful test than will use of the available parental-transmission information; however, these models generally have high heritability. Although we have shown that considerable power can be obtained by sampling more than the minimal number of children per family, it may be that current tests could be used, in conjunction with special reduction strategies, to reduce families or sibships to minimal size. One strategy would be to randomly sample the minimal number of children from each family and to compute the significance level on the basis of this reduced data set. Other strategies might sample the minimal number of children but might specify that they have extreme trait values (e.g., largest in family, a discordant pair, etc.). These methods should account for some of the power difference seen with use of all children compared with use of the minimal number of children needed (one for TR and two for TA). We are currently investigating these strategies.
For simplicity, we have assumed that all families have an equal number of children. If samples of families with an arbitrary number of children are available, then our statistics have the same algebraic form, and only the interpretation of the statistic changes. The mean of the random variable studied (Ui when parental information is available and Vi when sibship information is used) is now a weighted mean across family size, and the variance of the random variable is also a weighted variance across family size. The asymptotics rely on the number of families within each class, where class is defined by family size. If any of the classes have few families, then the normal approximation will be poor, and we would recommend the use of our permutation procedures.
The tests that we have proposed are for a diallelic marker; however, they are easily extended to multiallelic markers. There are two straightforward extensions. A statistic can be computed for each marker allele, and, as an overall statistic, the maximum of their absolute value or the sum of their squares can be used. The permutation procedures can then be used to measure significance. In addition to extensions to multiple alleles, it is possible to extend these methods to the use of multiple tightly linked markers. One could compute a statistic for each marker and then could define an overall statistic—perhaps the maximum or sum across markers. The permutation procedure can be used to measure significance, by simultaneously shuffling across the markers (Lazzeroni and Lange 1998; McIntyre et al., in press).
We have not included a technical discussion of costs. A researcher will have to weigh the genotyping and ascertainment costs, to determine whether resources should be spent on sampling additional children or on sampling parental-genotype information. In terms of power, for our models, larger families with parental-genotype information provide the greatest power; however, costs will determine the ‘‘optimal’’ sampling scheme. Allison (1997) has provided a discussion of costs associated with ascertainment, genotyping, and phenotyping.
Acknowledgments
The authors would like to thank Dr. Bruce S. Weir, for numerous helpful discussions. S.A.M. would particularly like to thank the National Institute of Environmental Health Sciences, for training through an Intramural Research Training Award. This material is also based on work supported under a National Science Foundation graduate fellowship (to S.A.M.).
Appendix A: Effect of Population Stratification on the Expectation of Ui
Consider a population that is composed of B subpopulations, where the probability of a random family from subpopulation b is φb. Suppose that there is random mating within each of the subpopulations, so that each subpopulation will have an expectation and variance of Ui specific to that subpopulation’s allele frequencies and linkage-disequilibrium coefficient. Denote the expectation and variance as μb and σ2b, respectively. The expectation and variance of Ui, for a random family, are therefore E(Ui)=ΣBb=1φbμb and
 |
Under the null hypothesis, there is no association, within each subpopulation, between alleles at the marker locus and the QTL. It follows, from equation (1) in the text, that the expectation of Ui, for each of the subpopulations, is 0—that is, μb=0 for b=1,...,B. Thus, the expectation and variance of Ui, for a random family, are 0 and 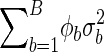 , respectively. The estimate of variance used in the construction of TQP is an estimate of this variance, so that TQP will be asymptotically standard normal under the null hypothesis. A discussion of the effects of population stratification and admixture is provided elsewhere (Ewens and Spielman 1995).
, respectively. The estimate of variance used in the construction of TQP is an estimate of this variance, so that TQP will be asymptotically standard normal under the null hypothesis. A discussion of the effects of population stratification and admixture is provided elsewhere (Ewens and Spielman 1995).
Appendix B: Derivation of the Expectation of Ui
Families with one or two heterozygous parents provide within-family information about association between alleles at the marker locus and QTL. We begin by considering families with one heterozygous parent. Without loss of generality, assume that the mother is heterozygous for the marker. Then the random variable Ui reduces to 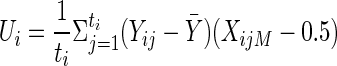 . Let pi|r be the conditional marker-allele probability Pr(Ai|Qr). Denote the mother’s marker/QTL haplotypes as HiM1 and HiM2. Let tr→Qr represent the event that allele Qr has been transmitted to the given individual. Denote the probability that a marker-homozygous parent transmits the QTL allele, Q1, to a child, by use of
. Let pi|r be the conditional marker-allele probability Pr(Ai|Qr). Denote the mother’s marker/QTL haplotypes as HiM1 and HiM2. Let tr→Qr represent the event that allele Qr has been transmitted to the given individual. Denote the probability that a marker-homozygous parent transmits the QTL allele, Q1, to a child, by use of  , where
, where  , and denote the probability that it transmits a Q2, by use of
, and denote the probability that it transmits a Q2, by use of 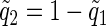 . Conditional on X*iM=1, X*iF=0 and with the assumption that
. Conditional on X*iM=1, X*iF=0 and with the assumption that 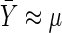 and ti=t,
and ti=t,
 |
We obtain the same derivation for families in which only the father is heterozygous. Thus, for families with one heterozygous parent:
For families with two heterozygous parents, we need the probability that a marker-heterozygous parent transmits the QTL allele, Q1, to a child. Denote this probability as  , where
, where 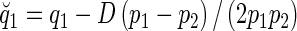 , and denote the probability that a Q2 is transmitted as
, and denote the probability that a Q2 is transmitted as 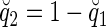 . We can write Ui as the sum of two components:
. We can write Ui as the sum of two components:
 |
Thus, conditional on X*iM=X*iF=1 and assuming that the parents are of the same genetic background, we have E(Ui)=2E(UiM). The above derivation can be used to compute the expectation for UiM, with one alteration. Transmissions from the other parent are now from a heterozygous parent, and, so,  (
(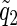 ) must be replaced by
) must be replaced by  (
( ). From this, we get
). From this, we get
Using equations (B1) and (B2), we can derive the expectation of Ui for a family with at least one heterozygous parent:
 |
Thus, we have
 |
Appendix C : Derivation of the Expectation of Vi for an Informative Family
As was the case for Ui, the random variable Vi is the sum of two components, one from the mother (ViM) and one from the father (ViF):
 |
where 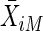 (
( ) is the part of
) is the part of 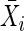 corresponding to the mother (father). There are three types of family defined by the number of heterozygous parents. Obviously, if family i has no heterozygous parents, then E(Vi)=0. Next, consider families with exactly one heterozygous parent. Without loss of generality, suppose that the mother is heterozygous. Then, conditional on X*iM=1, X*iF=0 and letting
corresponding to the mother (father). There are three types of family defined by the number of heterozygous parents. Obviously, if family i has no heterozygous parents, then E(Vi)=0. Next, consider families with exactly one heterozygous parent. Without loss of generality, suppose that the mother is heterozygous. Then, conditional on X*iM=1, X*iF=0 and letting  and ti=t, we have
and ti=t, we have
 |
 |
Thus, we have
For families with two heterozygous parents, we have E(Vi)=2E(ViM) (with the assumption that the parents are from the same population). However, E(ViM) is not that of equation (C1), since we are conditioning on two heterozygous parents. It can be shown that, conditional on there being two heterozygous parents,
The TQS test is recommended when parental information is not available, and, so, there will be no knowledge of how many of the family’s parents are heterozygous. All that can be determined is whether a sibship is informative for the marker. Thus, we need the expectation of Vi, conditional on a family being informative. Using equations (C1) and (C2), we get
 |
where
Electronic-Database Information
The URL for data in this article is as follows:
- University of Washington School of Public Health and Community Medicine Biostatistics, http://www.biostat.washington.edu/ steph/PROGRAMS/qtlassoc.html
References
- Allison DB (1997) Transmission-disequilibrium tests for quantitative traits. Am J Hum Genet 60:676–690 [PMC free article] [PubMed]
- Allison DB, Heo M, Kaplan N, Martin ER (1999) Sibling-based tests of linkage and association for quantitative traits. Am J Hum Genet 64:1754–1763 [DOI] [PMC free article] [PubMed]
- Bickeboller H, Clerget-Darpoux F (1995) Statistical properties of the allelic and genotypic transmission/disequilibrium test for multiallelic markers. Genet Epidemiol 12:865–870 [DOI] [PubMed]
- Boehnke M, Langefeld CD (1998) Genetic association mapping based on discordant sib pairs: the discordant-alleles test. Am J Hum Genet 62:950–961 [DOI] [PMC free article] [PubMed]
- Curtis D (1997) Use of siblings as controls in case-control association studies. Ann Hum Genet 61:319–333 [DOI] [PubMed]
- Ewens WJ, Spielman RS (1995) The transmission/disequilibrium test: history, subdivision, and admixture. Am J Hum Genet 57:455–464 [PMC free article] [PubMed]
- Falconer DS, Mackay TFC (1996) Introduction to quantitative genetics, 4th ed. Longman Group, Harlow, Essex, England [Google Scholar]
- Horvath S, Laird NM (1998) A discordant-sibship test for disequilibrium and linkage: no need for parental data. Am J Hum Genet 63:1886–1897 [DOI] [PMC free article] [PubMed]
- Lazzeroni LC, Lange K (1998) A conditional inference framework for extending the transmission/disequilibrium test. Hum Hered 48:67–81 [DOI] [PubMed]
- Martin ER, Kaplan NL, Weir BS (1997) Tests for linkage and association in nuclear families. Am J Hum Genet 61:439–448 [DOI] [PMC free article] [PubMed]
- McIntyre LM, Martin ER, Simonson KL, Kaplan NL. Circumventing multiple testing: a multi-locus Monte Carlo approach to testing for association. Genet Epidemiol (in press) [DOI] [PubMed] [Google Scholar]
- Monks SA, Kaplan NL, Weir BS (1998) A comparative study of sibship tests of linkage and/or association. Am J Hum Genet 63:1507–1516 [DOI] [PMC free article] [PubMed]
- Rabinowitz D (1997) A transmission disequilibrium test for quantitative trait loci. Hum Hered 47:342–350 [DOI] [PubMed]
- Schaid DJ (1996) General score tests for associations of genetic markers with disease using cases and their parents. Genet Epidemiol 13:423–449 [DOI] [PubMed]
- Schaid DJ, Rowland CR (1998) The use of parents, sibs, and unrelated controls to detection of associations between genetic markers and disease. Am J Hum Genet 63:1492–1506 [DOI] [PMC free article] [PubMed]
- Schaid DJ, Rowland CM (1999) Quantitative trait transmission disequilibrium test: allowance for missing parents. Genet Epidemiol 17:S307–S312 [DOI] [PubMed]
- Sham PC, Curtis D (1995) An extended transmission/disequilibrium test (TDT) for multi-allele marker loci. Ann Hum Genet 59:323–336 [DOI] [PubMed]
- Spielman RS, Ewens WJ (1996) The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet 59:983–989 [PMC free article] [PubMed]
- ——— (1998) A sibship test for linkage in the presence of association: the sib transmission/disequilibrium test. Am J Hum Genet 62:450–458 [DOI] [PMC free article] [PubMed]
- Spielman RS, McGinnis RE, Ewens WJ (1993) Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52:506–516 [PMC free article] [PubMed]
- Weir BS (1996) Genetic data analysis II. Sinauer, Sunderland, MA [Google Scholar]